

Abstract
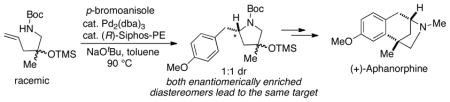
A concise asymmetric synthesis of (+)-aphanorphine has been achieved via a new enantioconvergent strategy. A racemic γ-aminoalkene derivative is transformed into a 1:1 mixture of enantiomerically enriched diastereomers using an asymmetric Pd-catalyzed carboamination. This mixture is then converted to an enantiomerically enriched protected aphanorphine derivative by a Friedel-Crafts reaction, which generates a quaternary all-carbon stereocenter. The natural product is obtained in three additional steps.
The tricyclic alkaloid aphanorphine was isolated from the blue-green alga Aphanizomenon flos-aquae in 1988 by Shimizu and Clardy.1 Although to date no biological activity of aphanorphine has been reported, aphanorphine shares common structural features with other bioactive (analgesic) benzomorphane alkaloids, such as morphine, pentazocine, and eptazocine (Figure 1). As such, aphanorphine has been the target of numerous synthetic efforts over the past twenty-three years.2,3,4
Figure 1.

Benzomorphane Alkaloids
Most approaches to the synthesis of enantiopure aphanorphine either employ chiral-pool starting materials or use stoichiometric amounts of a chiral reagent or auxiliary to set the absolute stereochemistry of the natural product. In contrast, asymmetric catalysis has only occasionally been used to control the absolute configuration in aphanorphine syntheses.2b,2g,3b,5 In these instances the stereocenter was generated via reduction or desymmetrization of a prochiral starting material.
We sought to explore an alternative, catalytic enantioconvergent route to the construction of aphanorphine, which could also potentially provide access to other interesting bicyclic alkaloids.6 Enantioconvergent transformations or strategies have significant potential synthetic utility, as they effect the conversion of both enantiomers of a racemic starting material to a single enantiomerically enriched product.7,8 We envisioned that a simple racemic starting material could be converted to enantiomerically enriched aphanorphine through use of two key transformations: an enantioselective Pd-catalyzed carboamination reaction that was recently developed in our laboratory,9 and an intramolecular Friedel-Crafts ring closure similar to that used in three prior aphanorphine syntheses.4b,10 As shown in Scheme 1, the catalyst controlled asymmetric Pd-catalyzed carboamination reaction of racemic substrate 5 could be used to set the C2 stereocenter of pyrrolidines 6a–b, thereby providing a mixture of enantiomerically enriched diastereomers. This mixture of diastereomers would then be converted to one product (8) in a Friedel-Crafts alkylation reaction, which would proceed via intermediate carbocation 7, and would generate the all-carbon quaternary stereocenter in an enantioconvergent C–C bond-forming step.11 A two-step sequence of reduction and demethylation could then be employed to transform 8 to aphanorphine.
Scheme 1.

Asymmetric Carboamination/Friedel-Crafts Alkylation Strategy
The enantioconvergent strategy outlined above has several attractive features: a) The substrate required for the asymmetric carboamination reaction could be prepared in a concise, straightforward manner; b) Analogs of the natural product could potentially be generated through use of different aryl halides in the carboamination step; c) Either enantiomer of the natural product should be accessible, as the absolute stereochemistry in the carboamination reaction would be controlled by the ligand; and d) This general strategy could potentially be adapted to provide access to a broad array of fused- or bridged bicyclic alkaloids that contain a benzylic all-carbon quaternary stereocenter.
