

Abstract
The mnd2 mutation on mouse chromosome 6 produces a progressive neuromuscular disorder. To determine the gene content of the 400-kb mnd2 nonrecombinant region, we sequenced 108 kb of mouse genomic DNA and 92 kb of human genomic sequence from the corresponding region of chromosome 2p13.3. Three genes with the indicated sizes and intergenic distances were identified: D6Mm5e (⩾81 kb)–787 bp–DOK (2 kb)–845 bp–LOR2 (⩾6 kb). D6Mm5e is expressed in many tissues at very low abundance and the predicted 526-residue protein contains no known functional domains. DOK encodes the p62dok rasGAP binding protein involved in signal transduction. LOR2 encodes a novel lysyl oxidase-related protein of 757 amino acid residues. We describe a simple search protocol for identification of conserved internal exons in genomic sequence. Evolutionary conservation proved to be a useful criterion for distinguishing between authentic exons and artifactual products obtained by exon amplification, RT–PCR, and 5′ RACE. Conserved noncoding sequence elements longer than 80 bp with ⩾75% nucleotide sequence identity comprise ∼1% of the genomic sequence in this region. Comparative analysis of this human and mouse genomic DNA sequence was an efficient method for gene identification and is independent of developmental stage or quantitative level of gene expression.
[The sequence data described in this paper have been submitted to the GenBank data library under the following accession numbers: AC003061, mouse BAC clone 245c12; AC003065, human BAC clone h173(E10); AF053368, mouse Lor2 cDNA; AF084363, 108-kb contig from mouse BAC 245c12; AF084364, mouse D6Mm5e cDNA.]
The mouse mutation mnd2 causes an autosomal recessive disorder characterized by muscle atrophy and wasting (Jones et al. 1993). Homozygous mice exhibit unsteady gait, growth retardation, and juvenile lethality. The mnd2 mutation is located on mouse chromosome 6 in a region corresponding to human chromosome band 2p13.3. We localized the mnd2 gene previously to a nonrecombinant interval of 0.2 cM and generated a 400-kb P1 and BAC contig of the region (Weber et al. 1998). Eight genes were identified in the nonrecombinant region and seven were eliminated from further consideration as candidate genes because of their normal expression pattern and coding sequence. Characterization of the gene D6Mm5e was complicated by the very low abundance of the transcript. To determine the complete structure of D6Mm5e and to identify additional candidate genes for mnd2, we initiated large-scale genomic sequencing of the nonrecombinant region.
Determining the complete gene content of the nonrecombinant interval in positional cloning is challenging because exons comprise a small fraction of genomic DNA and the available experimental methods for isolating exons are inefficient and labor intensive. Identification of genes that are expressed at a very low level is particularly difficult. Comparative large-scale sequence analysis is a newly feasible method for annotation of human genomic sequence. Comparison of 1196 orthologous mouse and human full-length mRNAs revealed an average of 85% nucleotide and protein sequence identity in the coding regions (Makalowski et al. 1996). Because coding sequences are among the most highly conserved in mammalian genomic DNA, they can be readily detected when the corresponding genomic sequences of human and mouse are compared (Hardison et al. l997). The effectiveness of this approach has been demonstrated in several recent studies (Galili et al. 1997; Gottlieb et al. 1997; Oeltjen et al. 1997; Ansari-Lari et al. 1998). We have combined exon amplification, cDNA isolation by RT–PCR, large-scale mouse genomic sequence analysis, and mouse/human comparative sequence analysis to identify genes in the mnd2 nonrecombinant region.
RESULTS
Identification of the Coding Exons of D6Mm5e
D6Mm5e was identified originally by exon amplification of a P1 clone in the mnd2 nonrecombinant region (Weber et al. 1998). Additional exons were isolated by RACE and RT–PCR using polyA+ RNA from muscle, brain, and testis. Three-prime RACE from the original exon consistently generated products containing the exons designated 7, 8, and 9 (Table 1). However, 5′ RACE experiments generated multiple products containing different combinations of 12 exons with a complex pattern of apparent alternative splicing (Jang 1998). The abundance of the D6Mm5e transcript is too low for detection on Northern blots. To identify additional exons using exon-prediction software, we obtained the sequence of mouse BAC clone 245 (Fig. 1). Analysis of the 108-kb sequence contig with the GENSCAN program predicted the position of the first coding exon, exon 1 (Table 1), which had not been recovered experimentally.
Table 1.
