

The crystallization and preliminary X-ray crystallographic analysis of the glpX-encoded class II fructose-1,6-bisphosphatase from M. tuberculosis in the apo form is reported.
Keywords: fructose-1,6-bisphosphatases; Mycobacterium tuberculosis
Abstract
Fructose-1,6-bisphosphatase (FBPase; EC 3.1.3.11), which is a key enzyme in gluconeogenesis, catalyzes the hydrolysis of fructose 1,6-bisphosphate to form fructose 6-phosphate and orthophosphate. The present investigation reports the crystallization and preliminary crystallographic studies of the glpX-encoded class II FBPase from Mycobacterium tuberculosis H37Rv. The recombinant protein, which was cloned using an Escherichia coli expression system, was purified and crystallized using the hanging-drop vapor-diffusion method. The crystals diffracted to a resolution of 2.7 Å and belonged to the hexagonal space group P6122, with unit-cell parameters a = b = 131.3, c = 143.2 Å. The structure has been solved by molecular replacement and is currently undergoing refinement.
1. Introduction
Mycobacterium tuberculosis (Mtb), which infects about one-third of the human population, is the leading cause of mortality arising from bacterial pathogens. In 2006, over nine million new cases and 1.7 million deaths occurred owing to TB (World Health Organization, 2008 ▶). Tuberculosis is a worldwide health threat in conjunction with the spread of HIV infection (Nunn et al., 2005 ▶). The pathogen is capable of surviving and replicating in the host macrophage, which is an important factor in the persistence of the disease.
Enzymatic pathways that are involved in growth and survival under nutritionally restrictive and stressful conditions in vivo represent attractive alternative targets for the development of novel antimicrobial agents. Although Mtb is capable of growth on a variety of substrates in vitro, there is evidence that suggests that during infection Mtb preferentially utilizes fatty acids as a source of energy (Boshoff & Barry, 2005 ▶). Moreover, genes involved in fatty-acid utilization (glyoxylate shunt, icl; gluconeogenesis, glpX and pckA) are required for growth and persistence in vivo (Sharma et al., 2000 ▶; Collins et al., 2002 ▶; Liu et al., 2003 ▶; Sassetti & Rubin, 2003 ▶; Muñoz-Elías & McKinney, 2005 ▶; Dunn et al., 2009 ▶; Marrero et al., 2010 ▶). Hence, the enzymes that catalyze the glyoxylate shunt and gluconeogenesis pathways are considered to be potential targets for the discovery of new drugs (Sharma et al., 2000 ▶; Smith et al., 2003 ▶; Anstrom & Remington, 2006 ▶; Purohit et al., 2007 ▶). A recent study (Marrero et al., 2010 ▶) demonstrated that phosphoenolpyruvate carboxykinase (PEPCK; EC 4.1.1.32) plays a pivotal role in the pathogenesis of Mtb. PEPCK is the enzyme encoded by pckA and connects the TCA cycle and gluconeogenesis by catalyzing the reversible decarboxylation and phosphorylation of oxaloacetate (OAA) to form phosphoenolpyruvate (PEP).
Furthermore, it has also been shown that PEPCK is essential for the growth and survival of Mtb during infection in mice, wherein the pathogen relies primarily on gluconeogenic substrates for in vivo growth and persistence (Marrero et al., 2010 ▶). Recently, efforts have been directed towards solving the three-dimensional structure of PEPCK in order to enhance our understanding of this important enzyme (Chim et al., 2011 ▶). Apart from these recent studies, the role of gluconeogenesis in Mtb pathogenesis remains largely unexplored. Therefore, understanding the key enzymes of the gluconeogenic pathway and obtaining key structural information about the enzymes involved in gluconeogenesis becomes important.
Fructose-1,6-bisphosphatase (FBPase; EC 3.1.3.11), a key enzyme in gluconeogenesis, catalyzes the hydrolysis of fructose 1,6-bisphosphate to form fructose 6-phosphate and orthophosphate. In glycolysis, the opposite reaction is catalyzed by phosphofructokinase whose product is fructose 6-phosphate, an important precursor in various biosynthetic pathways (Pontremoli et al., 1975 ▶). FBPases are members of the large superfamily of lithium-sensitive phosphatases, which includes both the inositol phosphatases and FBPases.
