

Factor H-binding protein B from T. denticola was expressed, purified and crystallized. A native data set was collected to 1.8 Å resolution.
Keywords: factor H-binding protein B, Treponema denticola, periodontal disease
Abstract
Treponema denticola is a primary etiological agent of periodontal disease. T. denticola evades complement-mediated killing by binding to the host’s factor H (FH), a negative regulator of the alternative complement pathway. The T. denticola FH-binding protein has been identified and designated as factor H-binding protein B (FhbB). Crystals of recombinant FhbB were obtained by the hanging-drop vapor-diffusion method using sodium citrate and 0.2 M sodium thiocyanate. FhbB crystals diffracted to 1.8 Å resolution and belonged to space group P43212 or P41212, with unit-cell parameters a = b = 46.76, c = 167.68 Å. Two FhbB molecules per asymmetric unit gave a Matthews coefficient of 2.2 Å3 Da−1 and a solvent content of 44%. FhbB is the smallest bacterially produced FH-binding protein identified to date. Determination of its structure will provide unique insight into the minimal structural determinants required for FH binding.
1. Introduction
Periodontal disease is a chronic disease that results from a ‘polymicrobial disruption of host homeostasis’ leading to the gradual destruction of tissue and bone supporting the teeth (Darveau, 2010 ▶). The bacterial species Treponema denticola, Tannerella forsythia and Porphyromonas gingivalis form the ‘red microbial complex’. The outgrowth of this complex correlates with disease severity (Socransky et al., 1998 ▶). To survive and thrive in crevicular fluid, which has high concentrations of complement, periopathogens must be able to circumvent complement-mediated destruction. The serum resistance of T. denticola correlates with its ability to bind human factor H (FH; McDowell et al., 2005 ▶, 2011 ▶), a key negative regulator of the alternative complement pathway (Zipfel & Skerka, 2009 ▶). Factor H regulates complement by functioning as a cofactor for the factor I-mediated cleavage of C3b and by accelerating the decay of the C3 convertase complex (Ruddy & Austen, 1971 ▶). The net effect of localization of FH on the bacterial cell surface is the inhibition of the alternative pathway C3 convertase preventing further C3b deposition. As a result, bacteria are protected against opsonization and phagocytosis.
T. denticola produces a single FH-binding protein designated FhbB (McDowell et al., 2005 ▶, 2007 ▶, 2009 ▶). FhbB is an 11.4 kDa (102-amino-acid) surface-exposed protein with a pI of 9.9. FhbB lacks a transmembrane domain but possesses a consensus lipidation motif, suggesting that it is tethered to the membrane through a lipid moiety (McDowell et al., 2007 ▶). It is the smallest known FH-binding protein produced by a human pathogen. Factor H-binding proteins identified to date have minimal primary sequence homology (McDowell et al., 2007 ▶; Dave et al., 2001 ▶; Hellwage et al., 2001 ▶; Kraiczy et al., 2001 ▶; Meri et al., 2002 ▶; Hovis et al., 2004 ▶; Jarva et al., 2004 ▶; Lu et al., 2006 ▶; Poltermann et al., 2007 ▶; Caswell et al., 2008 ▶; Madico et al., 2006 ▶). The ability of divergent proteins to bind a common ligand is postulated to rely upon conserved structural elements or physiochemical properties as opposed to a strict primary sequence (McDowell et al., 2005 ▶; Metts et al., 2003 ▶; Hovis et al., 2008 ▶). The molecular basis of the interaction of FH with bacterial FH-binding proteins remains an area of intensive research with implications for therapeutic and preventive approaches for infectious diseases. To date, structures have been determined for two bacterial FH-binding proteins: the 28 kDa Borrelia burgdorferi CspA protein (also referred to as BbCRASP-1 or BBA68; Cordes et al., 2005 ▶) and the 29 kDa Neisseria meningitidis Fhbp protein (Schneider et al., 2009 ▶). Since FhbB is the smallest identified FH-binding protein, determination of its structure will significantly advance our knowledge of the minimum molecular-binding requirements for interaction with FH. Here, we detail the expression, purification and crystallization of FhbB.
