

Structural studies revealed a proline switch as a novel mechanism for the multidrug-resistant nature of multidrug-resistant clinical isolate 769 HIV-1 protease variants.
Keywords: multidrug-resistance, HIV-1 protease, proline switch, 80s loop, expanded active-site cavity, wide-open flaps, docking
Abstract
The flexible flaps and the 80s loops (Pro79–Ile84) of HIV-1 protease are crucial in inhibitor binding. Previously, it was reported that the crystal structure of multidrug-resistant 769 (MDR769) HIV-1 protease shows a wide-open conformation of the flaps owing to conformational rigidity acquired by the accumulation of mutations. In the current study, the effect of mutations on the conformation of the 80s loop of MDR769 HIV-1 protease variants is reported. Alternate conformations of Pro81 (proline switch) with a root-mean-square deviation of 3–4.8 Å in the Cα atoms of the I10V mutant and a side chain with a ‘flipped-out’ conformation in the A82F mutant cause distortion in the S1/S1′ binding pockets that affects inhibitor binding. The A82S and A82T mutants show local changes in the electrostatics of inhibitor binding owing to the mutation from nonpolar to polar residues. In summary, the crystallographic studies of four variants of MDR769 HIV-1 protease presented in this article provide new insights towards understanding the drug-resistance mechanism as well as a basis for design of future protease inhibitors with enhanced potency.
1. Introduction
HIV-1 protease is a homodimeric aspartic protease that is critical for viral maturation (Peng et al., 1989 ▶) and infectivity (Kohl et al., 1988 ▶). Each monomer of the protease consists of 99 amino acids. Disabling the protease would significantly slow viral infection. HIV-1 protease has become one of the most important drug targets for the design of inhibitors to combat the HIV/AIDS problem owing to its critical role in the viral life cycle. Error-prone viral replication randomly incorporates multiple mutations, resulting in the emergence of multidrug-resistant strains under selection pressure from various treatment regimens. HIV-1 protease is one of the viral proteins that have been studied in detail in order to understand the drug resistance caused by various mutations. It is very important to study the structures of the apo protease in order to understand the overall stability of the protease dimer as well as the conformational flexibility of various critical domains of the protease such as the flaps, 80s loops (Pro79–Ile84) and active-site cavity. As summarized in Supplementary Table 11, a number of structures of HIV-1 protease variants are available (Lapatto et al., 1989 ▶; Navia et al., 1989 ▶; Wlodawer et al., 1989 ▶; Spinelli et al., 1991 ▶; Pillai et al., 2001 ▶; Kumar et al., 2002 ▶; Ishima et al., 2003 ▶; Logsdon et al., 2004 ▶; Martin et al., 2005 ▶; Liu et al., 2006 ▶; Heaslet et al., 2007 ▶), but only two (Logsdon et al., 2004 ▶; Martin et al., 2005 ▶; Heaslet et al., 2007 ▶) from a total of 12 structures of apo protease are crystal structures of MDR apo-protease strains (showing altered flap conformation). The first medium-resolution (Logsdon et al., 2004 ▶) and high-resolution (Martin et al., 2005 ▶) crystal structures of clinical isolate MDR769 (Palmer et al., 1999 ▶) HIV-1 protease with a wide-open conformation of the flaps was reported by our group; the second structure of an MDR strain with such an open conformation of the flaps was reported by another group (Heaslet et al., 2007 ▶).
Based on sequence analysis from the Stanford HIV database (http://hivdb.stanford.edu), it was found that among the mutations seen in the wild-type HIV-1 protease gene, 32.3% are constituted by natural polymorphisms (NPs; shown in Fig. 1 ▶ a). Such NPs are observed in strains isolated from treatment-naive patients. Once the patients are started on specific treatment regimens during HAART (highly active antiretroviral therapy), drug-resistant strains of protease emerge under clinical selection pressure. Depending on the combination cocktail used in HAART, some patients might eventually be selected for MDR strains of protease. Analysis of drug-resistance mutations (DMs) from the Stanford HIV database revealed that, based on more than 4000 clinical isolates, DMs constitute 60.6% of the protease gene. With more than 50% of the gene prone to DMs, designing inhibitors against such a drug target can be hypothetically compared with shooting a moving target.
Figure 1.

Mutations in the HIV-1 protease gene. (a) Amino-acid sequence of wild-type (NL4-3) HIV-1 protease aligned with isolate from treatment-naive patients showing the natural polymorphisms (green), the MDR769 clinical isolate (red) and isolate from patients that received treatment showing drug-resistance mutations (brown). (b) Crystal structure of MDR769 HIV-1 protease (PDB code 1tw7) showing color-coded domains: the dimerization domain is shown in brown, the active-site area is shown in red, the 80s loops are shown in green and the wide-open flaps are shown in yellow. (c) Domain diagram of the MDR769 HIV-1 protease showing the amino-acid residues that constitute the various domains. Color codes are the same as in (b). (d) The characteristic ten mutations of the MDR769 clinical isolate are highlighted as spheres; those shown as red spheres are the focus of the current study.
