

Abstract

A formal synthesis of berkelic acid is reported. The strategy employs the combination of a chiral exocyclic enol ether and an achiral isochromanone to afford the chroman spiroketal core via a base-triggered generation and cycloaddition of an o-quinone methide intermediate. Other key steps include equilibration of the spiroketal, intramolecular benzylic oxidation, and lactone addition/hemiketal reduction; all occur with good diastereoselectivity.
The natural product berkelic acid, isolated by Stierle et al. in 2006, is a secondary metabolite produced by a species of Penicillium that thrives in Berkely Pit Lake outside of Butte, Montana.1 The toxic lake, which is an abandoned copper mine that has since filled with over 40 billion gallons of contaminated groundwater, was designated in 1994 as an EPA superfund site.2 Given the proximity of other extreme environments, such as hotsprings of Yellowstone National Park some hundred miles to the south, it is doubtful that the extremophile evolved in the waste pit. However, researchers have speculated that some natural products produced in extreme environments serve to protect their progenitor. Berkelic acid, in particular, emerged from an assay-driven study aimed at identifying metal dependent enzyme inhibitors. The compound was reported to inhibit the cysteine protease caspase-1 (98 μM) and matrix metalloproteinase-3 (MMP-3, 1.87 μM). In further studies with the NCI-60 cancer cell panel, potent and selective cytotoxicity was observed toward a particularly aggressive ovarian cancer cell line (OVCAR-3, 91 nM).
Originally assigned as compound 1, berkelic acid belongs to a very select group of chroman spiroketal natural products of considerable synthetic interest and biological importance (Scheme 1).3 The unknown relative stereochemistry at the quaternary carbon, however, gave us pause. We first envisioned a diastereoselective plan that might address this question toward the end of the synthesis. Late stage introduction of the aryl acid utilizing an aryl-X substituent of compound 2, in principle, made this functionality more distinguishable from the methyl ester. Furthermore, researchers had theorized that MMP inhibitors less dependent upon metal chelation might prove more selective toward validated isoforms of the enzyme; this would allow for analog development along the synthetic pathway.4 We further imagined construction of the isobenzopyran ring by utilization of a stereocontrolled benzylic oxidation of the benzopyran with concomitant reduction of the ketone functionality within the R″ residue of compound 3. To construct the chroman spiroketal motif, we sought to employ chiral exocyclic enol ethers, such as 5, in a diastereoselective inverse demand Diels–Alder reaction with an o-quinone methide (o-QM) 4 generated at low temperature using our magnesium base-triggering methods in conjunction with various o-OBoc compounds 7.5
Scheme 1.
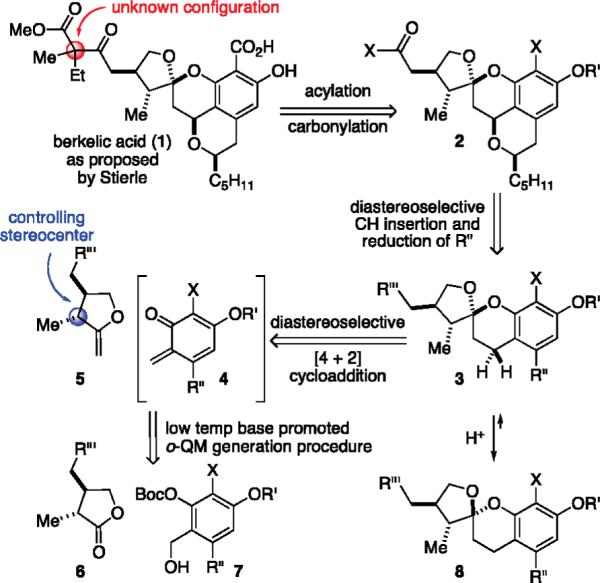
An Early Retrosynthetic Plan
Hence, we began by devising a short synthesis of exocyclic enol ethers (5a–c) derived from γ-lactone 9, which could be produced later in the desired enantiomeric form (Scheme 2).6 These enol ethers were then evaluated in conjunction with assorted o-QM precursors 7 (R″ = −H; X = −H, −Br; R′ = −Me, −Bn) using our standard magnesium base-triggering methodology.
Scheme 2.