2. Materials and methods
2.1. Generation and purification of recombinant FhbB
fhbB (T. denticola strain 35405; ORF tde0108; amino-acid residues 24–102) was PCR-amplified using primers that harbor ligase-independent cloning (LIC) tails and the amplicon was annealed with pET46 Ek/LIC (Novagen). The plasmid was propagated in Escherichia coli NovaBlue and then transferred into E. coli BL21 (DE3) for protein production as described previously (McDowell et al., 2009 ▶). The final recombinant protein, which carries an N-terminal hexa-His tag of 1.7 kDa, was purified (>95% homogeneity) by immobilized metal-affinity chromatography (IMAC; HisTrap, GE Healthcare) as per the manufacturer’s protocol and was concentrated using Amicon Ultra filters (molecular-weight cutoff 3 kDa; Millipore). The protein concentration was determined using the bicinchoninic acid (BCA) assay.
To derivatize FhbB with selenomethionine (SeMet), E. coli strain B834 (DE3) (pLysS) harboring pET46 Ek/LIC-fhbB was grown in LB supplemented with 100 mg l−1 ampicillin. To induce protein production, Overnight Express System 2 medium (Novagen) supplemented with 125 mg l−1 l-selenomethionine, 100 nM vitamin B12 and 100 mg l−1 ampicillin was employed. The cells were harvested and the His-tagged protein was purified as described above. All chromatography steps were performed on an ÄKTA Purifier (GE Healthcare). Incorporation of SeMet was confirmed by electrospray ionization mass spectrometry using VCU’s Mass Spectrometry Core Facility.
2.2. Crystallization
All crystals were grown using the hanging-drop vapor-diffusion method at 291 K. His-tagged protein was concentrated to 58 mg ml−1 and mixed in a 1:1 ratio with well solution to form a 2 µl drop. Initial FhbB crystals formed in 1.4 M sodium citrate tribasic dihydrate, 0.1 M HEPES pH 7.5 (Crystal Screen, Hampton Research) after seven weeks. The addition of 0.2 M sodium thiocyanate (Additive Screen, Hampton Research) yielded similar crystals in 2 d. The crystals were plate-like and diffracted poorly. The addition of 2.5% glycerol reduced the number of nucleation sites, slowed crystal growth and improved the morphology (sharp edges) and diffraction quality of the crystals.
Optimization of the original conditions was required to obtain high-quality SeMet FhbB crystals. The conditions employed consisted of 1.39 M sodium citrate tribasic dihydrate, 0.1 M HEPES pH 7.5, 10 mM DTT, 2.5% glycerol and 0.2 M sodium thiocyanate. The crystals grew as large half-moon-shaped crystals with soft edges. After transport of the trays at 305 K to the X-ray source, the drops became opaque. When the trays were returned to 291 K and left undisturbed for 19 d, the crystals reformed with sharp edges and improved diffraction properties.
2.3. Data collection and processing
Native and SeMet FhbB crystals were washed and flash-frozen in liquid nitrogen or directly in the cryostream. X-ray data were collected at 100 K using a Molecular Structure Corporation (MSC) X-Stream Cryogenic Crystal Cooler System and an R-AXIS IV++ image-plate detector with a Rigaku MicroMax-007 X-ray source equipped with MSC Varimax confocal optics operating at 40 kV and 20 mA. Data were processed and scaled with d*TREK (Pflugrath, 1999 ▶). Complete data-collection statistics are summarized in Table 1 ▶.
Table 1. Statistics of data collection and processing.
Values in parentheses are for the highest resolution shell.
| No. of crystals | 1 |
| Radiation source | Rotating anode |
| Wavelength (Å) | 1.54 |
| Detector | R-AXIS IV++ |
| Crystal-to-detector distance (mm) | 200 |
| Rotation/image (°) | 0.5 |
| Total rotation (°) | 100 |
| Resolution range (Å) | 28.46–1.81 (1.87–1.81) |
| Space group | P43212 or P41212 |
| Unit-cell parameters (Å) | a = b = 46.76, c = 167.68 |
| Mosaicity (°) | 0.929 |
| Total No. of measured reflections | 111360 (4291) |
| Unique reflections | 17573 (1618) |
| 〈I/σ(I)〉 | 17.6 (3.5) |
| Multiplicity | 5.7 (2.5) |
| Completeness (%) | 97.9 (92.2) |
| Rmerge (%)† | 5.2 (29.9) |
R
merge = 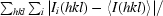
 , where Ii(hkl) is the ith used observation for unique hkl and 〈I(hkl)〉 is the mean intensity for unique hkl and summations are over all i observations and then over all hkl.