The MDR769 HIV-1 protease is one such challenging strain of HIV-1 protease that is resistant to almost all of the protease inhibitors available to date in HAART. The MDR769 HIV-1 protease gene consists of a set of ten mutations, all ten of which are known DMs and four of which are NPs (Fig. 1 ▶ a). Mutations such as V82A and I84V involving a change from a longer side chain to a shorter side chain cause an overall expansion of 3 Å in the active-site cavity, resulting in a loss of contacts with inhibitors. In addition to the active-site expansion, owing to conformational rigidity acquired by the MDR protease from other mutations the flaps show a wide-open conformation with an inter-flap distance (measured between the Cα atoms of Ile50 on either flap) of 12.3 Å (twice that of the wild-type protease). In combination, the expanded active-site cavity and the wide-open flaps cause a highly unstable binding of inhibitors. Thus, the MDR strains are propagated further even in the presence of the most potent inhibitors owing to compensatory mutations in substrate-cleavage sites.
The current study focuses on understanding the effects of additional point mutations on the MDR protease structure with MDR769 background (consisting of ten characteristic mutations). Fig. 1 ▶ shows a mapping of the characteristic MDR769 mutations and the additional point mutations further analyzed in this study. The active-site cavity, flaps, 80s loop and dimerization domains (Fig. 1 ▶) were mainly analyzed. Nine clinically relevant additional point mutations, I10R, I10V, I10F, L46I, V54M, V54L, A82F, A82S and A82T, were selected for further analysis with an MDR769 background. Of the nine point mutations chosen, four mutants (I10V, A82F, A82S and A82T) yielded diffraction-quality crystals. Crystallographic analysis of the four structures was mainly geared towards understanding (i) the capability of additional point mutations to cause further changes in the three-dimensional structure of the MDR769 HIV-1 protease that make the multidrug-resistance problem worse and (ii) the existence of a conserved trend among the four structures that can be targeted to design future inhibitors against an ensemble of MDR strains. The four crystal structures were further energy-minimized (in the absence of crystal contacts) using AMBER8 (Case et al., 2004 ▶) to clarify (Layten et al., 2006 ▶; Lexa et al., 2009 ▶) whether the wide-open conformation of the flaps in the structures would change to become similar to that of the wild-type protease. The inhibitor amprenavir was docked into the active-site cavities of each of the four mutant structures to determine the volume of chemical space and analyze the inhibitor-binding modes using the AutoDock Vina program (Trott & Olson, 2010 ▶).
2. Materials and methods
2.1. Protease expression and purification
Previously, the MDR769 HIV-1 protease gene with a D25N mutation was cloned into pRSET B using the clinical isolate obtained from the Center for AIDS Research, Stanford University, Stanford, California, USA to prevent autoproteolysis of the protease. In the current study, the D25N clone was used as a template to introduce additional point mutations by using mutagenic primers containing the point mutations. Mutagenesis was performed using the Multi-Site Directed Mutagenesis kit from Stratagene (La Jolla, California, USA). Protease expression and purification were performed as reported previously (Vickrey et al., 2003 ▶).
2.2. Protease crystallization
Crystallization trials were performed using the hanging-drop vapour-diffusion method at 295 K. Trials were set up using 1 µl purified protein solution (∼1.5–2 mg ml−1) mixed with an equal volume of well solution (precipitant 0.3–1.0 M sodium chloride) in the pH range 5.5–7.5. Diffraction-quality crystals of the MDR769 HIV-1 protease variants I10V, A82F, A82S and A82T were obtained within 1–3 d. The crystals have a bipyramidal morphology with dimensions of 0.1 × 0.1 × 0.05 mm and diffracted X-rays to beyond 2 Å resolution at the synchrotron (Advanced Photon Source, DND-CAT ID5B, Argonne National Laboratories, Argonne, Illinois, USA).
2.3. X-ray diffraction data collection and processing
Multiple crystals were used to collect diffraction data for each mutant. Data sets were initially obtained using the in-house Rigaku FRD system with R-AXIS HTC detector located in the department of Biochemistry and Molecular Biology, School of Medicine, Wayne State University. The A82F and A82T mutants diffracted to 1.6 Å resolution, while the I10V and A82S mutants diffracted to 2 Å resolution. All data sets were processed using MOSFLM (Leslie, 2006 ▶) and analyzed using SCALA (Evans, 2006 ▶) from the CCP4 (Winn et al., 2011 ▶; Potterton et al., 2003 ▶) suite of programs.
2.4. Structure solutions and refinement
The best data set for each mutant was selected to obtain a structure solution by the molecular-replacement method using MOLREP (Vagin & Teplyakov, 2010 ▶). The crystal structure of MDR769 HIV-1 protease (PDB entry 1tw7; Martin et al., 2005 ▶) was used as a search model to obtain the solutions. Each model was further refined using REFMAC5 (Murshudov et al., 2011 ▶). Solvent atoms were built using ARP/wARP (Lamzin & Wilson, 1993 ▶; Perrakis et al., 1999 ▶). Electron-density maps were generated using CCP4 and XtalView (McRee, 1999 ▶). Ramachandran plots were obtained by PROCHECK (Morris et al., 1992 ▶; Laskowski et al., 1993 ▶) analysis to validate the quality of the structures. Coordinate files for the four structures have been deposited in the RCSB Protein Data Bank with accession codes 3pj6, 3oqd, 3oqa and 3oq7 for the MDR769 I10V, A82F, A82S and A82T HIV-1 protease variants, respectively. Further analysis of the structures, such as calculation of dimer-interface contacts and the solvent-accessible surface area, was performed using CONTACT and AREAIMOL (Lee & Richards, 1971 ▶) from the CCP4 suite of programs. Superpositions were performed using LSQKAB (Kabsch, 1976 ▶) from CCP4. Dimer-interface surface area was calculated using the formula (2M − D)/2, where M is the total surface area of each monomer individually and D is the total surface of the biological dimer.
