

Abstract
Jacobsen syndrome is a haploinsufficiency disorder caused, most frequently by terminal deletion of part of the long arm of chromosome 11, with breakpoints in 11q23.3–11q24.2. Inheritance of an expanded p(CCG)n trinucleotide repeat at the folate-sensitive fragile site FRA11B has been implicated in the generation of the chromosome breakpoint in several Jacobsen syndrome patients. The majority of such breakpoints, however, map distal to this fragile site and are not linked with its expression. To characterize these distal breakpoints and ultimately to further investigate the mechanisms of chromosome breakage, a 40-Mb YAC contig covering the distal long arm of chromosome 11 was assembled. The utility of the YAC contig was demonstrated in three ways: (1) by rapidly mapping the breakpoints from two new Jacobsen syndrome patients using FISH; (2) by demonstrating conversion to high resolution PAC contigs after direct screening of PAC library filters with a YAC clone containing a Jacobsen syndrome breakpoint; and (3) by placing 23 Jacobsen syndrome breakpoints on the physical map. This analysis has suggested the existence of at least two new Jacobsen syndrome breakpoint cluster regions in distal chromosome 11.
Jacobsen syndrome is characterized by partial aneusomy of the distal long arm of chromosome 11, most frequently caused by terminal deletion of the chromosome, with breakpoints in the interval 11q23.3–q24.2. Patients exhibit a complex phenotype, but the syndrome is characterized typically by trigonocephaly, cardiac defects, thrombocytopaenia, dysmorphism, and mental retardation (for review, see Penny et al. 1995; Pivnick et al. 1996).
It is widely accepted that the chromosome breakage characteristic of certain genetic diseases, of which Jacobsen syndrome is one example, occurs de novo with no predisposition. We have presented evidence, however, that some chromosome deletions can result from the inheritance of defined sequence variants. In a proportion of Jacobsen syndrome patients, the chromosome deletion is associated with the expansion in one parent of a p(CCG)n trinucleotide repeat at a so-called folate-sensitive fragile site, FRA11B (Jones et al. 1994, 1995; and unpubl.).
Folate-sensitive fragile sites are cytogenetic phenomena observed in a small percentage of individuals during in vitro cell culture under conditions of folate deficiency. The fragile site manifests as a nonstaining gap in the metaphase chromosome that is susceptible to breakage (Sutherland and Hecht 1986). There is no evidence to suggest that such fragile sites are expressed in vivo, however. Folate-sensitive fragile sites have been shown to be attributable to the dramatic expansion of a p(CCG)n trinucleotide repeat (Kremer et al. 1991; Verkerk et al. 1991; for review, see Richards and Sutherland 1997). The p(CCG)n trinucleotide repeat at FRA11B is located in the 5′-untranslated region of the CBL2 proto-oncogene in chromosome sub-band 11q23.3. It is found most frequently in the population as an 11-copy repeat (i.e., n = 11) and almost always as a repeat of <50 copies, but is expanded to hundreds of copies in fragile site expressors (Jones et al. 1995).
In key Jacobsen syndrome patients, chromosome 11 is truncated very near to the site of FRA11B and the deleted chromosome derives from a parent carrying an expanded p(CCG)n trinucleotide repeat at FRA11B (Jones et al. 1995; and unpubl.). Chromosome truncation at a fragile site suggests a novel mechanism of chromosome breakage in vivo and challenges the dogma that congenital chromosome abnormalities are purely de novo events. It may also indicate a predisposition toward chromosome rearrangements in the offspring of certain individuals.
Although the fragile site FRA11B is linked with some Jacobsen syndrome breakpoints, this represents only ∼10% of cases (Michaelis et al. 1998; C. Jones, F.E. Cotter, and A. Tunnacliffe, unpubl.). Other breakpoints map more distally in chromosome 11q and are not apparently associated with FRA11B (Penny et al. 1995; Michaelis et al. 1998; C. Jones, F.E. Cotter, and A. Tunnacliffe, unpubl.). The question therefore arises as to whether these chromosome breakage events occur at random, or whether, like those mapping to FRA11B, there is evidence for involvement of specific sequences.
To address this question, it was critical to develop a physical mapping tool for distal chromosome 11q, which would allow us to rapidly and accurately map Jacobsen syndrome breakpoints from large numbers of patients; to search for sequences that could be implicated in the mechanism of chromosome breakage; and to clone out those sequences and surrounding regions. To this end, a high-resolution yeast artificial chromosome (YAC) contig was developed that covers the region of the Jacobsen syndrome breakpoints and other chromosome abnormalities and genetic diseases associated with the distal long arm of chromosome 11. In this paper the YAC contig, which extends over an estimated 40 Mb, has been used to map the breakpoints of two new Jacobsen syndrome patients, to rapidly convert a YAC clone into a high-resolution PAC contig covering one of these breakpoints, and to correlate mapping information for the breakpoints of 23 patients with the physical map.