Exon Structure of Mouse D6Mm5e
| Exon | Intron–EXON boundary | Exon size (bp) | EXON Intron boundary | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c | t | t | t | c | c | t | t | a | g | G | C | T | C | A | T | A | A | . | . | . | 298 | . | . | . | C | C | A | T | T | T | G | T | G | g | t | a | a | g | t | |||||||
| 2 | c | t | c | t | c | t | g | c | a | g | C | A | A | G | T | G | A | G | A | . | . | . | 186 | . | . | . | T | C | C | C | T | C | G | A | G | g | t | a | a | g | a | ||||||
| 3 | t | t | c | a | t | t | t | t | a | g | A | T | T | A | C | T | G | T | C | . | . | . | 169 | . | . | . | A | A | T | G | A | T | G | g | t | a | a | g | a | ||||||||
| 4 | t | c | t | t | c | t | g | c | a | g | A | G | A | G | T | T | C | T | . | . | . | 174 | . | . | . | T | C | A | T | C | C | A | A | g | t | a | c | a | c | ||||||||
| 5 | t | t | t | a | t | t | g | t | a | g | T | A | T | G | T | C | T | A | . | . | . | 163 | . | . | . | G | T | G | G | T | C | A | A | g | t | a | c | g | c | ||||||||
| 6 | t | c | t | t | t | t | c | c | a | g | G | G | C | T | C | T | G | A | . | . | . | 142 | . | . | . | T | G | C | T | T | G | C | T | G | g | t | g | a | g | t | |||||||
| 7 | t | t | t | g | t | t | g | c | a | g | A | A | G | A | G | A | G | A | C | . | . | . | 207 | . | . | . | A | T | C | A | T | A | G | A | G | g | c | a | a | g | t | ||||||
| 8 | t | c | c | a | c | c | c | c | a | g | A | G | C | A | C | C | C | T | A | . | . | . | 141 | . | . | . | C | T | T | A | G | A | A | A | G | g | t | a | c | c | t | ||||||
| 9 | t | t | c | t | t | t | g | c | a | g | G | G | G | G | G | T | C | A | G | . | . | . | 400 | . | . | . | A | C | A | T | A | A | T | T | A | A | A | ||||||||||
Figure 1.

Nonrecombinant region for positional cloning of mnd2. The distance between the closest recombinant markers, D6Mit164 and D6Mit211, is ∼400 kb (Weber et al. 1998). Mouse BAC 245c12 was sequenced by the random shotgun method to generate contigs of 42, 12, and 108 kb. The genes D6Mm5e, Dok, and Lor2 were identified in the 108 kb contig as described in the text (not shown to scale). Human BAC clone 173E10 was isolated by hybridization with a mouse D6Mm5e cDNA.
To determine which of the 12 internal exons isolated by 5′ RACE were evolutionarily conserved, we isolated a BAC clone containing the human ortholog, BAC 173 (E10) (Fig. 1). The sequences of the 108-kb mouse contig and the 92 kb human sequence were aligned as described in Methods. The alignment is presented as a percent identity plot (PIP) in Figure 2. This graphical representation facilitates identification of conserved exons and regulatory elements (horizontal bars) and demonstrates the arrangement of repeat elements and CpG islands.
Figure 2.
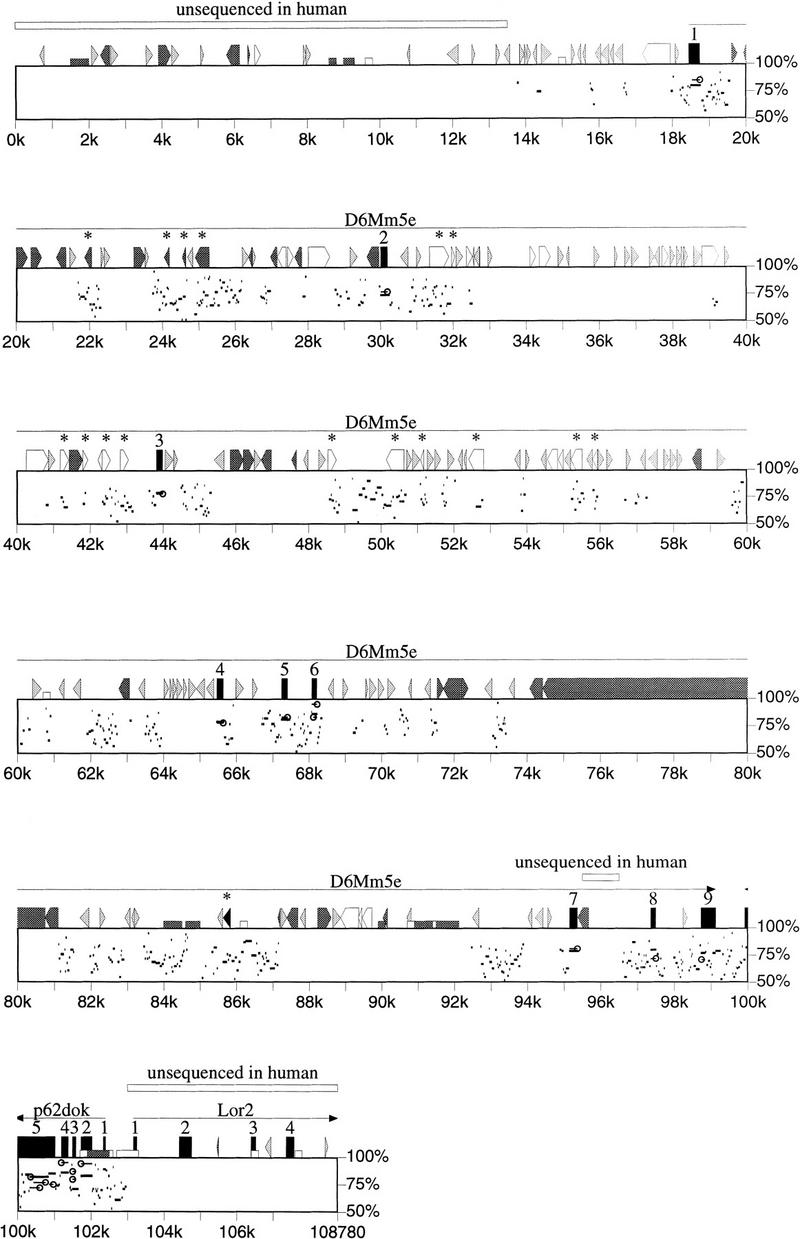
PIP of aligned mouse and human BAC sequences. The nucleotide position of the mouse sequence is shown on the horizontal axis and the percent identity of the nongapped alignment between mouse and human sequence in the range between 50% and 100% is plotted in the vertical axis. The length of the horizontal line indicates the size of the nongapped alignment. Lollipop symbols mark open reading frames that satisfy the criteria described in the text. (Numbered black boxes) Exons; (open arrows) LINE 1; (black triangles) MIR; (light gray triangle) SINEs other than MIR; (dark gray triangle) other interspersed repeats. Interspersed repeats that appear to have inserted prior to the divergence of human and mouse are marked with asterisks. CpG islands in the mouse sequence are represented by short boxes: (open) CpG/GpC ⩾ 0.6; (dark gray) CpG/GpC ⩾ 0.75.