2.5. Energy minimization of the structures
The crystal structures of the four mutants were energy-minimized using the SANDER module of AMBER8. The parameter and topology files were prepared using tLEaP. 1000 steps of energy minimization were performed using a steepest-descent followed by conjugate-gradient method in SANDER. Energy minimizations were performed in a vacuum.
2.6. Amprenavir-docking studies
Amprenavir was docked into the expanded active-site cavities of the four crystal structures of MDR769 HIV-1 protease variants using AutoDock Vina. The three-dimensional docking grid for each structure was generated covering the active-site area, flaps, 80s loops and dimerization domain carefully. Amprenavir was then docked into the grid using Vina. A positive-control docking experiment was performed using the crystal structure of wild-type HIV-1 protease in complex with amprenavir (PDB code 3ekv; N. M. King, M. Prabu-Jeyabalan, R. M. Bandaranayake, M. N. L. Nalam, A. Ozen, T. Haliloglu & C. A. Schiffer, unpublished work). Coordinates for the inhibitor were deleted from 3ekv and the inhibitor was then redocked into the protease active site from the crystal structure. The redocked pose of amprenavir was then compared with that of the actual crystal structure. Double-docking experiments were performed by two rounds of docking. The first round involves docking one molecule of amprenavir and the second round involves docking a second molecule of amprenavir into the active-site cavity of the MDR protease variant containing the first molecule docked previously. The binding-affinity values for amprenavir docked against each structure were analyzed and compared with that of the positive-control experiment. All figures in this article were created using the open-source program PyMOL v.0.99rc6 (http://www.pymol.org). Figs. 3(b), 3(c) and 3(d) were prepared using ARP Navigator through the CCP4 interface.
3. Results and discussion
3.1. Crystallization results, electron-density maps and structure analysis
Of the ten mutants with MDR769 background, only four yielded diffraction-quality crystals. Expression problems were encountered with the I10R and I10F mutants. The L46I, V54M and V54L mutants yielded poor-quality crystals that diffracted to low resolution. The crystal structures of MDR769 HIV-1 protease variants I10V, A82F, A82S and A82T were solved and analyzed. Diffraction data and refinement statistics are shown in Table 1 ▶. All four structures were solved in space group P41212. The asymmetric unit consists of one molecule of the protease monomer. Protease dimers for each structure were calculated by applying the crystallographic twofold symmetry. The 2|F o| − |F c| electron-density maps around the mutation area are shown in Fig. 2 ▶ for each mutant. Contiguous electron density was seen for all of the mutants except for A82F, in which partial electron density was seen for the side chain of Phe82. The Cα backbone of each mutant was superposed onto that of the wild-type HIV-1 protease around the mutation area to search for any conformational changes. No significant conformational changes were observed at the point of additional mutation in each of the corresponding four crystal structures. The four crystal structures were further analyzed for conformational changes in comparison with the native MDR769 HIV-1 protease (PDB entry 1tw7). Most of the conformational changes were seen in the flaps and the 80s loops of the four mutants when compared with the native MDR769 protease.
Table 1. Diffraction data and refinement statistics.
Values in parentheses are for the highest resolution shell.
| I10V | A82F | A82S | A82T | |
|---|---|---|---|---|
| PDB code | 3pj6 | 3oqd | 3oqa | 3oq7 |
| Crystal parameters | ||||
| Resolution range () | 27.662.25 | 27.621.71 | 27.282.25 | 27.231.71 |
| Unit-cell parameters (, ) | a = b = 44.85, c = 105.38, = = = 90.0 | a = b = 44.96, c = 104.95, = = = 90.0 | a = b = 45.13, c = 102.70, = = = 90.0 | a = b = 45.53, c = 101.92, = = = 90.0 |
| Space group | P41212 | P41212 | P41212 | P41212 |
| Solvent content (%) | 47.0 | 47.0 | 47.91 | 47.0 |
| Data processing | ||||
| No. of unique reflections | 5356 (768) | 12231 (1628) | 5466 (764) | 11367 (1108) |
| I/(I) | 14.2 (5.3) | 26.4 (2.8) | 20.3 (4.8) | 17.6 (2.5) |
| R merge † (%) | 7.4 (24.7) | 6.1 (45.9) | 7.1 (37.1) | 6.0 (36.8) |
| Data multiplicity | 3.4 (3.4) | 11.3 (4.4) | 6.4 (6.5) | 5.8 (3.0) |
| Completeness (%) | 97.6 (99.5) | 98.9 (93) | 99.7 (99.7) | 93.1 (65.2) |
| Refinement statistics | ||||
| No. of reflections used | 5099 | 11591 | 5196 | 10806 |
| R cryst ‡ (%) | 24.30 | 19.59 | 20.22 | 19.5 |
| R free (%) | 30.41 | 22.81 | 23.77 | 22.39 |
| No. of protein atoms | 787 | 760 | 755 | 756 |
| No. of water molecules | 10 | 119 | 55 | 114 |
| Mean temperature factors (2) | ||||
| Protein | 15.82 | 19.75 | 26.32 | 19.59 |
| Main chains | 15.40 | 18.63 | 25.80 | 18.57 |
| Side chains | 16.28 | 20.98 | 26.89 | 20.71 |
| Waters | 8.59 | 30.61 | 28.00 | 34.49 |
| R.m.s.d. bond lengths () | 0.021 | 0.013 | 0.019 | 0.013 |
| R.m.s.d. bond angles () | 1.94 | 1.34 | 1.82 | 1.40 |
| Ramachandran plot | ||||
| Most favored (%) | 87.2 | 96.2 | 96.2 | 94.9 |
| Additional allowed (%) | 10.3 | 3.8 | 3.8 | 5.1 |
| Generously allowed (%) | 2.6 | 0 | 0 | 0 |
| Disallowed (%) | 0 | 0 | 0 | 0 |
R
merge = 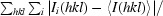
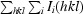 .