RESULTS
A YAC Contig for Distal Chromosome 11q
A high-resolution mapping tool for the characterization of Jacobsen syndrome breakpoints was generated by assembling a YAC contig for the distal long arm of chromosome 11. The CEPH mega-YAC library was screened exhaustively, with 269 STSs over the region from D11S923 (in band 11q22) to the long arm telomere. The full contig is shown in Figure 1 and, based on the fraction of chromosome 11 covered (as indicated by cytogenetic, radiation hybrid and genetic maps), it is estimated to span ∼40 Mb or ∼28% of chromosome 11. The contig consists of 254 YAC clones, ordered manually with reference to the 480-kb resolution radiation hybrid map (James et al. 1994). The contig resolves 169 positions with one to five STSs per position, with an average of six YACs for each resolved map position. The average resolution is ∼240 kb, whereas the STSs could provide for a theoretical resolution of ∼150 kb.
Figure 1.


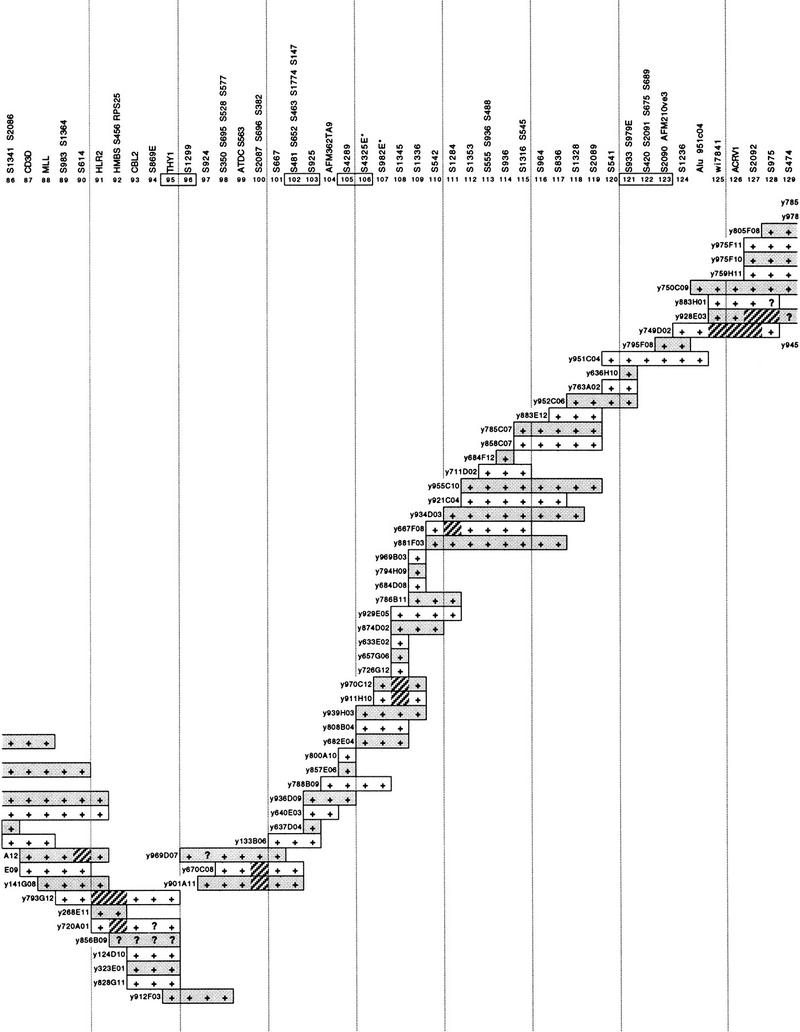
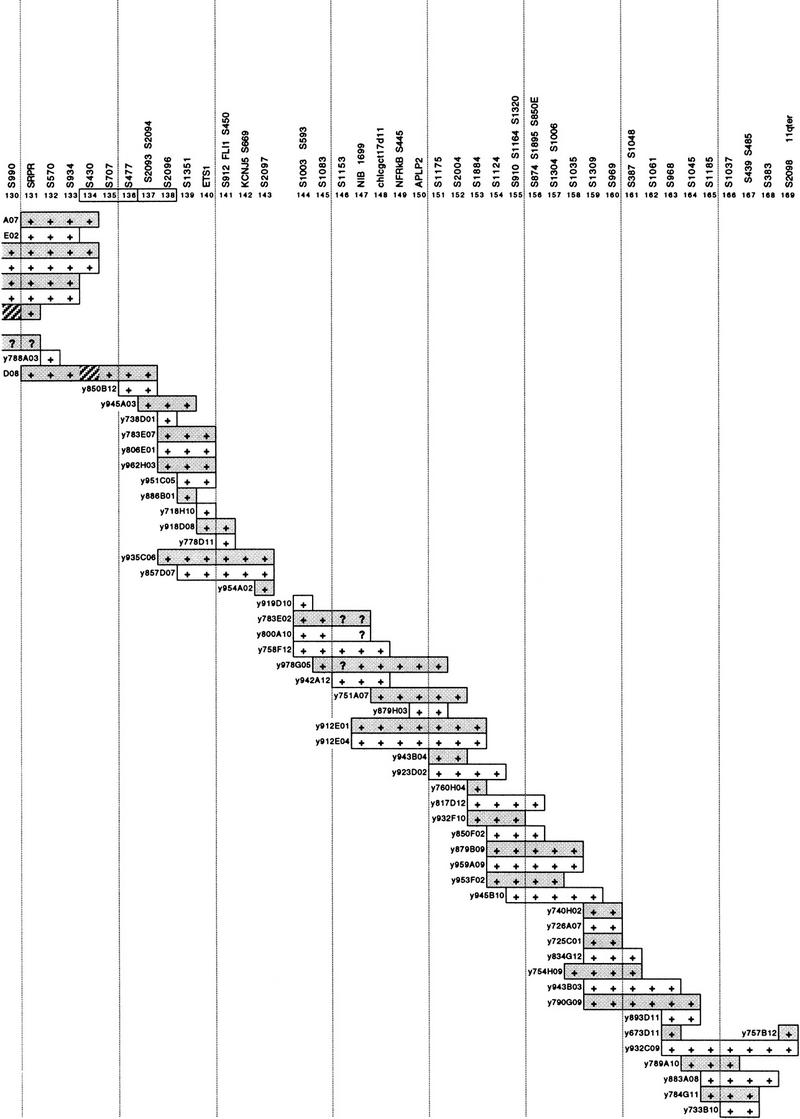
A 40-Mb YAC contig covering the distal long arm of chromosome 11. At top are the 169 resolved positions defined by the associated STSs. The prefix S in the name of STSs is an abbreviation of D11S. Alternate YACs are shaded for clarity. + and ? indicate a positive or uncertain PCR result for the STS at that position, respectively. Hatched segments indicate possible physical deletions or regions of instability in the YACs, consistently observed in different preparations of the same YAC. A discontinuity in the contig between positions 143 and 144 is indicated by a gap. Numbers of positions linked by a single YAC are boxed. Positions 106 and 107 (asterisks) had the reverse order in the PAC contig of Fig. 2.
Coverage is complete with the exception of one gap between positions 143 and 144 (Fig. 1), which we estimate to be at most a few hundred kilobases in size. The overlap between positions 124 and 125 relies on YAC-to-YAC Alu–PCR hybridization data (not shown) and is the only case in which a non-STS link is employed in the contig.
Mapping Jacobsen Syndrome Breakpoint Locations by FISH
The physical ordering of YAC clones into a contig encompassing the Jacobsen syndrome breakpoint region allows rapid physical mapping of unknown breakpoints. Metaphase chromosomes from a previously undescribed patient (RB) with the karyotype 46,XX del(11)(q23.3-qter) were analyzed by FISH using four YAC clones (Table 1): y133B06 (covering position numbers 101–103), y934D03 (positions 