, where Ii(hkl) is the ith used observation for unique hkl and 〈I(hkl)〉 is the mean intensity for unique hkl and summations are over all i observations and then over all hkl.
3. Results and discussion
Native FhbB was expressed as a soluble 6×His-tagged protein that yielded 78 mg recombinant protein per litre of culture (Fig. 1 ▶). Initial crystallization conditions were identified from a factorial screen (Crystal Screen, Hampton Research) around two months after setting up the crystallization drops. Sodium thiocyanate, identified from an additive screen, induced crystal growth under these conditions in 2 d (Fig. 2 ▶ a). The addition of glycerol slightly slowed crystal growth and yielded optimal diffraction-quality crystals (0.15 × 0.075 × 0.15 mm). A native data set to 1.8 Å resolution was collected using Cu Kα radiation (Fig. 3 ▶ a). The data obtained from the tetragonal-shaped crystals indicate either space group P43212 or P41212, with unit-cell parameters a = b = 46.8, c = 167.7 Å. Assuming two molecules per asymmetric unit, the Matthews coefficient was 2.22 Å3 Da−1 and the solvent content was 44%. SeMet-derivatized FhbB was produced (80 mg per litre of culture) in order to obtain single-wavelength anomalous dispersion data for phasing. Electrospray ionization mass spectrometry (ES-MS) analysis of SeMet FhbB showed the incorporation of two selenomethionines per 93-residue molecule compared with native FhbB. Although the sequence contained three methionines, including the initiator Met, the native FhbB ES-MS indicated that the N-terminal methionine was not present. This is most likely owing to processing during bacterial expression. The two selenomethionines per FhbB molecule would correspond to four SeMet residues per asymmetric unit. SeMet FhbB crystals were grown from the conditions used for native FhbB but resulted in soft half-moon-shaped crystals with poor diffraction (Fig. 2 ▶ b). Serendipitously, we discovered that a temperature jump (∼15 K) followed by re-equilibration at 291 K allowed diffraction-quality crystals to form (Figs. 2 ▶ c and 3 ▶ b). Collection of SAD data is currently under way. The final determination of the FhbB structure will provide valuable insight into the molecular determinants that mediate interactions with FH and contribute to FH-dependent complement-evasion strategies.
Figure 1.

Samples from recombinant FhbB purification steps as detailed in §2 were separated by SDS–PAGE (15% Tris–HCl) and the gel was stained with Coomassie Blue to visualize the proteins. Lane 1, Precision Plus molecular-weight marker (Bio-Rad); lane 2, crude cell lysate; lanes 3–5, buffer washes from the IMAC column; lane 6, pooled elution fractions.
Figure 2.

(a) Native FhbB crystals, (b) initial SeMet FhbB crystals and (c) SeMet FhbB crystals after temperature fluctuation.
Figure 3.

The crystal-to-detector distance, oscillation angle and exposure time for the frame in (a; native FhbB) were 150 mm, 0.5° and 421 s, respectively, and those for that in (b; SeMet FhbB) were 200 mm, 1° and 120 s, respectively.
Acknowledgments
This work was supported in part by a grant to RTM from NIAID-NIDCR (5R01DE017401) and by grants to JKB from the American Cancer Society (IRG9922504) and NIH (5K22CA122828-03). Dr Kristina Nelson at the VCU Mass Spectrometry Resource Center performed mass spectrometry with funding from the VCU College of Humanities and Science. We are grateful to Dr Faik Mosayev for his advice and help with cryoprotection.