.
R
cryst = 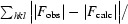
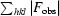 .
.
Figure 2.

Electron-density maps. Shown in red are the 2|F o| − |F c| electron-density maps of the four MDR769 HIV-1 protease variants: (a) A82F, (b) I10V, (c) A82S and (d) A82T. Residues Thr80–Val84 are shown for the A82F, A82S and A82T mutants. Residues Gln7–Thr12 are shown for the I10V mutant. All residues are shown as stick models within their corresponding density. Maps were contoured at 0.7σ for the A82F mutant and 1.0σ for the others.
3.2. The proline switch in the MDR769 I10V mutant HIV-1 protease
The crystal structure of the MDR769 I10V HIV-1 protease variant revealed unusual alternate conformations of Pro81 (proline switch) with a 3–4.8 Å root-mean-square difference in the Cα atoms of the two conformations. This is the first report of such a significant conformational change in the 80s loop. The I10V mutant protease dimer is shown in Fig. 3 ▶(a), showing the conformational change in the 80s loop arising from the proline switch (Fig. 3 ▶ b). The alternate conformations of Pro81 in the 80s loop with electron-density maps are shown in Figs. 3 ▶(c) and 3 ▶(d). Owing to the alternate conformation of Pro81, when the side chain is pointing towards the active-site cavity the protease structure looks normal and comparable to the native MDR769 HIV-1 protease, but when the side chain is pointing away (a less stable conformation; results from the energy-minimization studies discussed separately in this article) from the active-site cavity it exerts conformational stress on the 80s loop, leading to distortion of the S1/S1′ binding pockets. This results in unstable binding of the inhibitors owing to loss of contacts. This could be one of the most important mechanisms by which MDR769 and other similar MDR clinical isolates are selected during HAART. We propose that Leu23, Glu21, Asn83 and Val84 are key players in transmitting the conformational ripple effect of the mutation at codon 10 (with an MDR769 background) to the 80s loop, resulting in the proline switch. The intramonomer distortion is caused owing to an additional 3–4.8 Å increase in the distance measured between the Cα atoms of Ile50 and Pro81 within the same monomer. This shows that the S1/S1′ binding pocket is completely distorted owing to the proline switch, causing the loss of contacts with inhibitors. In addition to the original expansion of the active-site cavity of the native MDR769 strain, the I10V mutant with the proline switch shows an overall expansion of the active-site cavity by 6–7.8 Å compared with that of the wild-type HIV-1 protease. This represents the most expanded active-site cavity reported to date in the literature. The proline switch also causes a loss of intermonomer van der Waals interactions between the flap and the 80s loop. Loss of intermonomer contacts in combination with intramonomer distortions caused because of the proline switch may also weaken the protease dimer. Protease variants such as MDR769 I10V can be very challenging to the most potent inhibitors available to date, but on the other hand such variants are lethal to viral maturation unless there are compensatory mutations in the substrate for productive catalysis.
Figure 3.

The proline switch in the MDR769 I10V mutant. (a) MDR769 I10V HIV-1 protease dimer with alternate conformations of Pro81 (shown in the inset). Mutation I10V is highlighted as a stick model in magenta. (b) An |F o| − |F c| OMIT map (contoured at 1.5σ) is shown in red for the alternate conformations of Pro81 in the MDR769 I10V HIV-1 protease crystal structure. Stick models for the alternate conformations of Pro81 with a 3 Å shift in the Cα atoms are shown in the density. The conformation pointing towards the active-site cavity is shown in blue and that pointing away from the active-site cavity is shown in green. (c) 2|F o| − |F c| map (contoured at 1.0σ) for the 80s loop (Pro79–Asn83) including the alternate conformations of Pro81. (d) |F o| − |F c| map (contoured at 1.5σ) for the 80s loop showing the proline switch.
3.3. The flipped-out conformation of the Phe82 side chain in the MDR769 A82F mutant
It was hypothesized that a mutation from a short side chain to a bulky side chain at codon 82 should restore the lost contacts with inhibitors, but instead the crystal structure of the MDR769 A82F variant showed a flipped-out conformation of the Phe82 side chain as shown in Fig. 4 ▶. The bulky side chain of Phe82 increases the conformational stress on the flexible 80s loop, which causes its flipped-out conformation. This unusual conformation not only causes distortion in the S1/S1′ binding pockets but may also create a pulling-apart effect on the monomers, resulting in a weaker dimer. Such a protease variant can pose a threat to the most potent inhibitors as well as decrease viral replication/maturation unless the substrate molecule has compensatory mutations to accommodate the distorted active-site cavity of such an MDR protease.
Figure 4.