111–118), y951C04 (positions 120–124), and y918D08 (positions 140–141). The most proximal of these was retained on the deleted chromosome, whereas the three more distal clones were lost. This placed the breakpoint between D11S667 (position 101; the most proximal marker in y133B06) and D11S1328 (position 118; the most distal marker in y934D03). The location of the breakpoint was then defined further by FISH with y939H03 (positions 106–109), which gave a retained signal on the deleted chromosome, refining the proximal boundary of the breakpoint location to D11S4325E (position 106).
Table 1.
Summary of FISH Data
| Probe | Positions covereda | Patient RBb | Patient MCb |
|---|---|---|---|
| Initial screen | |||
| y133B06 | 101–103 | ++ | ++ |
| y934D03 | 111–118 | +− | ++ |
| y951C04 | 120–124 | +− | ++ |
| y918D08 | 140–141 | +− | +− |
| Secondary screens | |||
| y939H03 | 106–109 | ++ | N.D. |
| dJ213-J23 | 108 | ++ | N.D. |
| dJ104-A7 | 109 | ++ | N.D. |
| dJ101-N13 | — | ++ | N.D. |
| dJ261-O4 | — | +− | N.D. |
| dJ9-D18 | 111 | +− | N.D. |
| y975F11 | 127–134 | N.D. | ++ |
| dJ177-H4 | 135 | N.D. | +− |
Metaphase spreads from patients RB and MC were initially screened by FISH with a set of four YACs to give an approximate breakpoint location. This was followed by FISH experiments with YAC and PAC probes to give a higher resolution localization. YAC clones are prefixed by y; PAC clones by dJ.
The positions ordered in the YAC contig of Fig. 1 plus a location of each probe within it.
(+ +) Hybridization to both normal and deleted chromosomes; (+ −) hybridization to normal chromosome only. (N.D.) Experiment not done.
A second new patient (MC) had a 46,XY, del(11)(q23.3-qter) karyotype and was also subjected to FISH analysis with the same four YACs initially screened against patient RB (Table 1). In this case, however, the three most proximal YACs were retained on the deleted chromosome, whereas the most distal YAC, y918D08, was lost. This placed the breakpoint in this patient between D11S541 (position 120; the most proximal marker in y951C04) and D11S912/FLI1/D11S450 (position 141; the most distal markers in y918D08). The location of the breakpoint was then refined further by FISH with y975F11 (positions 127–134), which gave a retained signal on the deleted chromosome, and a PAC dJ177–h4 containing D11S707 (position 135), which was lost from the deleted chromosome. This placed the breakpoint between D11S2092 (position 127; the most proximal marker in y975F11) and D11S707.