Five different classes (or types) of FBPases have been identified based on their amino-acid sequences (FBPases I–V; Donahue et al., 2000 ▶; Nishimasu et al., 2004 ▶; Hines et al., 2006 ▶). Although the class I FBPases are most widely distributed among living organisms, many microorganisms such as Escherichia coli possess both class I and class II FBPases. The structural biology and the underlying regulatory mechanism of class I FBPases have been extensively studied and are well understood (Hines et al., 2006 ▶; Hines, Fromm et al., 2007 ▶; Hines, Kruesel et al., 2007 ▶).
The type II FBPase in E. coli is encoded by the glpX gene, which is part of the glycerol 3-phosphate regulon (Donahue et al., 2000 ▶).
Crystal structures of this enzyme have recently been reported in both the apo form and with substrate and catalytic products (PDB entries 3bih, 3big and 3d1r; Brown et al., 2009 ▶). In addition, structures of the same enzyme, previously referred to as ‘glycerol metabolism protein’, from E. coli are also available in the PDB (PDB entries 1ni9 and 2r8t; Brown et al., 2009 ▶; V. V. Lunin, T. Skarina, G. Brown, R. Sanishvili, A. Yakunin, A. Joachimiak, A. M. Edwards & A. Savchenko, unpublished work).
Initial genome sequencing and annotation of Mtb revealed the absence of a class I FBPase homolog (Cole et al., 1998 ▶). Later, results of genetic and biochemical analyses revealed that the Rv1099c gene of Mtb encodes the missing mycobacterial class II FBPase (Movahedzadeh et al., 2004 ▶). The protein encoded by the Mtb glpX (Rv1099c) gene has been shown experimentally to possess FBPase activity and is homologous to other experimentally verified class II FBPases from E. coli (GlpX; 42% identity) and Corynebacterium glutamicum (65% identity; Rittmann et al., 2003 ▶). Since the Mtb FBPase II has no human homolog, biochemical and structural studies may reveal certain characteristics that are unique to Mtb FBPase II and that may possibly be exploited for species-specific drug discovery targeting this enzyme. We have recently reported the cloning, expression, purification and initial biochemical characterization of the recombinant mycobacterial class II FBPase (Gutka et al., 2011 ▶). The crystallization, preliminary X-ray diffraction analysis and initial results of the structure determination of this enzyme are presented here.
2. Materials and methods
2.1. Expression and purification of Mtb FBPase
Unless mentioned otherwise, all reagents and chemicals were purchased from Fisher Scientific, Pittsburgh, Pennsylvania, USA. The glpX gene (Rv1099c) encoding the class II FBPase from Mtb H37Rv was PCR-amplified using gene-specific primers and cloned into the pET15b vector as described previously (Gutka et al., 2011 ▶). The expression and purification process followed was the same as that described elsewhere (Gutka et al., 2011 ▶). Briefly, E. coli BL21 (DE3) harboring pET15b-FBPase was grown at 310 K in 4 l LB broth containing 100 µg ml−1 ampicillin until the OD600 was about 0.5 and was then induced with isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.5 mM. The culture was grown for a further 8 h at 298 K and then centrifuged at 4000g for 20 min at 277 K. The cells were washed with phosphate-buffered saline and stored at 250 K until further use. The cell pellets collected from the 4 l culture were gently stirred at 277 K for 45 min with the addition of buffer A (50 mM sodium phosphate buffer pH 8.0 and 300 mM NaCl), 0.50 mg ml−1 lysozyme, 2 U ml−1 DNAse I (Sigma–Aldrich, St Louis, Missouri, USA) and Complete EDTA-free protease inhibitor (Roche Molecular Biochemicals, Indianapolis, Indiana, USA). The lysed cell pellet was subjected to sonication on ice for a total time of 20 min following a fixed sonication–lysis protocol. Insoluble cell debris was removed by centrifugation at 16 000g for 30 min at 277 K. The supernatant was filter-clarified through a 0.45 µm PVDF syringe filter (Millipore, Billerica, Massachusetts, USA) and loaded onto Ni–NTA Bind Resin (Novagen, Gibbstown, New Jersey, USA) pre-equilibrated in buffer A (50 mM sodium phosphate buffer pH 8.0 and 300 mM NaCl) with 10 mM imidazole. Mtb FBPase bound to the Ni–NTA column, which was then washed with 32 column volumes of buffer A plus 20 mM imidazole followed by 40 column volumes of