To test the feasibility of this strategy, we elected to pursue a synthesis of the non-natural (+)-enantiomer of aphanorphine; only a few prior studies have targeted (+)-aphanorphine,4 and the ligand needed to generate this compound, (R)-Siphos-PE,12 is commercially available.13 In order to obtain 8 in high ee it would be necessary to generate a 1:1 mixture of diastereomers in the asymmetric carboamination of (±)-5, as any degree of substrate control in the reaction of (±)-5 would lead to erosion of enantiomeric purity in the final product. However, prior studies have illustrated that diastereoselectivities are generally low in Pd-catalyzed carboamination reactions of substrates bearing a single homoallylic substituent,14 and the similarity in size of the homoallylic groups in (±)-5 (methyl vs. OTMS) suggested that there would be little substrate bias towards either diastereomer. Thus, we anticipated that this approach should afford 8 with a good degree of asymmetric induction. The precursor (±)-5 for the key asymmetric alkene carboamination reaction was prepared in three steps from commercially available N-Boc-1-amino-2-propanol 9 (Scheme 2). Oxidation of 9 to ketone 10 followed by addition of allylmagnesium bromide afforded racemic tertiary alcohol 11 in good yield. Protection of the tertiary alcohol with TMS-imidazole proceeded smoothly to afford aminoalkene (±)-5.
Scheme 2.

Synthesis of Carboamination Substrate
The coupling reaction between 5 and 4-bromoanisole was examined using our previously reported conditions for enantioselective alkene carboamination reactions (Scheme 3). After slight optimization of reaction conditions,15,16 the transformation afforded a 1:1 mixture of diastereomers 6a and 6b in 75% yield. We initially sought to separate the diastereomers and assay each individually to determine enantiomeric purity. Unfortunately, separation of the isomers was not possible. In addition, routes to prepare racemic samples of each diastereomer appeared to be cumbersome. As such, we elected to transform the mixture of isomers 6a–b to known compound 13,2b–c,4b,10 which we could then assay for enantiomeric purity. To this end, treatment of 6a–b with TFA led to cleavage of both the N-Boc and O-TMS groups, and tosylation of the resulting pyrrolidine derivative provided 12a–b in 83% yield (1:1 dr) over two steps. The enantioconvergent intramolecular Friedel-Crafts alkylation of the mixture of diastereomers 12a–b provided 13 in 63% yield,4b,10 and analysis by chiral HPLC indicated the molecule had been formed with 81% ee.17
Scheme 3.

Pd-Catalyzed Carboamination of 5 and Conversion to 13.
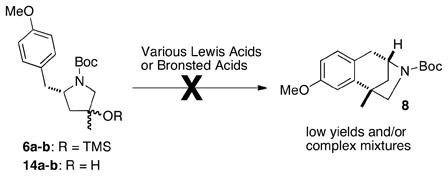
With a concise route to enantiomerically enriched diastereomers 6a–b in hand, we sought to complete the synthesis of (+)-aphanorphine. We envisioned that a Friedel-Crafts alkylation of 6a–b should provide 8, which could be transformed to the target in two steps (reduction and demethylation). Unfortunately, the Friedel-Crafts alkylation proved to be problematic (eq. 1). A variety of conditions were surveyed for reactions of 6a–b and desilylated analogs 14a–b, but all provided low yields of the desired product 8. Competing cleavage of the N-Boc group was frequently observed, along with formation of numerous side products.
 |
(1) |
Although the conversion of 6a–b to 8 was not successful, the N-tosylated derivative 13 has previously served as an intermediate in Zhai’s syntheses of aphanorphine.2b–c,4b,10 Thus, after some modification of literature procedures,16 13 was transformed to 15 in 64% yield via cleavage of the N-tosyl group with Red-Al followed by N-methylation under Eschweiler-Clarke conditions (Scheme 4). Although BBr3-mediated demethylation of 15 has been a key feature of prior syntheses,2–4 this step proved to be quite challenging.18 However, after some experimentation, we found that the conditions reported by Funk provided satisfactory results.19 With careful control of reaction temperature, and gradual warming from −30 °C to 0 °C in 10 °C increments over the course of 2 h, we were able to effect the conversion of 15 to 16 in 63% yield.
Scheme 4.