Nine exons of D6Mm5e were well conserved in the human sequence, with nucleotide identities of 74%–86% for the human and mouse exons (Fig. 3A). Except for the 5′ untranslated region (UTR), these exon alignments are gap-free and include coding sequences and splice sites. Splice sites for the conserved exons are in good agreement with consensus sequences (Wu and Krainer 1996) with the exception of the donor site for exon 7, which begins with the dinucleotide GC in the human and mouse gene. When RNA from mouse brain, muscle, and testis was amplified with a forward primer from exon 1 and reverse primer from exon 9, a single product containing the nine conserved exons and an open reading frame of 1644 bp was obtained. The combination of evolutionary conservation and experimental verification by RT–PCR indicate that this is the functional transcript of D6Mm5e.
Figure 3.

Amino acid sequences of D6Mm5e from mouse and human. The mouse protein sequence is indicated on the top line. The human protein sequence was predicted from genomic sequence. Comparison was performed using the GCG Bestfit program (Wisconsin Package 1997). The aligned proteins exhibit 72% sequence identity and 77% similarity.
The distance between exons 1 and 9, the first and last coding exons, is 81 kb in the mouse gene and 82 kb in the human gene. The positions of the nine conserved exons are indicated in Figure 2. The sequences around the conserved translation start site located in exon 1 (GCTGCCATGA in mouse, GCTGCCATGC in human) agree well with the Kozak consensus (Kozak 1989). Within 20 bp upstream of this initiation site, the conservation of the open reading frame is disrupted by gaps of 1 and 7 bp. A consensus splice-acceptor site is located within 50 bp upstream of the initiation methionine in both species (see Table 1), indicating that the rest of the 5′ UTR is contained in a nontranslated exon. The sequence of human BAC 173 extends upstream of exon 1 for only 6 kb. Comparison with additional human genomic sequence will be required for detection of the 5′ UTR and promoter.
Direct Prediction of Internal Exons of D6Mm5e by Human/Mouse Genomic Sequence Comparison
The 81-kb mouse gene and 82-kb human gene were analyzed together to predict exons that met the following criteria: (1) an ORF of ⩾80 bp that comprises ⩾50% of a gap-free region of alignment; (2) minimal amino acid similarity of 70%; and (3) minimal DNA identity of 50%. Sequences that satisfied these criteria were scanned for the dinucleotides AG and GT located at conserved positions within the ORF. Ten fragments within the D6Mm5e region met these criteria (Fig. 2, lollipop symbols). In 2 of the 10 predicted exons the ORF was present in reverse orientation. The seven internal exons of D6Mm5e described above were correctly predicted, and the 3′ exon was predicted with an incorrect splice donor site. This simple combined search of human and mouse genomic sequence thus efficiently identified the exons in the experimentally verified transcript.
Expression of D6Mm5e
The predicted nucleotide sequence of the human D6Mm5e transcript contains 1633 bp of coding sequence and 266 bp of 3′ UTR. There is 79% nucleotide sequence identity within the coding region of the human and mouse genes and 72% identity in the 3′ UTR. The encoded proteins demonstrate 72% amino acid sequence identity and 77% sequence similarity (Fig. 2). Analysis of the amino acid sequence with the programs Motif, BLOCK (Henikoff and Hennikoff 1994), and Profilescan (Gribskov et al. 1987; Bucher et al. 1996) did not identify any known protein domains. The TmPred program predicts one transmembrane domain containing amino acid residues 44–63, with equal probability for the two possible transmembrane orientations. There is a single matching EST in the public databases (GenBank accession no. W88205) isolated from a fetal cDNA library (E11.5–E14.5). This EST exhibits 98% sequence identity with D6Mm5e (396/402 nucleotides).
D6Mm5e appears to be expressed at very low abundance in a variety of tissues. Using two rounds of amplification for 40 cycles each with nested primers (see Methods), the complete ORF from exons 1 to 9 was amplified from polyA+ RNA from brain, muscle, testis, lung, stomach, thymus, and fetal RNA from day E10.5 to E14.5 (not shown). No hybridizing transcripts could be detected on Northern blots containing 20 μg of polyA+ RNA from mouse muscle and brain that were probed with the full-length mouse cDNA, although strong signals were obtained with several control probes (not shown). Expression in fetal RNA was also below the level required for detection on Northern blots. The failure to detect transcripts in several cDNA libraries, and the absence of related human ESTs in the databases, are consistent with the conclusion that D6Mm5e is expressed at a very low level in most tissues.