buffer A plus 50 mM imidazole. Mtb FBPase was eluted from the column with six column volumes of buffer A plus 250 mM imidazole. The eluate was immediately subjected to buffer exchange and concentration in an Amicon-15 Ultracel 100K (Millipore, Billerica, Massachusetts, USA) centrifuge concentrator against exchange buffer [20 mM tricine, 50 mM KCl, 1 mM MgCl2, 0.1 mM DTT, 15%(v/v) glycerol pH 7.7] prior to size-exclusion chromatography as described for FBPase II from C. glutamicum (Rittmann et al., 2003 ▶). Mtb FBPase was applied onto a Superdex 200 HiLoad 26/60 column (GE Healthcare Biosciences, Piscataway, New Jersey, USA) on an ÄKTApurifier system (GE Healthcare Biosciences, Piscataway, New Jersey, USA) pre-equilibrated with exchange buffer and eluted at a low flow rate of 1 ml min−1. 3 ml fractions were collected and were pooled according to the chromatogram and the observed purity profile on SDS–PAGE. The pooled fractions were concentrated and filtered through a 0.22 µm membrane filter. For analytical purposes, equal volumes of samples from different purification stages were analyzed by SDS–PAGE. All protein-purification steps were performed at 277 K. The protein concentration was estimated using the Bradford method (Bradford, 1976 ▶).
2.2. Crystallization
For crystallization, purified His-tagged recombinant Mtb FBPase was concentrated to 10 mg ml−1 in exchange buffer. Crystallization trials were performed by the hanging-drop vapor-diffusion method and suitable crystallization conditions were found from Index Screens I and II (Hampton Research, Aliso Viejo, California, USA). The crystals grew in the temperature range 295–300 K. Crystals were obtained by mixing 1 µl protein solution with 1 µl reservoir solution consisting of 1.8 M ammonium citrate tribasic pH 7.0. The volume of the reservoir solution was 300 µl per well.
2.3. Data collection and processing
The crystals were soaked in a cryoprotectant consisting of the reservoir solution (1.8 M ammonium citrate tribasic pH 7.0) plus 20%(v/v) glycerol. Subsequently, the crystals were flash-frozen in liquid nitrogen at 100 K. X-ray diffraction data for Mtb FBPase were collected from a single frozen crystal using a MAR 300 CCD detector on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory, Illinois, USA. A total of 360 frames were collected with 1° scan width and 1 s exposure time. The crystal-to-detector distance was set to 280 mm. The data were indexed and scaled with the HKL-2000 package (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
The recombinant His-tagged FBPase was purified to homogeneity from the soluble fraction by a combination of Ni–NTA and size-exclusion chromatography. The N-terminal hexahistidine tag was not removed from the protein. The Ni–NTA eluate was relatively impure, with some low-molecular-weight and high-molecular-weight impurities; hence, an additional step of size-exclusion chromatography was introduced to purify the protein to homogeneity (Gutka et al., 2011 ▶). Fractions 18–20 from the size-exclusion column were the most pure and had high protein content (Fig. 1 ▶ a); thus, they were pooled for further studies. The final purified Mtb FBPase protein appeared as a single band of approximately 37 kDa on SDS–PAGE (Fig. 1 ▶, fractions 15–22).
Figure 1.

(a) Size-exclusion profile of the affinity-captured protein showing several low-molecular-weight and high-molecular-weight impurities in addition to the main peak of Mtb FBPase. Only the fractions (18–20) corresponding to the main peak were used in the crystallization experiments. (b) Lanes 1–8, SDS–PAGE profile of SEC fractions 15–22. Lane 9, SeeBlue Plus2 molecular-weight standards (labeled in kDa).
Many hexagonal-looking crystals (Fig. 2 ▶) were found after 72 h using 1.8 M ammonium citrate tribasic pH 7.0 as the reservoir solution. The crystals grew to full size in about two weeks. Larger crystals were easily obtained by increasing the total drop volume to 4 µl (2 µl each of protein solution and precipitating agent) and 6 µl (3 µl each of protein solution and precipitating agent), indicating that the crystallization condition is scalable. The presence of Mtb FBPase protein in these crystals was confirmed by running an SDS–PAGE gel after dissolving these crystals in the gel-loading buffer.