References
- Caswell, C. C., Han, R., Hovis, K. M., Ciborowski, P., Keene, D. R., Marconi, R. T. & Lukomski, S. (2008). Mol. Microbiol. 67, 584–596. [DOI] [PubMed]
- Cordes, F. S., Roversi, P., Kraiczy, P., Simon, M. M., Brade, V., Jahraus, O., Wallis, R., Skerka, C., Zipfel, P. F., Wallich, R. & Lea, S. M. (2005). Nature Struct. Mol. Biol. 12, 276–277. [DOI] [PubMed]
- Darveau, R. P. (2010). Nature Rev. Microbiol. 8, 481–490. [DOI] [PubMed]
- Dave, S., Brooks-Walter, A., Pangburn, M. K. & McDaniel, L. S. (2001). Infect. Immun. 69, 3435–3437. [DOI] [PMC free article] [PubMed]
- Hellwage, J., Meri, T., Heikkilä, T., Alitalo, A., Panelius, J., Lahdenne, P., Seppälä, I. J. & Meri, S. (2001). J. Biol. Chem. 276, 8427–8435. [DOI] [PubMed]
- Hovis, K. M., Freedman, J. C., Zhang, H., Forbes, J. L. & Marconi, R. T. (2008). Infect. Immun. 76, 2113–2122. [DOI] [PMC free article] [PubMed]
- Hovis, K. M., McDowell, J. V., Griffin, L. & Marconi, R. T. (2004). J. Bacteriol. 186, 2612–2618. [DOI] [PMC free article] [PubMed]
- Jarva, H., Hellwage, J., Jokiranta, T. S., Lehtinen, M. J., Zipfel, P. F. & Meri, S. (2004). J. Immunol. 172, 3111–3118. [DOI] [PubMed]
- Kraiczy, P., Skerka, C., Brade, V. & Zipfel, P. F. (2001). Infect. Immun. 69, 7800–7809. [DOI] [PMC free article] [PubMed]
- Lu, L., Ma, Y. & Zhang, J.-R. (2006). J. Biol. Chem. 281, 15464–15474. [DOI] [PubMed]
- Madico, G., Welsch, J. A., Lewis, L. A., McNaughton, A., Perlman, D. H., Costello, C. E., Ngampasutadol, J., Vogel, U., Granoff, D. M. & Ram, S. (2006). J. Immunol. 177, 501–510. [DOI] [PMC free article] [PubMed]
- McDowell, J. V., Frederick, J., Miller, D. P., Goetting-Minesky, M. P., Goodman, H., Fenno, J. C. & Marconi, R. T. (2011). Mol. Oral Microbiol. 26, 140–149. [DOI] [PMC free article] [PubMed]
- McDowell, J. V., Frederick, J., Stamm, L. & Marconi, R. T. (2007). Infect. Immun. 75, 1050–1054. [DOI] [PMC free article] [PubMed]
- McDowell, J. V., Harlin, M. E., Rogers, E. A. & Marconi, R. T. (2005). J. Bacteriol. 187, 1317–1323. [DOI] [PMC free article] [PubMed]
- McDowell, J. V., Huang, B., Fenno, J. C. & Marconi, R. T. (2009). Infect. Immun. 77, 1417–1425. [DOI] [PMC free article] [PubMed]
- Meri, T., Hartmann, A., Lenk, D., Eck, R., Würzner, R., Hellwage, J., Meri, S. & Zipfel, P. F. (2002). Infect. Immun. 70, 5185–5192. [DOI] [PMC free article] [PubMed]
- Metts, M. S., McDowell, J. V., Theisen, M., Hansen, P. R. & Marconi, R. T. (2003). Infect. Immun. 71, 3587–3596. [DOI] [PMC free article] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Poltermann, S., Kunert, A., von der Heide, M., Eck, R., Hartmann, A. & Zipfel, P. F. (2007). J. Biol. Chem. 282, 37537–37544. [DOI] [PubMed]
- Ruddy, S. & Austen, K. F. (1971). J. Immunol. 107, 742–750. [PubMed]
- Schneider, M. C., Prosser, B. E., Caesar, J. J., Kugelberg, E., Li, S., Zhang, Q., Quoraishi, S., Lovett, J. E., Deane, J. E., Sim, R. B., Roversi, P., Johnson, S., Tang, C. M. & Lea, S. M. (2009). Nature (London), 458, 890–893. [DOI] [PMC free article] [PubMed]
- Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C. & Kent, R. L. (1998). J. Clin. Periodontol. 25, 134–144. [DOI] [PubMed]
- Zipfel, P. F. & Skerka, C. (2009). Nature Rev. Immunol. 9, 729–740. [DOI] [PubMed]