Evolutionarily Conserved Noncoding Sequences, a Processed Pseudogene, and Orthologous Repetitive Sequence Elements in the Introns of D6Mm5e
In addition to the exons, nine other conserved sequence blocks with lengths of ⩾80 bp and nucleotide sequence identity of ⩾75% (Fig. 4) were identified in the genomic DNA between exons 1 and 9. These sequence blocks do not contain open reading frames and are not recognized as exons by GRAIL or GENSCAN. In view of the much higher nucleotide mutation rate for nonfunctional human and mouse sequences, this degree of sequence conservation is evidence of function. These short conserved sequences accounted for ∼1% of the genomic sequence analyzed here.
Figure 4.
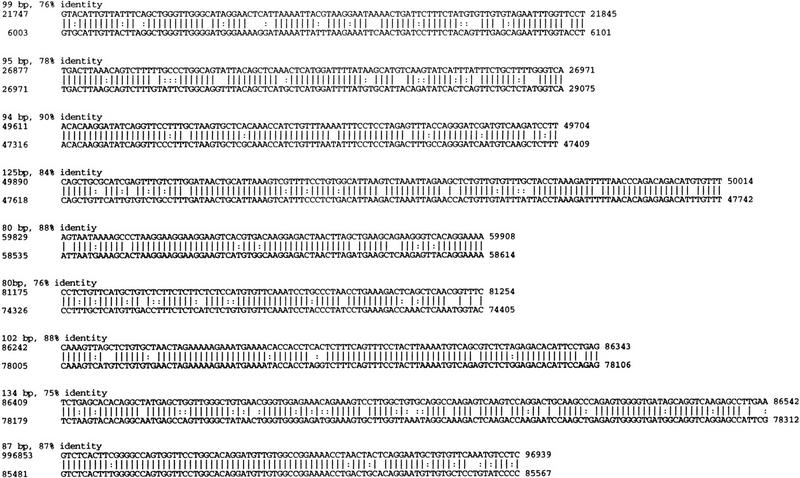
Evolutionarily conserved sequence blocks in the introns of D6Mm5e. Nine conserved elements ⩾80 bp in length with nucleotide sequence identity ⩾75% were identified. The sequences of the mouse elements (GenBank accession no. AF084363; top line) are aligned with the human sequence (GenBank accession no. AA003065). The first and last nucleotides of each element are numbered. Eight elements are located at corresponding positions in human and mouse. The coding sequence of mouse D6Mm5e begins in exon 1 at nucleotide 18441 and the polyadenylation signal begins at nucleotide 99125 in exon 9.
A 4-kb partially processed pseudogene is located between exons 1 and 2 of the human gene (nucleotides 24066–28039, GenBank accession no. AC003065). The pseudogene exhibits 85% nucleotide sequence identity with the human dystonia gene torsion B, including exon 2, intron 2, exon 3, exon 4, and exon 5 (GenBank accession no. AF007872; Laurie Ozelius, Massachusetts General Hospital, pers. comm.). The mouse gene does not contain any torsion B-related sequences.
To look for orthologous interspersed repetitive elements in the human and mouse DNA, we masked the human Alu elements and mouse B1/B2/B3/B4 elements before aligning the two genomic sequences. One LTR element, several LINE 1 elements, and one MIR appear to have inserted before human/mouse divergence (Fig. 2, asterisks). The LTR and the LINE 1 elements have been fragmented by more recent insertion events.
Complete Sequence of the Dok Gene from Mouse and Human
To identify other genes within the 108-kb mouse genomic contig, we carried out a BLAST search against the nonredundant nucleotide database. We identified the start site for the Dok gene encoding the RasGAP binding protein p62dok that functions in tyrosine-kinase signaling 787 bp downstream of D6Mm5e (Carpino et al. 1997; Yamanashi and Baltimore 1997). Comparison with cDNA sequences (mouse, GenBank accession no. U78818; human, GenBank accession no. U70987) identified 5 exons of Dok that span 2.4 kb of mouse genomic DNA (nucleotides 99,912–102,332 in GenBank accession no. AF084363) and 2.8 kb of human genomic DNA (nucleotides 88,774–91,560 in GenBank accession no. AC003065). Gene structure is conserved in the two species and all splice sites conform to the GT–AG consensus.
We determined Dok coding sequences from the mouse BAC clone that was isolated from strain 129/Sv, and from mouse cDNA isolated by RT–PCR from strain C57BL/6J. Both of these sequences differ from the published C57BL/6J cDNA sequence (GenBank accession no. U78818) at two codons. Our data indicate that residue 1148 is actually glutamic acid (not alanine), and residue 1157 is aspartate (not alanine).
Identification of the Novel Lysyl Oxidase-Related Gene Lor2
BLAST search of the expressed sequence tag (EST) database identified a mouse EST (GenBank accession no. AA522066 ) that matches three BAC segments with overall nucleotide identity of 568/569. The 2.8-kb EST clone was sequenced and found to contain an open reading frame of 2262 bp with 124 bp of 5′ UTR and 485 bp of 3′ UTR (GenBank accession no. AF053368). The context of the first ATG codon at nucleotide 125 (CCCGCCATGA) matches the Kozak consensus sequence for an optimal translation initiation site (Kozak 1989). The BAC clone contains the first 4 exons of Lor2 with 0.8 kb of cDNA sequence. The first exon of Lor2 begins 845 bp downstream of Dok and is transcribed in the opposite orientation (Fig. 1).