Figure 2.

(a) Hexagonal crystals of Mtb FBPase obtained at pH 7.0. (b, c) Larger crystals of Mtb FBPase obtained under similar conditions with increased drop volumes.
Diffraction data were collected using a single crystal cryoprotected with 1.8 M ammonium citrate tribasic pH 7.0 plus 20%(v/v) glycerol. The crystal diffracted to 2.7 Å resolution and a data set with an overall completeness of 98.5% was collected. Symmetry and systematically absent reflections suggested that the Mtb FBPase crystals belonged to space group P6122 (or its enantiomer P6522), with unit-cell parameters a = b = 131.3, c = 143.2 Å, even though the weak reflections in the higher resolution shells had a high R merge (Table 1 ▶) which could indicate a lower symmetry space group. The cross-rotation function was calculated using the program Phaser from the CCP4 suite (Winn et al., 2011 ▶).
Table 1. Data-collection and processing statistics.
Values in parentheses are for the last shell.
| Synchrotron-radiation source | 22-ID, SER-CAT, APS |
| Wavelength (Å) | 1.0000 |
| Space group | P6122 |
| Unit-cell parameters (Å) | a = b = 131.3, c = 143.2 |
| Total No. of reflections | 212726 |
| No. of unique reflections | 20561 |
| Multiplicity | 10.4 (7.2) |
| Resolution (Å) | 40–2.7 (2.75–2.70) |
| Completeness (%) | 98.5 (88.1) |
| Rmerge† | 0.092 (0.686) |
| 〈I/σ(I)〉 | 5.3 (2.0) |
| Mosaicity (°) | 0.43 |
| Wilson plot (B overall) (Å2) | 71.5 |
R
merge = 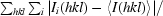
 .
.
Determination of the Matthews coefficient suggested the presence of 50.3% solvent content in the unit cell (V M = 2.47 Å3 Da−1) with a dimer in the asymmetric unit of the higher symmetry space group (Matthews, 1968 ▶). The data-collection statistics are shown in Table 1 ▶.
Initially, the possibility of the space group being P61 could not be excluded. Thus, an initial search was undertaken using the Phaser program (McCoy et al., 2007 ▶) from the CCP4 suite (Winn et al., 2011 ▶) using the E. coli GlpX monomer as a probe (PDB entry 3d1r; Brown et al., 2009 ▶) in space group P61 and looking for four monomers in the asymmetric unit. No complete solution was found; however, a reproducible dimer was recognized among the partial solutions. This dimer differed from that suggested previously as a functional unit for the E. coli enzyme (Brown et al., 2009 ▶) and was used as a search model in space group P6122. A solution was found with MOLREP as implemented in the CCP4 suite (Winn et al., 2011 ▶). The crystallographic statistics for P6122 with one dimer in the asymmetric unit were an R factor of 0.504 and a correlation coefficient of 0.574; those for P6522 were an R factor of 0.632 and a correlation coefficient of 0.308, indicating P6122 to be the correct solution.
This preliminary model allowed the construction of a revised Mtb FBPase model with the correct sequence and initial refinement (using REFMAC as implemented in the Coot software) proceeded normally [R free = 0.390, R value (working + test) = 0.332 and R value (working) = 0.329; Murshudov et al., 2011 ▶]. Further refinement of this structure is in progress and the final structural results will be reported in due course.
Acknowledgments
The authors are grateful to the American Lung Association (Grant No. RG-82534-N) for their support. Drs S. Mehboob and B. Santarsiero are acknowledged for their help with crystallization, data collection and computer support, and Professor M. Johnson and the Center for Pharmaceutical Biotechnology are acknowledged for providing the facilities for crystallization and data processing. Data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
References
- Anstrom, D. M. & Remington, S. J. (2006). Protein Sci. 15, 2002–2007. [DOI] [PMC free article] [PubMed]
- Boshoff, H. I. & Barry, C. E. III (2005). Nature Rev. Microbiol. 3, 70–80. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem. 72, 248–254. [DOI] [PubMed]
- Brown, G., Singer, A., Lunin, V. V., Proudfoot, M., Skarina, T., Flick, R., Kochinyan, S., Sanishvili, R., Joachimiak, A., Edwards, A. M., Savchenko, A. & Yakunin, A. F. (2009). J. Biol. Chem. 284, 3784–3792. [DOI] [PMC free article] [PubMed]
- Chim, N. et al. (2011). Tuberculosis, 91, 155–172. [DOI] [PMC free article] [PubMed]
- Cole, S. T. et al. (1998). Nature (London), 393, 537–544.