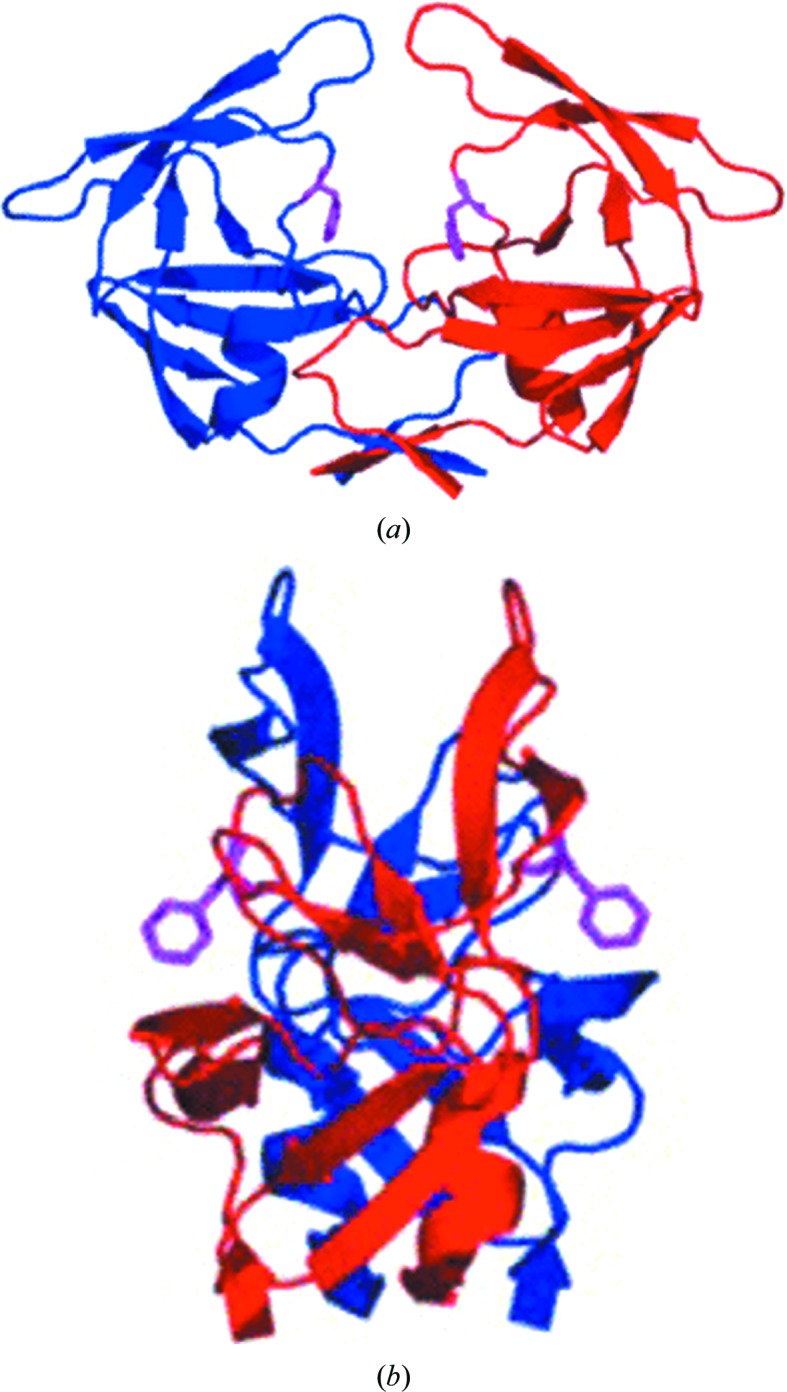
Crystal structure of the MDR769 A82F HIV-1 protease. Shown here are the front view (a) and the side view (b) of the MDR769 A82F HIV-1 protease dimer. The two monomers of the protease are shown as red and blue cartoon representations. Residue Phe82 is highlighted as a magenta stick model to show the characteristic flipped-out conformation of the side chain pointing away from the active-site cavity.
3.4. The A82S and A82T mutants
The crystal structures of the MDR769 A82S and A82T HIV-1 protease variants show that the hydroxyl group on the side chain is pointing away from and towards the active-site cavity, respectively. Owing to the presence of the Pro81 side chain next to Thr82 in the A82T mutant, the hydroxyl group of Thr82 is repelled away from Pro81 towards Leu23, which is at a farther distance from Thr82. Crystallographic analysis of the MDR769 A82S and A82T structures suggests that if residue 82 is mutated from a hydrophobic residue such as Val (wild type) or Ala (native MDR769) to a polar residue such as Ser or Thr some of the critical hydrophobic contacts with the inhibitor, especially in the P1/P1′ region, are disrupted. The introduction of polarity at position 82 results in a local change in the electrostatics of the S1/S1′ binding pockets. Considering the architecture of the side chain, since Thr mimics Val (wild-type protease) more than Ser, the A82T mutant has a less distorted S1/S1′ pocket compared with that of the A82S mutant. Nevertheless, both mutants challenge the hydrophobic P1 and P1′ functional groups of inhibitors, with A82S being relatively severe compared with A82T. Thus, decreased hydrophobicity in the S1/S1′ binding pockets together with the wide-open conformation of the flaps result in unstable binding of the inhibitors, leading to drug resistance. Unfortunately, protease variants such as MDR769 A82T can be productive in substrate catalysis, aiding successful propagation of MDR strains.