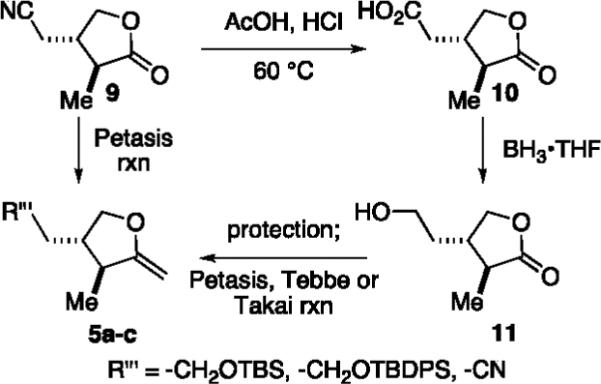
Construction of Enol Ether Coupling Partners
Our early experiments showed that the low temperature cycloaddition afforded what we believed to be the desired relative stereochemistry, whereby the methyl residue in the enol ether had controlled the spiroketal stereocenter (>4:1) in compound 3.7 We further observed and reported that this kinetic spiroketal epimer could be equilibrated to the corresponding thermodynamic epimer spiroketal 8 (>1:2) with a protic acid.8 A short time thereafter, Fürstner reported the synthesis of berkelic acid methyl ester 12 by coupling three separate chiral components together. Their observations led to the revision of three stereocenters and an assignment of the relative stereochemistry including the quaternary stereocenter (Figure 1).9 This landmark effort paved the way for future synthetic successes, and soon afterward, Snider reported the first total synthesis establishing the absolute structure of berkelic acid as its enantiomer (−)-1'.10 The Snider synthesis, which involved an acid-catalyzed coupling of two chiral components followed by late-stage chromatographic resolution of the quaternary center, is masterful in its overall efficiency. Sometime later, De Brabander reported a biomimetic total synthesis combining spicifernin and pulvilloric acid surrogates.11
Figure 1.
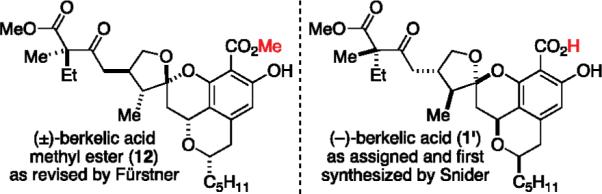
Subsequent revisions by Fürstner and Snider.
These successes and structural revisions forced us to reevaluate our strategy with the prime intention of preserving our initial diastereoselective approach. While we remained confident that enol ethers 5a–c would react to form chroman spiroketals,12 we needed to redesign our o-QM precursor so that a new substituent could be elaborated into the desired tetracycle. We postulated that compound 17 might prove to be a novel o-QM precursor participating in our base-triggering procedure and thereby lead to the o-QM carboxylate 16 and the spiroketal 15 (Scheme 3). Unfortunately, the cycloaddition was expected to afford the kinetic diastereomer. Although our prior equilibration studies were encouraging, the final (2:1) ratio would be a challenge to improve. However, we hoped that the added carboxylic acid residue and aryl iodide might bolster the ratio afforded by a thermodynamic equilibration. While the specifics of the sequence needed for isobenzopyran formation remained undetermined, it mandated stereoselective benzylic oxidation, etherification, and stereoselective incorporation of the pentyl residue to produce the fully elaborated tetracycle 13. Acylation could then be used to introduce the quaternary center and methyl ester. The exact timing for carboxylation, however, depended on the concluding events and surrounding functionality.
Scheme 3.