- Collins, D. M., Wilson, T., Campbell, S., Buddle, B. M., Wards, B. J., Hotter, G. & De Lisle, G. W. (2002). Microbiology, 148, 3019–3027. [DOI] [PubMed]
- Donahue, J. L., Bownas, J. L., Niehaus, W. G. & Larson, T. J. (2000). J. Bacteriol. 182, 5624–5627. [DOI] [PMC free article] [PubMed]
- Dunn, M. F., Ramírez-Trujillo, J. A. & Hernández-Lucas, I. (2009). Microbiology, 155, 3166–3175. [DOI] [PubMed]
- Gutka, H. J., Rukseree, K., Wheeler, P. R., Franzblau, S. G. & Movahedzadeh, F. (2011). Appl. Biochem. Biotechnol., doi:10.1007/s12010-011-9219-x. [DOI] [PubMed]
- Hines, J. K., Fromm, H. J. & Honzatko, R. B. (2006). J. Biol. Chem. 281, 18386–18393. [DOI] [PubMed]
- Hines, J. K., Fromm, H. J. & Honzatko, R. B. (2007). J. Biol. Chem. 282, 11696–11704. [DOI] [PubMed]
- Hines, J. K., Kruesel, C. E., Fromm, H. J. & Honzatko, R. B. (2007). J. Biol. Chem. 282, 24697–24706. [DOI] [PubMed]
- Liu, K., Yu, J. & Russell, D. G. (2003). Microbiology, 149, 1829–1835. [DOI] [PubMed]
- Marrero, J., Rhee, K. Y., Schnappinger, D., Pethe, K. & Ehrt, S. (2010). Proc. Natl Acad. Sci. USA, 107, 9819–9824. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Movahedzadeh, F., Rison, S. C., Wheeler, P. R., Kendall, S. L., Larson, T. J. & Stoker, N. G. (2004). Microbiology, 150, 3499–3505. [DOI] [PubMed]
- Muñoz-Elías, E. J. & McKinney, J. D. (2005). Nature Med. 11, 638–644. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nishimasu, H., Fushinobu, S., Shoun, H. & Wakagi, T. (2004). Structure, 12, 949–959. [DOI] [PubMed]
- Nunn, P., Williams, B., Floyd, K., Dye, C., Elzinga, G. & Raviglione, M. (2005). Nature Rev. Immunol. 5, 819–826. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pontremoli, S., De Flora, A., Salamino, F., Melloni, E. & Horecker, B. L. (1975). Proc. Natl Acad. Sci. USA, 72, 2969–2973. [DOI] [PMC free article] [PubMed]
- Purohit, H. J., Cheema, S., Lal, S., Raut, C. P. & Kalia, V. C. (2007). Infect. Disord. Drug Targets, 7, 245–250. [DOI] [PubMed]
- Rittmann, D., Schaffer, S., Wendisch, V. F. & Sahm, H. (2003). Arch. Microbiol. 180, 285–292. [DOI] [PubMed]
- Sassetti, C. M. & Rubin, E. J. (2003). Proc. Natl Acad. Sci. USA, 100, 12989–12994. [DOI] [PMC free article] [PubMed]
- Sharma, V., Sharma, S., Hoener zu Bentrup, K., McKinney, J. D., Russell, D. G., Jacobs, W. R. & Sacchettini, J. C. (2000). Nature Struct. Biol. 7, 663–668. [DOI] [PubMed]
- Smith, C. V., Huang, C., Miczak, A., Russell, D. G., Sacchettini, J. C. & Höner zu Bentrup, K. (2003). J. Biol. Chem. 278, 1735–1743. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- World Health Organization (2008). Global Tuberculosis Control: Surveillance, Planning, Financing. WHO Report WHO/HTM/TB/2008.393. Geneva: World Health Organization.