3.5. Dimer calculation and analysis
The crystal structures of all four mutants were solved as monomers in space group P41212. Crystallographic twofold symmetry was applied to obtain the dimers for each mutant. These dimers are perfectly symmetrical and represent the functional biological unit. Calculation and analysis of dimer-interface contacts and surface areas suggested that all four mutants and native MDR769 show fewer contacts compared with the wild-type protease. Among the four mutants, the I10V and A82T mutants show a relatively larger number of contacts at the dimer interface compared with native MDR769, while the A82F and A82S mutants show relatively fewer contacts compared with native MDR769. Although all of the mutants, including native MDR769, showed a similar molecular weight to surface area ratio compared with the wild-type protease at the dimer interface, the distribution of the surface area was found to be different. The wild-type and A82F mutant protease dimers showed a relatively larger surface area at the dimer interface. The inter-flap distance was calculated between the Cα atoms of Ile50 of the two flaps for all the mutants, as shown in Table 2 ▶. All four mutants showed a conserved trend of wide-open flaps with varying inter-flap distances. The overall dimer stability and wide-open flaps were comparable to those of native MDR769 for all four mutants.
Table 2. Inter-flap distance measurements between C atoms of Ile50 on the two flaps.
3.6. Energy-minimization studies
The crystal structures of all four mutants show a conserved wide-open conformation of the flaps with varying inter-flap distances (Table 2 ▶). To dissect the dilemma of whether the wide-open conformation of the flaps arises from the effect of crystal contacts or the conformational rigidity acquired by the accumulation of mutations, 1000 steps of energy minimization were performed for each of the four crystal structures in the absence of crystal contacts using the SANDER module of AMBER8 (the steepest-descent method followed by a conjugate-gradient method). Energy-minimized structures still showed wide-open flaps, with the inter-flap distance being twice that of the wild-type protease with closed flaps. Analysis of the minimized structures showed that the I10V mutant possesses the highest total energy even after 1000 steps of minimization. The I10V mutant was then further tested with the alternate conformations of Pro81 one at a time. The mutant without the proline switch exhibited a total energy of −22 064 kJ and that with the proline switch exhibited a total final energy of −21 922 kJ. This indicates that the proline switch is a less stable conformation with an energy difference of 142 kJ compared with its alternate conformation. Although the energy minimization relaxed the overall structure in each case, the flaps were still wide open, indicating that the conformational rigidity acquired by MDR769 arises from accumulation of mutations and cannot be easily counteracted by simple energy-minimization studies; instead, highly potent inhibitors with enhanced enthalpic and entropic factors are needed. Mutation-induced conformational flexibility of the 80s loops in concert with flap opening results in unstable binding of inhibitors, leading to the selection of multidrug-resistant strains of HIV-1 protease.
3.7. Docking studies with amprenavir
In order to confirm the conserved trend of an expanded active-site cavity among the four structures, the inhibitor amprenavir was docked against each structure using the AutoDock Vina program. The positive control showed that the redocked pose of amprenavir aligns with the actual crystal structure (PDB entry 3ekv) with an average root-mean-square deviation of less than 0.5 Å. Preliminary docking studies using amprenavir against the four mutant structures showed that the expanded active-site cavity can accommodate two molecules of the inhibitor. The secondary docking proved that the expanded active-site cavity can indeed accommodate two molecules of amprenavir without any steric clashes between the two docked molecules. The binding affinities of the first and second molecules of amprenavir docked against the A82F mutant suggested that the second molecule of APV shows better binding affinity than the first one because the first molecule encounters a large chemical space compared with the second one when binding in the expanded active-site cavity. As shown in Fig. 5 ▶, the I10V mutant with the proline switch accommodated two molecules of amprenavir with some space in between them. This indicates that even two molecules that are bound are free to move within the active-site cavity without any steric clashes. The A82F, A82S and A82T mutants accommodated two molecules of amprenavir each (Supplementary Figs. 1a, 1b and 1c 1), but the two docked molecules of amprenavir were closely packed within the active-site cavity. Redocked amprenavir in the positive control shows better binding affinity (−37.7 kJ mol−1) compared with the four mutants (−24.7 to −26.4 kJ mol−1). This suggests that the inhibitor is highly unstable in the expanded active-site cavity of each of the four mutants. All four mutants show similar binding profiles as well as binding affinities. Detailed combined QM/MM (quantum mechanics/molecular mechanics) studies of all the available protease inhibitors docked against the four mutant structures presented in this article are currently in progress.
Figure 5.