By BLASTP search, amino acid residues 531–730 of the predicted protein demonstrated 50% amino acid identity and 65% similarity to the enzyme lysyl oxidase [GenBank accession nos. M94054 (human), M65142 (mouse), and M97881 (chicken)]. The gene was designated Lor2, lysyl oxidase-related protein 2. Alignment of Lor2 with two human proteins, lysyl oxidase and lysyl oxidase-related protein WS9-14, is shown in Figure 5. The domain structure of mouse Lor2 is similar to that of WS9-14 (Saito et al. 1997). Cleavage of the predicted 26 residue amino-terminal signal peptide sequence of Lor2 (von Heijne 1986) would generate a mature protein of 726 amino acids. Ten cysteine residues from the lysyl oxidase domain and eight out of 10 residues of the putative copper-binding site (WEWHSCHQHYH) are conserved in Lor2, including the four histidine residues involved in copper-binding coordination (Krebs and Krawetz 1993). Analysis of mouse Lor2 with ProfileScan identified four copies of the 100-amino acid speract receptor cysteine-rich domain (SRCR) (Resnick et al. 1994) (overlined in Fig. 5).
Figure 5.
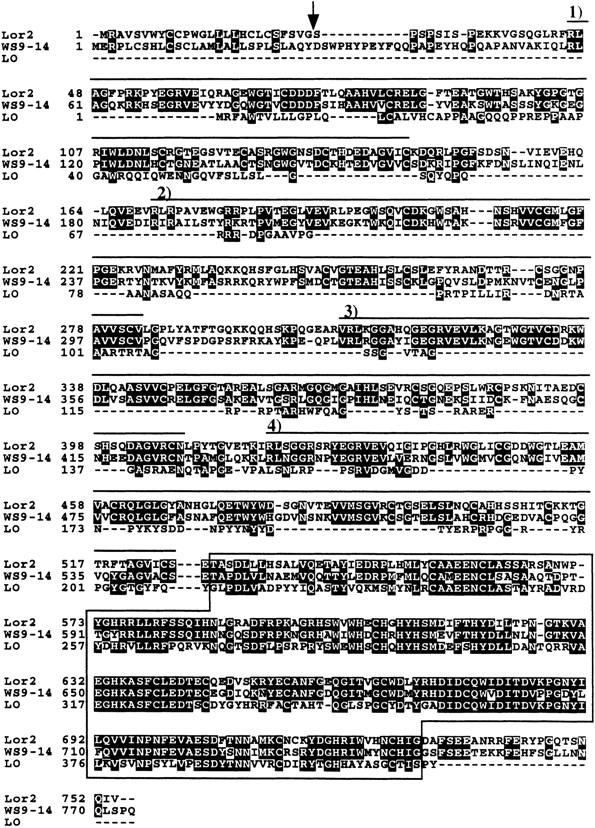
Amino acid sequence alignment of mouse Lor2 and two related human proteins. The predicted sequence of mouse Lor2 (GenBank accession no. AF053368) is aligned with the human enzyme lysyl oxidase (GenBank accession no. M94054) and the human lysyl oxidase related protein WS9-14 (GenBank accession no. U89942). SRCR domains are overlined and numbered. A region with 50% sequence identity to lysyl oxidase is boxed. (Arrow) Predicted signal sequence cleavage site.
DISCUSSION
Our experience demonstrates the effectiveness of large-scale sequence analysis for gene identification in the context of positional cloning. The genes LOR2 and DOK were represented by multiple ESTs in public databases, but their chromosomal locations had not been determined previously and the intron/exon structure of the genes was not known. Database searching with genomic sequence from the nonrecombinant region generated information about gene organization that permitted these genes to be evaluated as positional candidates for mnd2.
Genes that are expressed at very low abundance are represented poorly in the databases and require additional methods for determining gene structure. The value of human/mouse comparative sequence for this purpose was demonstrated by the analysis of D6Mm5e. A simple search strategy for conserved open reading frames with splice sites was effective in identifying all of the experimentally verified coding exons. The GENSCAN exon prediction program, using mouse genomic sequence, correctly predicted 8 of the 9 exons of D6Mm5e. However, GENSCAN made one incorrect prediction and missed exon 7. The nonconsensus splice donor site of exon 7, with GC in positions +1 and +2 rather than the standard GT or AC (Wu and Krainer 1996), is probably responsible for this error. Our prior experimental analysis of D6Mm5e was more time consuming and less effective than the genomic sequence comparison. Extensive application of 5′ RACE and RT–PCR failed to identify the first coding exon of D6Mm5e and amplified several exons incorrectly that contained in-frame stop codons. These products may have been amplified from partially spliced or incorrectly spliced nuclear transcripts in the polyA+ RNA template, and might have been avoided by preliminary purification of cytoplasmic RNA.
In addition to coding sequences, human/mouse comparison can identify evolutionarily conserved transcriptional regulatory elements (Hardison et al. 1997). D6Mm5e is a large gene, spanning more than 81 kb in human and mouse. Approximately 1% of the intronic sequences of D6Mm5e comprise conserved sequences longer than 80 bp with >75% sequence identity in human and mouse. It is not clear whether this degree of conservation is typical for mammalian genomes. The conserved elements (Fig. 4) could encode small RNAs or function as regulatory elements in RNA processing, transcription, or chromatin structure. There is an 87-bp element in intron 7 that demonstrates 87% nucleotide identity in human and mouse, and may contribute to recognition of the nearby nonconsensus splice donor site.