Conversion of YACs to PAC Contigs: Further Refinement of Breakpoint Location
As a first-order physical map, the YAC contig can be used for the development of higher resolution contigs in BAC, PAC, and cosmid vectors. To demonstrate this, YACs y939H03 and y934D03, defining the breakpoint of patient RB, were used for direct hybridization screening of gridded PAC library filters. Hybridization was performed using a crude yeast miniprep of each YAC clone as a probe. Although this method does identify some PAC clones that hybridize to yeast sequences, these clones were easily recognizable, as they were consistently positive in hybridizations with YAC clones from different regions of the genome and could therefore be rejected. A total of 45 PAC clones corresponding specifically to y939H03, and 24 PAC clones corresponding to y934D03, were isolated. These were initially rescreened with STSs from the interval D11S667–D11S1328 to confirm their provenance. Selected PACs were then chosen for FISH experiments on metaphase chromosomes from patient RB to begin to refine the breakpoint location. One PAC clone, dJ9-D18, containing D11S1284 (position 111) and therefore corresponding to the proximal end of y934D03, was lost from the 11q− chromosome. Therefore, a contig was only constructed from the PAC clones corresponding to the more proximal YAC, y939H03.
Assembly of the isolated PACs into an ordered contig was performed first by screening of clones by PCR using the chromosome 11 STS markers contained within the YAC, which identified 10 positive clones and allowed an initial ordering after reference to the YAC contig and radiation hybrid map. A second stage of contig assembly was carried out by hybridization analysis of the isolated PACs. Southern blots were prepared of all 45 PAC clones digested with EcoRI, and these were sequentially probed with individual PAC clones. PAC-to-PAC hybridization generates two bands of hybridization corresponding to vector, and two bands corresponding to end fragments, between nonoverlapping PACs. Overlapping clones are clearly distinguished by the presence of additional, similar-sized bands of hybridization, and the extent of overlap can easily be determined.
This method was used to generate a PAC contig, corresponding to the region of the YAC contig covering the interval from D11S4325E (position 106) to D11S1336 (position 109), which consisted of 23 PAC clones extending over ∼750 kb (Fig. 2). FISH analysis of YAC y939H03 suggested that it was chimeric (data not shown) because in addition to signals in 11q23.3, additional signal was observed on chromosome 3p. Mapping data from the CEPH Web site (http://www.cephb.fr) confirms this, as y939H03 also contains STSs from chromosome 3p. As a measure of the efficacy of this technique, 19 PACs corresponding to the chromosome 3p sequences contained within y939H03 were also assembled into a contig (data not shown). Therefore, of the 45 PACs initially isolated, 42 clones mapped to one of the two regions contained within the YAC and could be assembled into contigs, whereas only two PACs were found not to contain inserts, and one clone was not linked to either contig.
Figure 2.

A PAC contig corresponding to YAC clone y939H03 covering the chromosome breakpoint of Jacobsen syndrome patient RB. STS markers within the contig are listed without the D11 prefix for clarity; e.g., D11S982E is shown as S982E; the prefix dJ is omitted from PAC names. Alternate PACs are shaded. YAC contig position numbers are in parentheses. STSs were demonstrated within PACs by PCR. PAC end clones are given the prefix ec, together with the clone name and end from which they are derived; they were screened against PACs by hybridization. (+) Positive results for PCR of STSs or hybridization with PAC end clones. Overlap of PAC clones in the contig was also determined by cross-hybridization of PACs against each other. (?) An unclear hybridization.
Several clones from the 11q PAC contig were then chosen for further FISH mapping to define PACs flanking and containing the RB breakpoint. PAC clones dJ213–J23, dJ104–A7, and dJ101–N13 were retained on both chromosomes, whereas clone dJ261–O4 was lost from the deleted chromosome (Table 1). These data suggest that the deletion breakpoint in patient RB is within PACs dJ101–N13 or dJ33–N1, probably distal to the marker D11S1336 (Fig. 2).
This exercise highlights potential inaccuracies in estimating spacing and relative orientation of markers in both radiation hybrid (RH) mapping and the assembly of a large YAC contig. It is clear from the assembly of our PAC contig that the relative order of markers in YAC y939H03 is D11S982E–D11S4325E–D11S1345–D11S1336. The apparent order of markers in the YAC contig, however, shows D11S982E and D11S4325E in the opposite orientation. In addition, the RH map suggests that the markers are relatively evenly spaced, whereas the PAC contig suggests otherwise. Where there is an apparent conflict in the mapping data, the higher resolution map, that is, the PAC map, is more likely to be correct.
