

Abstract
There is considerable interest in the discovery and characterization of single nucleotide polymorphisms (SNPs) to enable the analysis of the potential relationships between human genotype and phenotype. Here we present a strategy that permits the rapid discovery of SNPs from publicly available expressed sequence tag (EST) databases. From a set of ESTs derived from 19 different cDNA libraries, we assembled 300,000 distinct sequences and identified 850 mismatches from contiguous EST data sets (candidate SNP sites), without de novo sequencing. Through a polymerase-mediated, single-base, primer extension technique, Genetic Bit Analysis (GBA), we confirmed the presence of a subset of these candidate SNP sites and have estimated the allele frequencies in three human populations with different ethnic origins. Altogether, our approach provides a basis for rapid and efficient regional and genome-wide SNP discovery using data assembled from sequences from different libraries of cDNAs.
[The SNPs identified in this study can be found in the National Center of Biotechnology (NCBI) SNP database under submitter handles ORCHID (SNPS-981210-A) and debnick (SNPS-981209-A and SNPS-981209-B).]
The development of methods for the in vitro amplification of specific target sequences, such as the polymerase chain reaction (PCR), and the discovery of highly informative genome-wide polymorphic markers such as microsatellites, or short tandem repeat (STR) sequences, have enabled the creation of low-density genetic maps for all human chromosomes (Dib et al. 1996; Broman et al. 1998). These resources have enabled the location of genes through positional cloning techniques for many different traits, such as cystic fibrosis and Huntington’s disease, which are inherited in a simple Mendelian fashion (Collins 1995). Increasingly, however, studies have focused on mapping susceptibility genes for more complex and common disorders such as diabetes (Mein et al. 1998), breast cancer (Miki et al. 1994), atherosclerosis, obesity, and hypertension (Schork 1997; Cl’ement et al. 1998), and autoimmune disorders (Becker et al. 1998). It has been suggested that the identification of genes with low penetrance or modest effects on human traits, like those generating susceptibility or resistance to common disorders, may require the use of genetic maps containing 100,000 to 300,000 randomly spaced markers from the human genome (Kruglyak 1997; Wang 1998).
To generate sufficiently dense genetic maps for complex trait mapping, efforts have focused on identifying the most common type of DNA sequence variation, single nucleotide polymorphisms (SNPs). These variations are estimated to occur once every 500 to 1000 bp when any two chromosomes are compared (Cooper et al. 1985; Li and Sadler 1991; Harding et al. 1997). Thus, scanning the human genome for this form of sequence variation (Collins et al. 1997) could identify millions of potentially informative genetic markers. The development of genetic maps composed of SNPs has many other advantages with respect to population-based analysis of the human genome. First, SNPs are diallelic in populations, and their allele frequencies can be estimated easily in any population through a variety of techniques (Kwok et al. 1994). Second, SNPs are highly stable genetic markers compared to tandem repeat markers where the high mutation rates can confound genetic analysis in populations (Hastbacka et al. 1992; Marshall et al. 1993). Last, many technologies have been developed to type SNPs in an automated fashion, and many of these yield simple positive or negative outcomes that can be interpreted easily by a computer (Nikiforov et al. 1994a; Hacia et al. 1996; Head et al. 1997; Wang et al. 1998).
Although SNPs can be typed rapidly when identified, the process of genome-wide SNP discovery has been initiated only recently (Wang et al. 1998). To identify new gene-associated SNPs, we have taken advantage of the rapidly developing databases of partial cDNA sequences, expressed sequence tags (ESTs), that have been generated from many different human tissues. Because the majority of these libraries have been obtained from different individuals, assembly of overlapping sequences for the same region can lead to the identification of new SNPs. Furthermore, even within a single library, SNPs can be identified as differences between the sequences of the contributed maternal and paternal chromosomes. In this report, we describe a strategy for rapidly identifying candidate SNPs within assembled cDNA sequences and present data that suggests that at least 50% of these candidates are highly polymorphic SNPs. A significant risk in such an analysis is that many sequence variations are the result of poor quality sequence data typically found in single-pass EST data sets. Therefore, the success of any strategy using EST data sets hinges on the strategies used to cull sequence errors from the candidate pool.