For complete evaluation of candidate genes in positional cloning, localization of the transcription start site remains problematic. Programs that effectively recognize start sites are not yet available, and experimental approaches such as primer extension and 5′ RACE are laborious, especially for low-abundance transcripts.
A Simple Method for Exon Prediction in Aligned Human and Mouse Genomic Sequence
A new program for analysis of alignments generated by the Sim program was used to predict conserved internal protein-coding exons. The criteria were set to identify open reading frames of >80 bp bounded by splice sites in both species, with ⩾70% amino acid similarity and ⩾50% nucleotide sequence identity. Ten regions that fit these criteria were identified by automatic search of 81 kb of genomic DNA. Cases of overlapping ORFs were resolved in favor of the higher similarity match. Elimination of the two exons on the opposite strand led to precise identification of all internal coding exons of D6Mm5e by this simple procedure. In the future, the search criteria may be further optimized and extended to include prediction of first and last coding exons and to incorporate GENSCAN exon predictions and database searches for large scale identification of coding and regulatory elements.
Evaluation of D6Mm5e, Lor2, and Dok as Candidate Genes for the mnd2 Mutation
The 108-kb region analyzed here represents 25% of the nonrecombinant region for the neuromuscular disease gene mnd2 (Weber et al. 1998). D6Mm5e, Lor2, and Dok were tested as candidates by Southern blotting of genomic DNA with cDNA probes and by sequencing the open reading frame and 5′ and 3′ UTRs from RT–PCR products. To compare the size and abundance of mRNAs in mutant and wild-type tissues, the Dok and Lor2 transcripts were analyzed on Northern blots. Because the D6Mm5e mRNA could not be detected by Northern blotting, we amplified each exon from genomic DNA and sequenced the splice sites. No differences between homozygous mnd2 mice and the wild-type strain C57BL/6J were detected by any of these assays, indicating that the mutant gene lies elsewhere in the nonrecombinant region. We plan to sequence the remainder of the nonrecombinant region from human and mouse clones to complete the gene inventory of this gene-rich region and identify the gene responsible for this fatal neuromuscular disorder.
Genome Annotation by Human/Mouse Comparison
The rate of sequencing of the human genome is accelerating and it is likely that most of the genomic sequence will become available in public databases during the next 5 years. Extraction of information about gene structure and function for the estimated 100,000 genes in the human genome will require methods that are efficient and generally applicable. Sequencing the corresponding mouse genomic sequences in parallel with the human provides such a method. The divergence of nonfunctional sequences during the 80 million years of evolutionary separation between mouse and human genomes is sufficient to permit the recognition of conserved sequences in exons and other functional elements. The PIP software (W. Miller, unpubl.) provides graphic representation of conserved elements that is easy to interpret. In addition to providing the complete exon content of genes, comparative sequence analysis identifies conserved sequences of currently unknown function. Annotation of these conserved sequences provides the basis for future experimental analysis of these potential functional elements.
METHODS
Isolation of Human and Mouse BAC Clones
Mouse BAC clone 245c12 was isolated by hybridization with a cDNA fragment from D6Mm5e (Weber et al. 1998). Random shotgun sequencing generated contigs of 108, 12, and 48 kb (GenBank accession no. AC003061) (Hua 1998). Analysis of the 108 kb contig (GenBank accession no. AF084363) with the RepeatMasker program indicated that nucleotide composition is 44% GC with 28% interspersed repeats and 2.3% simple sequence. Human BAC clone 173 (E10) was isolated by hybridization with a cDNA probe containing exons 5–9 of mouse D6Mm5e (Genome Systems, St. Louis, MO). Random shotgun sequencing generated 92 kb of sequence with one gap (GenBank accession no. AC003065).
RACE and RT–PCR
Rapid amplification of 3′ cDNA ends (3′ RACE) was carried out as described (Frohman 1993). Rapid amplification of 5′ cDNA ends (5′ RACE) was performed using RACE-ready cDNA from mouse muscle and testis (Clontech) with nested primers. Reverse transcriptase–polymerase chain reaction (RT–PCR) was carried out using 1-μg aliquots of poly(A)+ RNA. RNA was converted to first-strand cDNA using oligo(dT) primer or random primers with SuperScript II reverse transcriptase (GIBCO/BRL). For amplification of the mouse D6Mm5e transcript exons 1–9, first-strand cDNA was diluted with water to 50 μl. PCR was carried out using 1 μl of first strand cDNA as template with the forward primer S154F from exon 1 (CTGCCATGAACCGAAGGAAAACTAC) and the reverse primer G5-2R from exon 9 (GAAGAGGAAGAGCTGTTTCTTAGCC). The reaction was carried out in a volume of 50 μl with 40 cycles of 40 sec at 94°C, 40 sec at 65°C, and 2 min at 72°C. The second round of amplification contained 1 μl of the first-round product as template with the same forward primer and the reverse primer G261R (GCTCGAACTCTGTTGGTCTG). All other primer sequences are available by request from the authors. The EST clones W88205 and AA522066 were purchased from Research Genetics (Huntsville, AL).