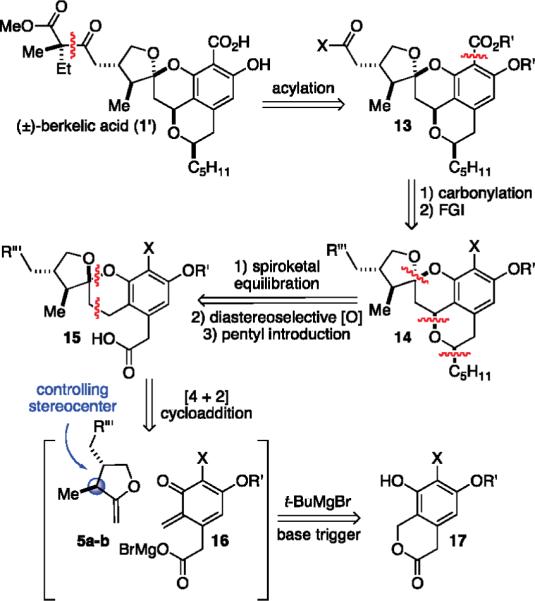
Revised Diastereoselective Strategy
Proceeding in the forward direction, tert-butyl magnesium chloride (1.05 equiv) is added to the lactone 18 (0.1 M in THF, −78 °C), which we had previously prepared in seven steps from commercial materials,13 followed by the addition of the enol ether 5a or 5b (neat, 2.1 equiv). Subsequent warming of the mixture to room temperature affords the respective spiroketals 19a–b in 60–65% yield with a 3:1 ratio of spiroketal epimers favoring the endo isomer (Scheme 4). Treatment of the mixture 19a (0.01 M in CHCl3,65 °C) with DDQ afforded the spiroketal 20a of opposite configuration with a 1:1 ratio at the oxidized benzylic carbon atom. This result indicated to us that, under the reaction conditions, equilibration of the spiroketal was occurring more slowly than benzylic oxidation. To circumvent this issue, equilibration was carried out before exposure to the oxidant. Relative energy calculations for all of the possible diastereomers of spiroketal 19a indicated that the desired spiroketal epimer 19b is more stable by greater than 4.5 kcal/mol.14 Gratifyingly, treatment of the 3:1 mixture of 19a or 19b (0.06 M in CHCl3, rt) with TFA (0.3% in CHCl3) confirms these calculations and results in a nearly quantitative yield of the corresponding thermodynamic spiroketals in a 10:1 diastereomeric ratio. Separation and intramolecular benzylic oxidation using the prior conditions produced the respective lactones 21a–b as single diastereomers.
Scheme 4.
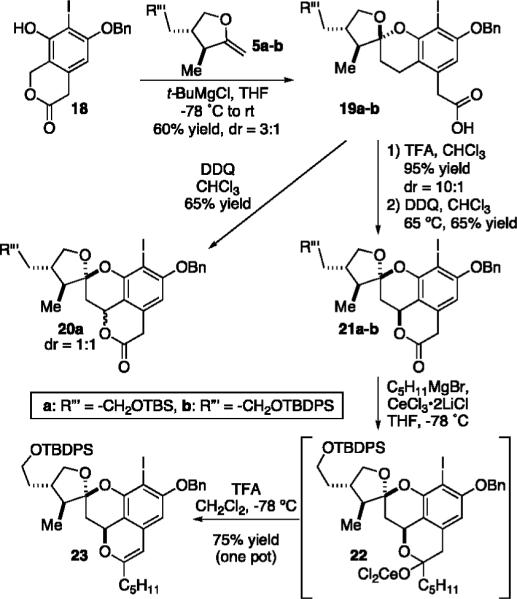
Diastereoselective Construction of Enol Ether 23
Our next hurdle involved selective mono addition of the desired pentyl alkyl chain to the lactone scaffold 21. The methylene adjoining the lactone carbonyl in compound 21 was problematic—addition of most organometallic species resulted in deprotonation and returned starting material upon workup. Weinreb amides derived from 21 were also investigated in the hope that an acyclic system would prove less acidic and undergo mono addition to afford the corresponding ketone. However, we eventually uncovered observations by Cohen et al.15 that describe the selective mono addition of organocerium species, prepared by the normal Imamato process, to lactones.16 After confirming the integrity of the desired pentylcerium reagent (generated by the method of Knochel)17 by testing its reaction with 1,3-diphenylacetone, we were pleased to observe its selective mono addition to lactone 21 produces the corresponding hemiketal 22 in greater than 80% yield. The hemiketal 22, however, exists in equilibrium with its ketone counterpart, which thwarted its complete spectroscopic identification. We therefore sought to reduce it in situ. Conventional reduction tactics, such as the combined action of triethylsilane and protic or Lewis acids, afforded a mixture of isobenzopyran diastereomers favoring the undesired stereochemistry. In the absence of a reductant, however, treatment of the cerium alkoxy hemiketal 22 (0.02 M in CH2Cl2, −78 °C) in situ with TFA resulted in smooth formation of the corresponding enol ether 23 in 75% yield over the one-pot operation from lactone 21.