Completion of the Synthesis
In conclusion, we have developed a concise synthesis of (+)-aphanorphine 16 that affords the target molecule in 10 steps and 13% overall yield from commercially available starting materials. Importantly, this synthesis demonstrates the viability of a new, enantioconvergent strategy for the construction of benzomorphane-type alkaloids. The four-step sequence in which 5 is transformed to 13 not only effects construction of two C–C bonds, one C–N bond, and two rings, but also converts a racemic substrate to an enantiomerically enriched product. A quaternary all-carbon stereocenter is generated during the enantioconvergent C–C bond-forming step, and the product is formed with a synthetically useful level of stereocontrol. Further studies on the expansion of this strategy and its extension to other targets of interest are currently underway.
Supplementary Material
Acknowledgments
The authors acknowledge the NIH-NIGMS for financial support of this work (GM-071650). Additional funding was provided by GlaxoSmithKline, Amgen, and Eli Lilly. The authors thank Professors R. L. Funk (Department of Chemistry, The Pennsylvania State University) and J. R. Fuchs (Department of Medicinal Chemistry, The Ohio State University) for helpful discussions.
Footnotes
Supporting Information Available Experimental procedures, characterization data for all new compounds, descriptions of stereochemical assignments, and copies of 1H and 13C NMR spectra for all new compounds reported in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gulavita N, Hori A, Shimizu Y, Laszlo P, Clardy J. Tetrahedron Lett. 1988;29:4381–4384. [Google Scholar]
- 2.For recent syntheses of (−)-aphanorphine, see: Medjahdi M, González-Gómez JC, Foubelo F, Yus M. Eur Org Chem. 2011:2230–2234.Donets PA, Goeman JL, Van der Eycken J, Robeyns K, Van Meervelt L, Van der Eycken EV. Eur J Org Chem. 2009:793–796.Yang X, Cheng B, Li Z, Zhai H. Synlett. 2008:2821–2822.Yang X, Zhai H, Li Z. Org Lett. 2008;10:2457–2460. doi: 10.1021/ol800737d.Grainger RS, Welsh EJ. Angew Chem Int Ed. 2007;46:5377–5380. doi: 10.1002/anie.200701055.Li M, Zhou P, Roth HF. Synthesis. 2007:55–60.Bower JF, Szeto P, Gallagher T. Org Biomol Chem. 2007;5:143–150. doi: 10.1039/b614999e.
- 3.For syntheses of (−)-aphanorphine prior to 2007, see: Takano S, Inomata K, Sato T, Takahashi M, Ogasawara K. J Chem Soc, Chem Commun. 1990:290–292.Bower JF, Szeto P, Gallagher T. Chem Commun. 2005:5793–5795. doi: 10.1039/b510761j.and references cited therein.
- 4.For syntheses of (+)-aphanorphine, see reference 2a and: Ma Z, Zhai H. Synlett. 2007:161–163.Ma Z, Hu H, Xiong W, Zhai H. Tetrahedron. 2007;63:7523–7531.Katoh M, Inoue H, Honda T. Heterocycles. 2007;72:497–516.Katoh M, Inoue H, Suzuki A, Honda T. Synlett. 2005:2820–2822.Meyers AI, Schmidt W, Santiago B. Heterocycles. 1995;40:525–529.Takano S, Inomata K, Sato T, Ogasawara K. J Chem Soc, Chem Commun. 1989:1591–1592.
- 5.a) Fadel A, Arzel P. Tetrahedron: Asymmetry. 1995;6:893–900. [Google Scholar]; b) Kita Y, Futamura J, Ohba Y, Sawama Y, Ganesh JK, Fujioka H. J Org Chem. 2003;68:5917–5924. doi: 10.1021/jo034573g. [DOI] [PubMed] [Google Scholar]; c) Taylor SK, Ivanovic M, Simons LJ, Davis MM. Tetrahedron: Asymmetry. 2003;14:743–747. [Google Scholar]
- 6.One prior synthesis of (−)-aphanorphine employed an enantioconvergent strategy in which a racemic intermediate was transformed to an enantiopure intermediate via lipase-catalyzed resolution followed by a subsequent Wharton rearrangement. This synthesis afforded the natural product in 23 steps (longest linear sequence) from commercially available materials. See: Shimizu M, Kamikubo T, Ogasawara K. Heterocycles. 1997;46:21–26.