Positioning of Jacobsen Syndrome Breakpoints on the Physical Map
Several recent reports have used haplotype analysis to map Jacobsen syndrome breakpoints. Microsatellite analysis of patient DNA can define the extent of the hemizygous region corresponding to the deletion, ideally with reference to parental DNA (Penny et al. 1995; Michaelis et al. 1998; T. Mattina, A. Tunnacliffe, and C. Jones, in prep.). The availability of the YAC contig for distal chromosome 11q gives these breakpoints a physical location and allows an initial assessment of the likelihood of breakpoint clustering at sites distal to fragile site FRA11B.
Figure 3 shows the location on the physical map of the breakpoints from 23 Jacobsen syndrome patients. Although in many cases the precision of the physical mapping is limited by the informativeness of the haplotype analysis achieved, it is apparent that a number of breakpoints can be localized to small physical intervals. For example, the breakpoint of patient VH reported by Michaelis et al. (1998) is located distal to FRA11B between D11S924 (position 97) and D11S2087/D11S696/D11S382 (position 100). This interval is contained within a single YAC, y969D07, and therefore would be amenable to conversion to a PAC contig, as demonstrated above, with a view to further refinement of the breakpoint location and its subsequent cloning. The breakpoint of AD, reported by the same authors, is between D11S924 (position 97) and D11S925 (position 103) and is contained within either the same YAC as patient VH, that is y969D07, or its overlapping neighbor, y133B06 (Fig. 1). Several patients from the group reported by Penny et al. (1995), that is, patients 4, 5, 6, 7, and 9, although not mapped with the same accuracy as patients VH and AD, could also have breakpoints in this region.
Figure 3.

Localization of 23 Jacobsen syndrome chromosome breakpoints on the YAC contig of distal chromosome 11q. (Solid bar) The intact region of the deleted chromosome with the most distal marker known to be retained (+). (Shaded bar) The maximum extent of the region in which the breakpoint must be located with the most proximal deleted marker (usually bounded by −). (X) A breakpoint is defined with the resolution of a single position on the YAC contig, i.e., to FRA11B for patients 3 and 8, and close to D11S1336 for patient RB. Patients numbered 1–17 are from Penny et al. (1995), of which patients 3 and 8 were shown to have breakpoints at fragile site FRA11B (Jones et al. 1995); patients VH and AD are from Michaelis et al. (1998); patient C is from Breton-Gorius et al. (1995); patient GS is from T. Mattina, C. Jones, and A. Tunnacliffe (in prep.); and patients RB and MC are described in this paper.
A second region that could evidence Jacobsen syndrome breakpoint clustering is represented by patient RB (this paper) and patients 11, 12, and 13 of Penny et al. (1995). For these four patients, the breakpoints fall in the interval between D11S1345 (position 108) and D11S933 (position 121; Fig. 3), although for patient RB, the best-mapped case, the breakpoint probably falls between D11S1336 (position 109) and D11S542 (position 110). If the apparent breakpoint clustering in these two regions is confirmed by fine scale mapping, it would strongly implicate previously undescribed fragile sites or other specific sequences in nonrandom chromosome breakage.
DISCUSSION
It has long been the dogma within the medical genetics community that the chromosome abnormalities typical of many birth defects initially arise de novo without predisposing factors (e.g., Gardner and Sutherland 1989). Certain chromosome instability syndromes, including ataxia telangiectasia, Fanconi anemia, and Bloom syndrome, are recognized exceptions. The chromosome rearrangements seen in such cases, however, usually occur randomly throughout the genome and are attributable to defects in DNA repair; recurring rearrangements at specific loci are unusual. The location of terminal deletions from several patients with Jacobsen syndrome within a few kilobases of each other (Jones et al. 1995; and unpubl.) was therefore surprising and led to the implication of the closely linked folate-sensitive fragile site FRA11B in the genesis of these breakpoints.
Clearly, however, only a fraction of Jacobsen syndrome breakpoints are associated with FRA11B (Jones et al. 1995; Penny et al. 1995; Michaelis et al. 1998; C. Jones, F.E. Cotter, and A. Tunnacliffe, unpubl.), with the majority being located distal to the fragile site. The question therefore arises as to the etiology of these distal breakpoints—whether they also have resulted from the inheritance of particular sequence variants, or whether they are in fact purely sporadic phenomena. To address this question, a large series of breakpoints must be mapped precisely and, where appropriate, breakpoint regions cloned and sequenced. This requires a powerful and accurate physical mapping tool for distal chromosome 11q, where the breakpoints are located. To this end, and to facilitate the genetics of the distal long arm of chromosome 11 generally, a YAC contig has been constructed extending ∼40 Mb to the long arm telomere.
The YAC contig represents a twofold increase in resolution over the RH map of the same region (James et al. 1994), with 169 positions spaced on average 240 kb apart, compared with 480 kb. It therefore demonstrates a further refinement of marker ordering for distal chromosome 11q and also provides the first-order cloning tools (i.e., the individual YACs themselves) for focusing on particular regions of interest. The contig is anchored by a large number of Généthon microsatellite markers that allow easy reference to genetic mapping data for disease cloning projects. We have also demonstrated how the YAC map can be rapidly and easily converted to a higher resolution map in PAC vectors. A 750-kb PAC contig covering a Jacobsen syndrome breakpoint region was assembled after direct screening of PAC library filters with a crude preparation of YAC DNA.