We had planned to conclude our synthesis with carbonylation, and compound 23 enabled us to investigate a variety of plausible, mild conditions that might prove compatible with the β-keto ester functionality. Unfortunately, all palladium- and zinc-mediated coupling processes failed in our hands. In addition, we found that all homogeneous and heterogeneous hydrogenations of the enol ether in 23 caused hydrogenolysis of the aryl iodo functionality. These problems forced us to proceed with carbonylation before reduction of the enol ether, much earlier than originally planned. Lithium-halogen exchange proceeds with iodide 23 (0.01 M in THF, −110 °C) upon addition of t-BuLi (2 equiv), whereupon cannulation onto carbon dioxide affords the desired acid upon acidic workup (Scheme 5). Subsequent hydrogenation of the crude acid (0.01 M in EtOAc) over palladium on carbon (10%, 1 atm H2) occurs from the less hindered face to afford salicylic acid 24 in a 64% yield over two steps with >10:1 dr.18
Scheme 5.
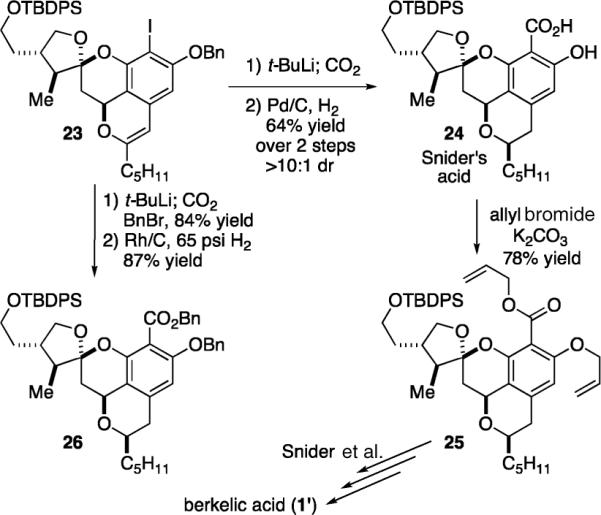
Formal Syntheis of (±)-Berkelic Acid (1′)
The salicylic acid 24 had been a key intermediate in the Snider synthesis, and it is positioned six steps from the natural product. However, this acid had not been fully characterized. We therefore constructed the published allyl derivative 25 from acid 24. Spectroscopic comparison of the 1H NMR showed our synthetic material to be the same as Snider's, thereby confirming all of the stereochemical assignments and resulting in a formal synthesis of berkelic acid with the synthesis of the acid 24.
Nevertheless, we sought to shorten the potential endgame for our route by one additional step. Thus, the iodo compound 23 was subjected to lithium-halogen exchange and addition of carbon dioxide as before. In this instance, however, the carboxylate anion was intercepted with benzyl bromide to produce the corresponding benzyl ester in 84% yield. We further discovered that this material (0.025 M in toluene) could be selectively reduced with rhodium on carbon at 65 psi of hydrogen pressure with the benzyl ether and ester remaining intact, thereby yielding compound 26 in 87% yield. In view of the newly reported synthesis of (−)-1'by Fürstner in which a benzyl ester is used for orthogonal deprotection in the presence of a methyl ester, the benzylated material 26 should prove as viable in the Snider endgame as ester 25.
In conclusion, we have successfully accomplished a diastereoselective formal synthesis of berkelic acid by interception of Snider's previously uncharacterized salicylic acid 24. We believe that our strategy has merit since our tactical construction of the isobenzopyran ring is orthogonal to previous syntheses; all the prior strategies utilize the combination of various chiral components together with various chiral isobenzopyran derivatives to arrive at the tetracycle. In our case, the relative stereochemistry of the tetracyclic core is efficiently relayed from the stereocenters derived from the original enol ether o-QM coupling partner. Furthermore, the flexibility of this route allows for the introduction of various scaffold modifications of the tetracylic core (acid removal, spiroketal epimierization, alkyl derivatization) that may shed light on the mechanism of action of this interesting natural product.19
Supplementary Material
Acknowledgment
This work was funded with support from the National Institute of General Medical Sciences (GM064831).