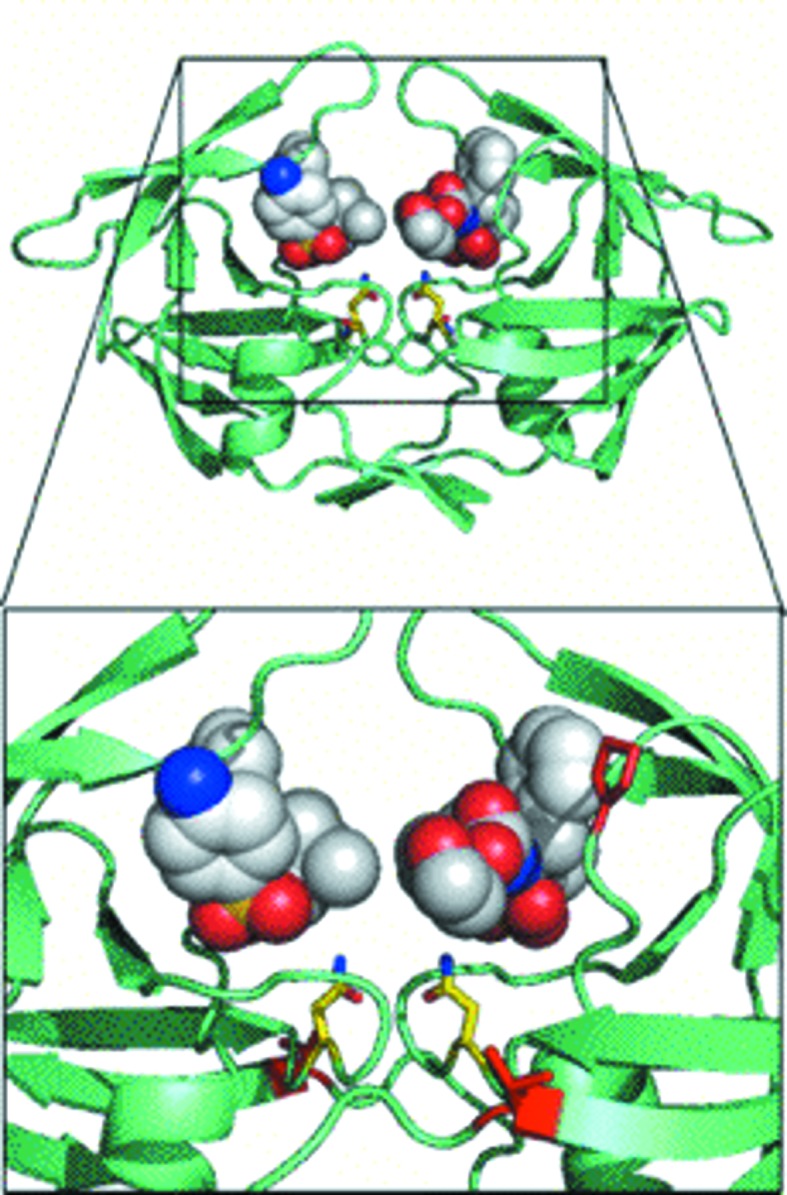
Docking studies with amprenavir. The MDR769 I10V mutant is shown in a greenish cyan color (cartoon representation) with two molecules of amprenavir (shown as spheres) docked into the expanded active-site cavity with the proline switch. Asn25 and Asn125 are highlighted as yellow stick models. Residues Val10 and Pro81 are highlighted as red stick models. The active-site cavity is magnified in the inset for clarity.
3.8. Clinical significance of the four mutants
The clinical significance of the four mutants (I10V, A82F, A82S and A82T) was analyzed in detail using the Stanford HIV database. The frequency of various DMs seen in a pool of clinical isolates that consist of a representative mutation (I10V, A82F, A82S or A82T) was analyzed. The A82F mutant is associated with at least eight other DMs based on 177 clinical isolates from treated patients. Similarly, A82S and A82T mutations are associated with at least six and seven other DMs, respectively, within their pools of clinical isolates from treated patients. On the other hand, the I10V mutation was only associated with two other mutations (L63P and L90M) based on 495 clinical isolates (1.5 to five times the size of the isolate pools for the other three mutants). This indicates that the mutation I10V is very potent in inducing drug resistance, probably by changing the three-dimensional structure of the protease. Thus, the current study provides new insights into the multidrug-resistance mechanisms that can be utilized in future drug design for designing potent inhibitors against ensembles of MDR strains of HIV-1 protease.
4. Summary and conclusions
In summary, we have crystallized, solved and analyzed the structures of four MDR769 HIV-1 protease variants. The crystal structure of the I10V mutant showed that mutation at codon 10 causes a conformational ripple effect leading to alternate conformations of Pro81 (proline switch). The crystal structures of the A82F, A82S and A82T mutants indicate that mutations at codon 82 may not necessarily restore the lost contacts and that the decreased hydrophobicity of the S1/S1′ pockets further causes loss of contacts with inhibitors. Energy-minimization studies of the crystal structures suggested that the wide-open nature of the flaps is a consequence of the conformational rigidity inherited from the MDR769 background and not of the crystal contacts. Additionally, we also found that the I10V mutant with the proline switch has the highest energy, indicating that such a conformation is less stable. Docking studies with amprenavir show that all four mutants show a conserved trend of an expanded active-site cavity, as a consequence of which two molecules of the inhibitor are able to bind in the active-site cavity.
Based on the four crystal structures presented in this article, we conclude that the flexibility caused in the 80s loop (either directly owing to mutations such as A82F, A82S and A82T or indirectly from those such as I10V) not only causes a loss of contacts with the inhibitors, resulting in the multidrug-resistance problem, but also results in a weaker HIV-1 protease dimer that is functionally not as active as the wild-type enzyme. However, such protease variants can still function when there are compensatory mutations in the substrate-cleavage site to accommodate the conformational distortions in the protease binding pockets and the active-site cavity. This study provides further insight relevant to the design of future inhibitors with enhanced potency.