There are 15 YACs that exhibited possible instability, which are shown in Figure 1 with the problem segments highlighted. In several cases, it is clear that these problem segments are clustered, as they are observed in overlapping or neighboring YACs. Of note are the regions spanning positions 90–100 and 125–134, which together account for over half of all the problem sites. Two YACs, y738D01 and y800A10, occur in two widely separated parts of the contig (positions 59 and 138, and positions 105 and 144, respectively). These presumably represent chromosome 11–chromosome 11 chimeras and is within expectation given the overall high chimerism of the CEPH mega-YAC library.
With the exception of local inversion of order at a few positions, the agreement in order between the YAC contig, the RH map (James et al. 1994), and the genetic map (Dib et al. 1996) is very good. Furthermore, there is no conflict in order with the independently constructed contig of Arai et al. (1996) that overlaps substantially with the centromeric half of our contig. In addition to the present studies, large parts of this contig have been available to the scientific community to support studies on genetic disease (van Schothorst et al. 1996; Baysal et al. 1997) and disease-associated cytogenetic phenomena (Cherif et al. 1994; Stilgenbauer et al. 1996; Döhner et al. 1997; Koreth et al. 1999). Comparison of the terminal 10 Mb of the contig with RH and genetic maps has also been made by Yousry et al. (1999).
Where a chromosomal region of interest is represented by a breakpoint, as in Jacobsen syndrome, the most effective initial mapping approach is FISH. When used with the YAC contig, this is extremely rapid—the location of the breakpoints of two new patients was refined from 40 Mb to <1 Mb in just two FISH experiments. Further refinement of breakpoint map position is facilitated by the conversion of YACs to a PAC contig followed by further FISH experiments with PAC clones. The location of the breakpoint of patient RB within two overlapping PACs will result in rapid cloning of this breakpoint region and will allow examination of the surrounding sequence for clues to the mechanism of chromosome breakage.
The 23 Jacobsen syndrome breakpoints positioned on the physical map represented by the YAC contig all fall within the interval D11S1356/D11S976 (position 85) to D11S912/FLI1/D11S450 (position 141), an estimated distance of ∼13.5 Mb in 11q23.3–q24. Although terminal deletions may occur more distally, it is probable that the resulting phenotype would be extremely mild and therefore likely to be missed. Furthermore, it is unlikely that such cases could strictly be included under the Jacobsen syndrome umbrella in the absence of its major features. The wide variation in breakpoint location in Jacobsen syndrome is no doubt largely responsible for the high degree of phenotypic variability. There are probably >100 genes in the 13.5-Mb interval, a significant proportion of which might exhibit haploinsufficiency. It is therefore arguable that Jacobsen syndrome is not a single, clearly defined disease but a collection of genetic disorders with overlapping phenotypes.
Although only two of the patients represented in Figure 3 have breakpoints at FRA11B, two new patients have recently been identified where this fragile site is also implicated (C. Jones, F.E. Cotter, and A. Tunnacliffe, unpubl.). Nevertheless, the majority of Jacobsen syndrome breakpoints map distal to FRA11B, and an initial examination of the data for breakpoint clustering, which would be indicative of hot spots of chromosome breakage, suggests the possibility of at least two new breakpoint cluster regions: between D11S924 and D11S925, and between D11S1345 and D11S933. Further examination of these regions is under way to produce a precise definition of breakpoint locations and to search for sequences such as p(CCG)n trinucleotide repeats, which may be implicated in the structural lability of the chromosome.
METHODS
YAC Library Screening
The CEPH mega-YAC library was screened by PCR according to standard methods (Cohen et al. 1993). All primer sequence and PCR conditions for the STSs have been described (James et al. 1994; Shows et al. 1996) and are accessible from several electronic sources (ftp://ftp.well.ox.ac.uk/pub/genetics/rh11/; http://shows.med.buffalo.edu/; http://mcdermott.swmed.edu/). Clones were verified using individual YAC agarose plug DNA, and after preliminary ordering, YACs were assembled into a putative order in microtiter format and subject to repeated PCR screening for all markers until consistent results were obtained. PCR was performed as described in James et al. (1994).
Fluorescence In Situ Hybridization
EBV-transformed lymphoblastoid cell lines derived from Jacobsen syndrome patients were used as a source of metaphase chromosomes. Cells were grown in RPMI/10% fetal calf serum (GIBCO BRL), and metaphase chromosomes and spreads were prepared using standard techniques. Probes were labeled with either biotin-16–dUTP or digoxygenin-11–dUTP using a nick-translation protocol (Sambrook et al. 1989). Labeled PAC DNA (200 ng) or 500-ng labeled YAC DNA was preannealed with 20 μg of Cot1 DNA (GIBCO BRL) at 37°C for 90 min in a hybridization solution (2× SSC; 50% formamide; 10% dextran sulfate). Metaphase spreads were denatured in 70% formamide/2× SSC at 70°C for 3 min and preannealed probe was hybridized in a moist chamber at 37°C overnight. A Texas Red-conjugated chromosome 11 α-satellite probe (Oncor) was cohybridized to specifically detect both chromosomes 11. Detection of labeled probes was achieved using “sandwiches” of FITC-conjugated avidin and biontinylated anti-avidin (Vector Laboratories), or Texas Red-conjugated anti-digoxygenin (Boehringer Mannheim). Slides were mounted in antifadant solution (AF1; Citifluor) containing 10 μg/ml DAPI. Staining and hybridization signals were analyzed under a computer-controlled Nikon epifluorescence microscope equipped with a cooled charge-coupled device (CCD) camera. Computer images were merged and Q-banding (generated by DAPI staining) converted to G-banding, using IP Lab Spectrum software (Digital Scientific).