Footnotes
Supporting Information Available: Experimental details for the preparation of new compounds and complete spectral data for new compounds accessible and stable as single entities (5a–c, 18, 19a–b, 21a–b, 22–26). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Stierle AA, Stierle DB, Kelly K. J. Org. Chem. 2006;71:5357. doi: 10.1021/jo060018d. [DOI] [PubMed] [Google Scholar]
- (2). EPA ID: MTD980502777.
- (3).Sperry J, Wilson ZE, Rathwell DCK, Brimble MA. Nat. Prod. Rep. 2010;27:1117. doi: 10.1039/b911514p. [DOI] [PubMed] [Google Scholar]
- (4).(a) Itoh Y, Nagase H. Essays Biochem. 2002;38:21. doi: 10.1042/bse0380021. [DOI] [PubMed] [Google Scholar]; (b) Zucker S, Cao J, Chen WT. Oncogene. 2000;19:6642. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- (5).(a) Selenski C, Pettus TRR. J. Org. Chem. 2004;69:9196. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]; (b) Jones RM, Van de Water RW, Lindsey CC, Pettus TRR. J. Org. Chem. 2001;66:3435. doi: 10.1021/jo001752e. [DOI] [PubMed] [Google Scholar]
- (6).See Supporting Information and Pirrung MC, Dunlap SE, Trinks UP. Helv. Chim. Acta. 1989;72:1301.
- (7).Marsini MA, Huang Y, Lindsey CC, Wu K-L, Pettus TRR. Org. Lett. 2008;10:1477. doi: 10.1021/ol8003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Huang Y, Pettus TRR. Synlett. 2008:1353. doi: 10.1055/s-2008-1072750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Buchgraber P, Snaddon TN, Wirtz C, Mynott R, Goddard R, Fürstner A. Angew. Chem., Int. Ed. 2008;47:8450. doi: 10.1002/anie.200803339. [DOI] [PubMed] [Google Scholar]; (b) Snaddon TN, Buchgraber P, Schulthoff S, Wirtz C, Mynott R, Fürstner A. Chem.–Eur. J. 2010;16:12133. doi: 10.1002/chem.201001133. [DOI] [PubMed] [Google Scholar]
- (10).(a) Wu X, Zhou J, Snider BB. Angew. Chem., Int. Ed. 2009;48:1283. doi: 10.1002/anie.200805488. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu X, Zhou J, Snider BB. J. Org. Chem. 2009;74:6245. doi: 10.1021/jo901221a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bender CF, Yoshimoto FK, Paradise CL, De Brabander JK. J. Am. Chem. Soc. 2009;131:11350. doi: 10.1021/ja905387r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Compound 5b was made by Snider and coworkers in enantiopure form by 1,4-addition of metallated chiral phosphoramides: see ref 10a and Hanessian S, Gomtsyan A, Malek N. J. Org. Chem. 2000;65:5623. doi: 10.1021/jo000388g.
- (13). See the Supporting Information for experimental details.
- (14). Calculations were performed using Macromodel MM3 Monte Carlo conformational analysis.
- (15).(a) Ahn Y, Cohen T. J. Org. Chem. 1994;59:3142. [Google Scholar]; (b) Mudryk B, Shook CA, Cohen T. J. Am. Chem. Soc. 1990;112:6389. [Google Scholar]
- (16).Imamoto T, Kusumoto T, Tawarayama Y, Sugiura Y, Mita T. J. Org. Chem. 1984;49:3904. [Google Scholar]
- (17).Krasovskiy A, Kopp F, Knochel P. Angew. Chem., Int. Ed. 2006;45:497. doi: 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- (18).Reduction of hemiacetal analogs is reported to stereoselectively provide the corresponding 2,6-cis-tetrahydropyran. See: Nicolaou KC, Cole KP, Frederick MO, Aversa RJ, Denton RM. Angew. Chem., Int. Ed. 2007;119:9031. doi: 10.1002/anie.200703742.
- (19).Fischbach MA, Walsh CT. Science. 2009;325:1089. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.