DNA Sequencing
PCR products were gel purified using QIAEX (Qiagen) and both strands were sequenced using the DyeDeoxy terminator cycle sequencing kit (Perkin Elmer Applied Biosystem) with analysis on an ABI model 373A DNA Automated Sequencer in the University of Michigan Sequencing Core (R. Lyons, Director) and assembly with Sequencher software (GENCODE, Ann Arbor, MI). Large-scale sequencing was carried out as described previously (Chissoe et al. 1995; Hua 1998). BAC DNA was purified with a cleared-lysate diatomaceous earth method (Pan et al. 1994) and sequenced using the double-stranded, shotgun-based approach (Bodenteich et al. 1994). Sequences were screened to eliminate vector, assembled into contiguous fragments, and proofread using the Phred/Phrap/Consed system developed by P. Green (http://chimera.biotech.washington.edu/uwgc/). Contigs larger than 1 kb were deposited before publication in the unfinished division of the high-throughput genome sequencing (HTGS) GenBank database with no restriction on public access. Accession numbers are AC003061 for mouse BAC clone 245c12 and AC003065 for human BAC clone h173. Completion of the BAC sequences is in progress.
Computer Software and Sequence Analysis
Database searches were performed using the BLAST network service of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/BLAST/). The RepeatMasker program (A.F.A. Smit and P. Green, http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) was used to block the repeats prior to submitting the sequence to the BLAST server using PowerBLAST (Zhang and Madden 1997). For protein-coding-region recognition in the genomic sequences, GRAIL2 (Xu et al. 1994) and GENSCAN (Burge and Karlin 1997) were used. The GRAIL2 program was implemented through the e-mail server located at grail@grailsrv.lsd.ornl.gov. The GENSCAN program was accessed through the World Wide Web site http://gnomic.stanford.edu/GENSCANW.html. Prediction of transmembrane regions was performed using the TMpred program (http://ulrec3.unil.ch/software/TMPRED_form.html). The Profile search was carried out using the ProfileScan server (http://ulrec3.unil.ch/software/PFSCAN_form.html). Protein sequence motifs were searched for using the MOTIF program (http://www.genome.ad.jp/SIT/MOTIF.html). Prediction of signal peptides was carried out using WWW server (http://psort.nibb.ac.jp).
Mouse/Human Sequence Comparison
Repeat elements were masked by the RepeatMasker program and genomic sequences were aligned using a modified version of the Sim program (Huang et al. 1990) with the default parameters (+1 for a match, −1 for a mismatch, and −6–0.2 k for a gap of length k). For another view of the alignment, regions between successive gaps were converted into segments of percent identity relative to positions in the mouse sequence, and the resulting data were drawn as a PIP using local alignment to postscript (LAPS) (Fig. 2). Only segments with an identity of 50% or more were plotted, so regions that match poorly appear blank (Fig. 2). A pairwise alignment of the cDNA sequences was performed using the GCG Bestfit program (Wisconsin Package 1997).
Acknowledgments
We thank Jane Santoro and Emily B. Harkins for manuscript preparation. We are grateful to Laurie Ozelius for providing unpublished information about the human torsion B gene structure and for pointing out the pseudogene in the intron of D6Mm5e. A Northern blot of mouse embryonic RNA was provided by Douglas Mortlock and Jeffrey Innis. This work was supported by a grant from the Muscular Dystrophy Association (M.H.M.), U.S. Public Health Service grants GM24872 (M.H.M.), HG00313 (B.A.R.), and National Library of Medicine grant LM05110 (W.M.). W.J. was recipient of a predoctoral fellowship from the Center for Organogenesis, University of Michigan.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL meislerm@umich.edu; FAX (734) 763-9691.
REFERENCES
- Ansari-Lari MA, Oeltjen JC, Schwartz S, Zhang Z, Muzny DM, Gorrell Lu, JH, Chinault AC, Belmont JW, Miller W, Gibbs RA. Comparative sequence analysis of a gene-rich cluster at human chromosome 12p13 and its syntenic region in mouse chromosome 6. Genome Res. 1998;8:29–40. [PubMed] [Google Scholar]
- Bodenteich A, Chissoe S, Wang YF, Roe BA. Shotgun cloning as the strategy of choice to generate templates for high throughput dideoxynucleotide sequencing. In: Adams MD, Fields C, Venter C, editors. Automated DNA sequencing and analysis techniques. London, UK: Academic Press; 1994. pp. 42–50. [Google Scholar]
- Bucher P, Karplus K, Moeri N, Hofmann K. A flexible search technique based on generalized profiles. Comput Chem. 1996;20:3–24. doi: 10.1016/s0097-8485(96)80003-9. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Carpino N, Wisniewski D, Strife A, Marshak D, Kobayashi R, Stillman B, Clarkson B. p62dok: A constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 1997;88:197–204. doi: 10.1016/s0092-8674(00)81840-1. [DOI] [PubMed] [Google Scholar]