Supplementary Material
PDB reference: MDR769 HIV-1 protease variants, I10V, 3pj6
PDB reference: A82F, 3oqd
PDB reference: A82S, 3oqa
PDB reference: A82T, 3oq7
Supporting information file. DOI: 10.1107/S0907444911011541/mn5004sup1.pdf
Acknowledgments
We thank the National Institutes of Health for funding to LCK (AI65294). Portions of this work were performed at the DuPont–Northwestern–Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E. I. DuPont de Nemours & Co., The Dow Chemical Company and Northwestern University. Use of the APS, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: MN5004). Services for accessing this material are described at the back of the journal.
References
- Case, D. A. et al. (2004). AMBER8. University of California, San Francisco, USA.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Heaslet, H., Rosenfeld, R., Giffin, M., Lin, Y.-C., Tam, K., Torbett, B. E., Elder, J. H., McRee, D. E. & Stout, C. D. (2007). Acta Cryst. D63, 866–875. [DOI] [PubMed]
- Ishima, R., Torchia, D. A., Lynch, S. M., Gronenborn, A. M. & Louis, J. M. (2003). J. Biol. Chem. 278, 43311–43319. [DOI] [PubMed]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923.
- Kohl, N. E., Emini, E. A., Schleif, W. A., Davis, L. J., Heimbach, J. C., Dixon, R. A., Scolnick, E. M. & Sigal, I. S. (1988). Proc. Natl Acad. Sci. USA, 85, 4686–4690. [DOI] [PMC free article] [PubMed]
- Kumar, M., Kannan, K. K., Hosur, M. V., Bhavesh, N. S., Chatterjee, A., Mittal, R. & Hosur, R. V. (2002). Biochem. Biophys. Res. Commun. 294, 395–401. [DOI] [PubMed]
- Lamzin, V. S. & Wilson, K. S. (1993). Acta Cryst. D49, 129–147. [DOI] [PubMed]
- Lapatto, R., Blundell, T., Hemmings, A., Overington, J., Wilderspin, A., Wood, S., Merson, J. R., Whittle, P. J., Danley, D. E. & Geoghegan, K. F. (1989). Nature (London), 342, 299–302. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Layten, M., Hornak, V. & Simmerling, C. (2006). J. Am. Chem. Soc. 128, 13360–13361. [DOI] [PMC free article] [PubMed]
- Lee, B. & Richards, F. M. (1971). J. Mol. Biol. 55, 379–400. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Lexa, K. W., Damm, K. L., Quintero, J. J., Gestwicki, J. E. & Carlson, H. A. (2009). Proteins, 74, 872–880. [DOI] [PMC free article] [PubMed]
- Liu, F., Kovalevsky, A. Y., Louis, J. M., Boross, P. I., Wang, Y.-F., Harrison, R. W. & Weber, I. T. (2006). J. Mol. Biol. 358, 1191–1199. [DOI] [PubMed]
- Logsdon, B. C., Vickrey, J. F., Martin, P., Proteasa, G., Koepke, J. I., Terlecky, S. R., Wawrzak, Z., Winters, M. A., Merigan, T. C. & Kovari, L. C. (2004). J. Virol. 78, 3123–3132. [DOI] [PMC free article] [PubMed]
- Martin, P., Vickrey, J. F., Proteasa, G., Jimenez, Y. L., Wawrzak, Z., Winters, M. A., Merigan, T. C. & Kovari, L. C. (2005). Structure, 13, 1887–1895. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Morris, A. L., MacArthur, M. W., Hutchinson, E. G. & Thornton, J. M. (1992). Proteins, 12, 345–364. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Navia, M. A., Fitzgerald, P. M., McKeever, B. M., Leu, C.-T., Heimbach, J. C., Herber, W. K., Sigal, I. S., Darke, P. L. & Springer, J. P. (1989). Nature (London), 337, 615–620. [DOI] [PubMed]
- Palmer, S., Shafer, R. W. & Merigan, T. C. (1999). AIDS, 13, 611–667. [DOI] [PMC free article] [PubMed]
- Peng, C., Ho, B. K., Chang, T. W. & Chang, N. T. (1989). J. Virol. 63, 2550–2556. [DOI] [PMC free article] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Mol. Biol. 6, 458–463. [DOI] [PubMed]
- Pillai, B., Kannan, K. K. & Hosur, M. V. (2001). Proteins, 43, 57–64. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Prabu-Jeyabalan, M., Nalivaika, E. & Schiffer, C. A. (2002). Structure, 10, 369–381. [DOI] [PubMed]
- Spinelli, S., Liu, Q. Z., Alzari, P. M., Hirel, P. H. & Poljak, R. J. (1991). Biochimie, 73, 1391–1396. [DOI] [PubMed]
- Trott, O. & Olson, A. J. (2010). J. Comput. Chem. 31, 455–461. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vickrey, J. F., Logsdon, B. C., Proteasa, G., Palmer, S., Winters, M. A., Merigan, T. C. & Kovari, L. C. (2003). Protein Expr. Purif. 28, 165–172. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wlodawer, A., Miller, M., Jaskólski, M., Sathyanarayana, B. K., Baldwin, E., Weber, I. T., Selk, L. M., Clawson, L., Schneider, J. & Kent, S. B. (1989). Science, 245, 616–621. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: MDR769 HIV-1 protease variants, I10V, 3pj6
PDB reference: A82F, 3oqd
PDB reference: A82S, 3oqa
PDB reference: A82T, 3oq7
Supporting information file. DOI: 10.1107/S0907444911011541/mn5004sup1.pdf