- 7.For recent reviews on enantioconvergent synthesis, see: Turner NJ. In: Asymmetric Organic Synthesis with Enzymes. Gotor V, Alfonso I, Garcia-Urdiales E, editors. Wiley VCH; Weinheim: 2008. pp. 115–132.Mohr JT, Ebner DC, Stoltz BM. Org Biomol Chem. 2007;5:3571–3576. doi: 10.1039/b711159m.Huerta FF, Minidis ABE, Bäckvall J-E. Chem Soc Rev. 2001;30:321–331.Strauss UT, Felfer U, Faber K. Tetrahedron: Asymmetry. 1999;10:107–117.
- 8.For selected recent examples of enantioconvergent strategies in natural product syntheses, see: Bender CF, Yoshimoto FK, Paradise CL, De Brabander JK. J Am Chem Soc. 2009;131:11350–11352. doi: 10.1021/ja905387r.Petrova KV, Mohr JT, Stoltz BM. Org Lett. 2009;11:293–295. doi: 10.1021/ol802410t.Ueberbacher BJ, Osprian I, Mayer SF, Faber K. Eur J Org Chem. 2005:1266–1270.Fehr C, Galindo J, Etter O. Eur J Org Chem. 2004:1953–1957.Yoshida N, Ogasawara K. Org Lett. 2000;2:1461–1463. doi: 10.1021/ol005805q.Ducray P, Rousseau B, Mioskowski C. J Org Chem. 1999;64:3800–3801.
- 9.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157–12159. doi: 10.1021/ja106989h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Zhai H, Luo S, Ye C, Ma Y. J Org Chem. 2003;68:8268–8271. doi: 10.1021/jo0348726. [DOI] [PubMed] [Google Scholar]; b) Hu H, Zhai H. Synlett. 2003:2129–2130. [Google Scholar]
- 11.For examples of enantioconvergent processes that generate C–C bonds, see: Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018.Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2943. doi: 10.1021/cr020027w.
-
12.(R)-Siphos-PE = (11aR)-(+)-10,11,12,13-Tetrahydrodiindeno[7,1-de:1′,7′-fg][1,3,2]dioxaphosphocin-5-bis[(R)-1-phenylethyl]amine
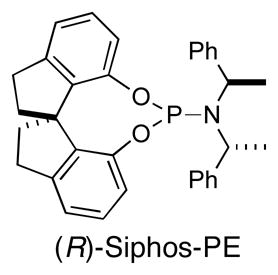
- 13.The compound sold as (S)-Siphos-PE is not the enantiomer of (R)-Siphos-PE, but is instead a diastereomer with the opposite configuration of the spirocyclic bis-phenoxide moiety but the same configuration of the bis(phenethyl)amide) group.
- 14.a) Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; b) Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447–6459. [Google Scholar]; c) Jepsen TH, Larsen M, Nielsen MB. Tetrahedron. 2010;66:6133–6137. [Google Scholar]
- 15.In our prior studies, optimal results were achieved with substrate concentrations of 0.2 M. However, best results were obtained in reactions of 5 using 0.33 M concentrations of substrate.
- 16.See the Supporting Information for complete experimental details.
- 17.The enantioselectivity of this transformation is consistent with our prior results on Pd-catalyzed enantioselective alkene carboamination reactions that generate pyrrolidine products. See reference 9.
- 18.Many of the previously described approaches to aphanorphine are formal syntheses that terminate prior to the challenging demethylation step.
- 19.Fuchs JR, Funk RL. Org Lett. 2001;3:3923–3925. doi: 10.1021/ol016795b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.