- Chissoe SL, Bodenteich A, Wang YF, Wang YP, Burian D, Clifton SW, Crabtree J, Freeman A, Iyer K, Jian L, et al. Sequence and analysis of the human ABL gene, the BCR gene, and regions involved in the Philadelphia chromosomal translocation. Genomics. 1995;27:67–82. doi: 10.1006/geno.1995.1008. [DOI] [PubMed] [Google Scholar]
- Frohman MA. Rapid amplification of complementary DNA ends for generation of full length complementary DNAs: Thermal RACE. Methods Enzymol. 1993;218:340–356. doi: 10.1016/0076-6879(93)18026-9. [DOI] [PubMed] [Google Scholar]
- Galili N, Baldwin HS, Lund J, Reeves R, Gong W, Wang Z, Roe BA, Emanuel BS, Nayak S, Mickanin C, et al. A region of mouse chromosome 16 is syntenic to the DiGeorge, velocardiofacial syndrome minimal critical region. Genome Res. 1997;7:17–26. doi: 10.1101/gr.7.1.17. [DOI] [PubMed] [Google Scholar]
- Gottlieb S, Emanuel BS, Driscoll DA, Sellinger B, Wang Z, Roe B, Budarf ML. The DiGeorge syndrome minimal critical region contains a goosecoid-like homeobox gene which is expressed early in human development. Am J Hum Genet. 1997;60:1194–1201. [PMC free article] [PubMed] [Google Scholar]
- Gribskov M, McLachlan AD, Eisenberg D. Profile analysis: Detection of distantly related proteins. Proc Natl Acad Sci. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997;7:959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Protein family classification based on searching a database of blocks. Genomics. 1994;19:97–107. doi: 10.1006/geno.1994.1018. [DOI] [PubMed] [Google Scholar]
- Hua A. “Sequencing the distal end of the DiGeorge syndrome critical region on human chromosome 22.” Ph.D. thesis. Norman, OK: University of Oklahoma; 1998. [Google Scholar]
- Huang X, Hardison R, Miller W. A space-efficient algorithm for local similarities. Comput Appl Biosci. 1990;6:373–381. doi: 10.1093/bioinformatics/6.4.373. [DOI] [PubMed] [Google Scholar]
- Jang W. “Transcript characterization in the region of the mouse neuromuscular mutation mnd2.” Ph.D. thesis. Ann Arbor, MI: University of Michigan; 1998. [Google Scholar]
- Jones JM, Albin RL, Feldman EL, Simin K, Schuster TG, Dunnick WA, Collins JT, Chrisp CE, Taylor BA, Meisler MH. mnd2: A new mouse model of inherited motor neuron disease. Genomics. 1993;16:669–677. doi: 10.1006/geno.1993.1246. [DOI] [PubMed] [Google Scholar]
- Krebs CJ, Krawetz SA. Lysyl oxidase copper-talon complex: A model. Biochem Biophys Acta. 1993;1202:7–12. doi: 10.1016/0167-4838(93)90056-w. [DOI] [PubMed] [Google Scholar]
- Kozak M. The scanning model for translation: An update. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makalowski W, Zhang J, Boguski MS. Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res. 1996;6:846–857. doi: 10.1101/gr.6.9.846. [DOI] [PubMed] [Google Scholar]
- Oeltjen JC, Malley TM, Muzny DM, Miller W, Gibbs RA, Belmont JW. Large-scale comparative sequence analysis of the human and murine Bruton’s tyrosine kinase loci reveals conserved regulatory domains. Genome Res. 1997;7:315–329. doi: 10.1101/gr.7.4.315. [DOI] [PubMed] [Google Scholar]
- Pan HQ, Wang YP, Chissoe SL, Bodenteich A, Wang Z, Iyer K, Clifton SW, Crabtree JS, Roe BA. The complete nucleotide sequences of the pSacBII P1 cloning vector and three cosmid cloning vectors: pTCF, svPHEP, and LAWRIST16. Genet Anal Techniques Appl. 1994;11:181–186. doi: 10.1016/1050-3862(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Resnick D, Pearson A, Krieger M. The SRCR superfamily: A family reminiscent of the Ig superfamily. Trends Biochem Sci. 1994;19:5–8. doi: 10.1016/0968-0004(94)90165-1. [DOI] [PubMed] [Google Scholar]
- Saito H, Papaconstantinou J, Sato H, Goldstein S. Regulation of a novel gene encoding a lysyl oxidase-related protein in cellular adhesion and senescence. J Biol Chem. 1997;272:8157–8160. doi: 10.1074/jbc.272.13.8157. [DOI] [PubMed] [Google Scholar]
- von Heijne G. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, Jang W, Simin K, Yu J, Meisler MH. High resolution genetic, physical and transcript map of the mnd2 region of mouse chromosome 6. Genomics. 1998;54:107–115. doi: 10.1006/geno.1998.5496. [DOI] [PubMed] [Google Scholar]
- Wisconsin Package Version 9.1. 1997. Genetics Computer Group (GCG), Madison, Wisconsin.
- Wu Q, Krainer AR. U1-mediated exon definition interactions between AT-AC and GT-AG introns. Science. 1996;274:1005–1008. doi: 10.1126/science.274.5289.1005. [DOI] [PubMed] [Google Scholar]
- Xu Y, Mural RJ, Shah MB, Uberbacher EC. Recognizing exons in genomic sequence using GRAIL II. Principles Methods Genet Eng. 1994;16:241–253. [PubMed] [Google Scholar]
- Yamanashi Y, Baltimore D. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 1997;88:205–211. doi: 10.1016/s0092-8674(00)81841-3. [DOI] [PubMed] [Google Scholar]
- Zhang J, Madden TL. PowerBLAST: A new network BLAST application for interactive or automated sequence analysis and annotation. Genome Res. 1997;7:649–656. doi: 10.1101/gr.7.6.649. [DOI] [PMC free article] [PubMed] [Google Scholar]