RESULTS
cDNA Assembly
Sequence data from 19 normalized cDNA libraries from the Washington University EST database (Hillier et al. 1996) were assembled into contigs using Phred, a base-calling program (Ewing and Green 1998; Ewing et al. 1998), and Phrap, a sequence alignment and contig assembly program (P. Green, http://genome.washington.edu). Phrap groups sequence reads into contigs based on their Smith–Waterman sequence similarity and stores the relative alignment information of each sequence. It also classifies sequences that do not have sufficient similarity with any other sequence as “singlets” and excludes them from the set of assembled contigs. In this analysis we assembled the 5′ and 3′ ESTs independently. This was done to simplify the subsequent identification of coding SNPs (cSNPs) that could be abundant among the 5′ contigs. It also simplified the molecular confirmation of a group of SNPs in the 3′ set, particularly in the absence of data on exon/intron boundaries, as SNPs in these contigs were likely to be located in contiguous, noncoding sequences. After assembly with Phrap, the 5′ set of 113,497 sequences yielded 21,447 contigs containing two or more sequence reads, whereas the 3′ set of 109,305 sequences assembled into 19,198 contigs of two or more reads. Seventy-eight thousand total sequences were classified as singlets and were excluded from the contigs. The mean number of sequence reads per contig was 5.3 and 5.7 for the 5′ and 3′ assemblies, respectively, and followed an exponential distribution (data not shown).
Identifying Candidate SNPs Among ESTs
Although biases in the number of unique alleles represented per contig is likely based on tissue specificity of the ESTs and the number of ESTs contributed per library, the description of the 19 assembled libraries suggested that at least 10 individuals (20 chromosomes) were possibly represented among the various contigs. Therefore, nucleotide mismatches in overlapping sequence reads could represent common sequence variations. In fact, >6000 mismatches were identified in these assemblies; however, some of these mismatches were probably attributable to base-calling errors or errors generated during cDNA synthesis or propagation in Escherichia coli (polymerase or reverse transcriptase errors; Cooper and Krawczak 1995). To address these potential biases in the data set we devised a strategy to sort mismatches and select high-quality candidate SNPs through a series of four filters.
Filter 1 eliminated clusters of mismatches that often occur in regions of low-quality trace data. This filter searched for three window sizes of perfectly matched sequences of 5, 10, or 20 bp around each candidate single base-pair mismatch (Fig. 1). Filter 2 identified sequence mismatches by either base substitution type or insertion/deletion type. Because the program Phrap is designed to align many sequences, the software often inserts a space in one sequence to align it with another sequence. As these types of insertion/deletion mismatches are typically an artifact of the software and attributable to low-quality sequence data, we concentrated on substitution types for the resequencing and confirmation of candidate SNP sites later in this project.
Figure 1.
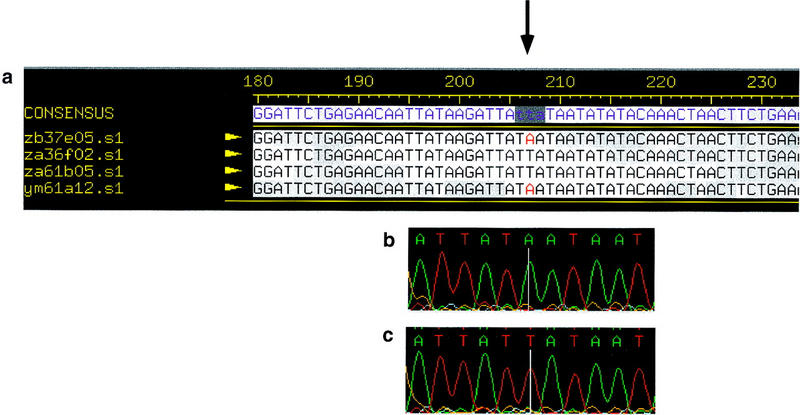
An example of a contig containing a high quality mismatch. (a) A Consed view of a contig containing sequences from the 3′-untranslated region of the erythroblastosis virus oncogene homolog 2 (ETS2). The arrow indicates the location of the high-quality mismatch (A vs. T, position 207). Examples of sequence traces show an A at position 207 (b) and a T at position 207 (c). The mismatch has been confirmed as a common SNP by DNA sequence analysis.