Assembly of the PAC Contig
High-density filters of the de Jong PAC library [supplied by the Human Genome Mapping Project (HGMP) Resource Centre, Hinxton, Cambridge, UK; Ioannou et al. 1994)] were hybridized with 150 ng of YAC miniprep DNA, labeled with [α-32P]dCTP (ICN) using a previously described oligo-labeling protocol (Feinberg and Vogelstein 1983). YAC-specific PAC clones were grown in 15 ml of “terrific broth” containing kanamycin (25 μg/ml), and DNA was prepared by alkaline lysis, followed by a modified protocol of purification with Hybaid midiprep columns. Primary screening was performed by PCR of 1 ng of PAC DNA using appropriate STS markers and standard protocols (Sambrook et al. 1989). Positive clones were chosen for the preliminary rounds of Southern analysis by PAC-to-PAC hybridization. Ordering of some PACs was refined by hybridization of PAC end probes, derived using a previously described PCR-based technique, bubble–PCR (Riley et al. 1990).
Acknowledgments
We thank Drs. Laura Penny and Jane Hurst for providing new patient samples, and patients RB and MC and their parents for the generous gift of blood samples. The enthusiastic support of Mrs. Annet van Betuw and the European Chromosome 11q Network is also gratefully acknowledged. We thank the CEPH YAC team for generous provision of materials and encouragement for this project; Ms. Helen Steingruber for technical assistance; and the UK HGMP Resource Centre for computing facilities and primer synthesis; special thanks go to Barbara Gorick of the HGMP Resource Centre for PAC library filters and pools. A sabbatical visit by C.J. to the Women’s and Children’s Hospital, Adelaide, Australia, was generously hosted by Professor Grant Sutherland, Dr. Rob Richards, and Ms. Liz Baker and was supported by the Reach Foundation of Great Ormond Street Hospital, London. Parts of this work were supported by European Commission grants BMH4-CT95-0914 and BMH4-CT97-2240, and by the Wellcome Trust (M.R.J.); by the Leukaemia Research Fund and the Sydney and Phyllis Goldberg Research Trust (C.J. and F.E.C.); and by the Medical Research Council (grants G9206115 and G9113915), the Isaac Newton Trust, and the Royal Society (A.T.).
Footnotes
E-MAIL at10004@biotech.cam.ac.uk; FAX 44-1223-334162.
E-MAIL james@well.ox.ac.uk; FAX 44-1865-742196.
REFERENCES
- Arai Y, Hosoda F, Nakayama K, Ohki M. A yeast artificial chromosome contig and NotI restriction map that spans the tumor suppressor gene(s) locus, 11q22.2-q23.3. Genomics. 1996;35:195–206. doi: 10.1006/geno.1996.0339. [DOI] [PubMed] [Google Scholar]
- Baysal BE, van Schothorst EM, Farr JE, James MR, Devilee P, Richard CW., III A high resolution STS, EST, and gene-based physical map of the hereditary paraganglioma region on chromosome 11q23. Genomics. 1997;44:214–221. doi: 10.1006/geno.1997.4880. [DOI] [PubMed] [Google Scholar]
- Breton-Gorius J, Favier R, Guichard J, Cherif D, Berger R, Debili N, Vainchenker W, Douay L. A new congenital dysmegakaryopoietic thrombocytopenia (Paris-Trousseau) associated with giant platelet alpha-granules and chromosome 11 deletion at 11q. Blood. 1995;85:1805–1814. [PubMed] [Google Scholar]
- Cherif D, Bernard O, Paulien S, James MR, Le Paslier D, Berger R. Hunting 11q23 deletions with fluorescence in situ hybridization (FISH) Leukemia. 1994;8:578–586. [PubMed] [Google Scholar]
- Cohen D, Chumakov I, Weissenbach J. A first generation physical map of the human genome. Nature. 1993;366:398–400. doi: 10.1038/366698a0. [DOI] [PubMed] [Google Scholar]
- Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Döhner H, Stilgenbauer S, James MR, Benner A, Weilguni T, Bentz M, Fischer K, Hunstein W, Lichter P. 11q deletions identify a new subset of B-cell chronic lymphocytic leukemia characterized by extensive nodal involvement and inferior prognosis. Blood. 1997;89:2516–2522. [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Gardner RJM, Sutherland GR. Chromosome abnormalities and genetic counseling. New York, NY: Oxford University Press; 1989. [Google Scholar]
- Ioannou PA, Amemiya CT, Garnes J, Kroisel PM, Shizuya H, Chen C, Batzer MA, De Jong PJ. A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nat Genet. 1994;6:84–89. doi: 10.1038/ng0194-84. [DOI] [PubMed] [Google Scholar]
- James MR, Richard CW, III, Schott JJ, Yousry C, Clark K, Bell J, Terwilliger JD, Hazan J, Dubay C, Vignal A, et al. A radiation hybrid map of 506 STS markers spanning human chromosome 11. Nat Genet. 1994;9:70–76. doi: 10.1038/ng0994-70. [DOI] [PubMed] [Google Scholar]
- Jones C, Slijepcevic P, Marsh S, Baker E, Langdon WY, Richards RI, Tunnacliffe A. Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum Mol Genet. 1994;3:2123–2130. doi: 10.1093/hmg/3.12.2123. [DOI] [PubMed] [Google Scholar]
- Jones C, Penny L, Mattina T, Yu S, Baker E, Voullaire L, Langdon WY, Sutherland GR, Richards RI, Tunnacliffe A. Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene CBL2. Nature. 1995;376:145–149. doi: 10.1038/376145a0. [DOI] [PubMed] [Google Scholar]
- Koreth, J., C.J. Bakkenist, Z. Larin, N.C.A. Hunt, M.R. James, and J.O.D. McGee. 1999. 11q23.1 and 11q25-qter YACs suppress tumor growth in vivo. Oncogene (in press). [DOI] [PubMed]
- Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- Michaelis RC, Velagaleti GVN, Jones C, Pivnick EK, Phelan MC, Boyd E, Tarleton J, Wilroy RS, Tunnacliffe A, Tharapel AT. Most Jacobsen syndrome deletion breakpoints occur distal to FRA11B. Am J Med Genet. 1998;76:222–228. [PubMed] [Google Scholar]
- Penny LA, Dell’Aquila M, Jones MC, Bergoffen J, Cunniff C, Fryns J-P, Grace E, Graham JM, Jr, Kousseff B, Mattina T, et al. Clinical and molecular characterization of patients with distal 11q deletions. Am J Hum Genet. 1995;56:676–683. [PMC free article] [PubMed] [Google Scholar]
- Pivnick EK, Velagaleti GVN, Wilroy RS, Smith ME, Rose SR, Tipton RE, Tharapel AT. Jacobsen syndrome: Report of a patient with severe eye anomalies, growth hormone deficiency, hypothyroidism associated with deletion 11(q23q25) and review of 52 cases. J Med Genet. 1996;33:772–778. doi: 10.1136/jmg.33.9.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards RI, Sutherland GR. Dynamic mutation: Possible mechanisms and significance in human disease. Trends Biochem Sci. 1997;22:432–436. doi: 10.1016/s0968-0004(97)01108-0. [DOI] [PubMed] [Google Scholar]
- Riley J, Butler R, Ogilvie D, Finniear R, Jenner D, Powell S, Anand R, Smith JC, Markham AF. A novel, rapid method for the isolation of terminal sequences from yeast artificial chromosome (YAC) clones. Nucleic Acids Res. 1990;18:2887–2890. doi: 10.1093/nar/18.10.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shows TB, Alders M, Bennet S, Burbee D, Cartwright P, Chandrasekharappa S, Cooper P, Courseaux A, Davies C, Devignes M-D, et al. Report of the fifth international workshop on human chromosome 11 mapping. Cytogenet Cell Genet. 1996;74:1–56. doi: 10.1159/000134383. [DOI] [PubMed] [Google Scholar]
- Stilgenbauer S, Liebisch P, James MR, Schröder M, Schlegelberger B, Fischer K, Bentz M, Lichter P, Döhner H. Molecular cytogenetic delineation of a novel critical genomic region in chromosome bands 11q22.3-23.1 in lymphoproliferative disorders. Proc Natl Acad Sci. 1996;93:11837–11841. doi: 10.1073/pnas.93.21.11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GR, Hecht F. Fragile sites on human chromosomes. New York, NY: Oxford University Press; 1985. [Google Scholar]
- van Schothorst EM, Jansen JC, Bardoel AFJ, van der Mey AGL, James MR, Sobol H, Weissenbach J, van Ommen G-JB, Cornelisse CJ, Devilee P. Confinement of PGL, an imprinted gene causing hereditary paragangliomas, to a 2-cM interval on 11q22-q23 and exclusion of DRD2 and NCAM as candidate genes. Eur J Hum Genet. 1996;4:267–273. doi: 10.1159/000472213. [DOI] [PubMed] [Google Scholar]
- Verkerk AJMH, Pieretti M, Sutcliffe JS, Fu Y-H, Kuhl DPA, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Yousry, C., D. Browne, D. Cherif, C.W. Richard III, N. Janin, V. Baud, M. Lipinski, U. Claussen, B. Horsthemke, M. Birdsall et al. 1999. Mapping of human chromosome 11qter. Cytogenet. Cell Genet. (in press).