Filters 3 and 4 addressed the quality of each base call relative to its position and frequency in a contig. Because the quality of base calls in the early portion of a read are more prone to error (Koop et al. 1993; Ewing and Green 1998; Ewing et al. 1998), we eliminated the base-substitution mismatches in the first 100 bases of a sequence in Filter 3. In Filter 4 we stipulated that a sequence mismatch must occur in more than one sequence in a contig before it could be considered a high-quality candidate SNP. This final filter limited the number of mismatches that could arise from random copying or cloning errors, as these are unlikely to occur at the same base twice.
To test the filtering scheme, we randomly selected 100 contigs (fifty 5′ and fifty 3′ contigs) and visually inspected the mismatches using the Consed program (Gordon et al. 1998). A candidate SNP was considered verified if the trace data was of high quality as determined by Phred base-calling software and if the candidate SNP passed through all four filters. Filter 1 eliminated many sequence mismatches, mainly attributable to base-calling errors in the test data set. These errors often occurred in regions of low sequence quality and tended to cluster together. Results from this filter indicated that ≤44% of the mismatches were candidate SNPs (Table 1). Window sizes >5 bp did not proportionally increase the number of candidate mismatches that could be verified visually; however, during the resequencing and SNP confirmation portion of this project we concentrated mostly on contigs containing mismatches with windows of 20 bp.
Table 1.
Percent Verified EST–SNPs
| Direction | Window size | Substitutions verified (%) | Ins/Del verified (%) | Total verified (%) |
|---|---|---|---|---|
| 5′ | 5 | 92 | 10 | 26 |
| 10 | 100 | 23 | 40 | |
| 20 | 100 | 7 | 44 | |
| 3′ | 5 | 80 | 11 | 34 |
| 10 | 81 | 37 | 32 | |
| 20 | 80 | 18 | 38 |
The total number of EST–SNPs in the entire data set was estimated for both 5′ and 3′ contigs. Three window frame sizes (5, 10, and 20 bp), corresponding to the number of matching base pairs 5′ and 3′ to a sequence mismatch, were tested.
The results from Filter 2, which screened for polymorphism type, indicated that mismatches attributable to base substitution types were of high quality and more likely to be SNPs than insertion/deletion types. More than 80% of the substitution mismatches in the test set were verified with Consed for 5, 10, and 20 bp window frame sizes (Table 1). Thus, during the SNP confirmation portion of this project we chose to concentrate primarily on substitution-type mismatches, namely A/G or T/C base substitutions as these types make up >60% of all base substitutions (Table 2). These base substitution results agree with the proportion of transition type substitutions previously reported to occur in human DNA (Harding et al. 1997; Nickerson et al. 1998).
Table 2.
Distribution of Substitution Types among the Candidate SNPs
| Type | Contigs | Total | Percent | |
|---|---|---|---|---|
| 3′ | 5′ | |||
| A/C | 38 | 24 | 62 | 7.3 |
| G/T | 70 | 47 | 117 | 13.8 |
| A/G | 128 | 119 | 247 | 29 |
| C/T | 158 | 123 | 281 | 33 |
| A/T | 46 | 25 | 71 | 8.4 |
| C/G | 50 | 22 | 72 | 8.5 |
Distribution of SNPs. Together, A/G and C/T substitution types totaled >60% of the substitution types.
Fewer candidate SNP sites were identified in the 5′ contigs (360 candidates) than the 3′ contigs (490 candidates). This result may be by chance, or may be related to the coding potential of the 5′ and 3′ sequences as lower nucleotide diversities may be found in coding sequences that could be more prevalent among the 5′ sequences (Li and Stadler 1991; Nickerson et al. 1998). Higher quality libraries should allow for more accurate coding versus non-coding frequency analysis in the future.
The percentage of verified SNPs in the test data set dropped by half after runs through Filters 3 and 4. These filters eliminated mismatches in the first 100 bases of a contig as well as eliminated mismatches that were seen in only one sequence of a contig. On the basis of the filtering results of the test data set, we estimated that the candidate pool of SNPs in the entire data set of 6000 mismatches was 850 sites.
Confirmation of Candidate SNPs
To verify that the 850 candidate SNPs were potentially polymorphic, ∼10% of the candidates were randomly selected for molecular confirmation after analysis with the viewing software Consed. A total of 88 high-quality mismatches (candidate SNPs) from 73 different contigs were tested. Primers for PCR amplification of the candidate SNP sites were designed based on the contig data for each site. Seventeen of the 88 sites were analyzed initially by fluorescence-based DNA sequencing of PCR products. An example of two common but silent base substitutions confirmed by sequence analysis in the coding region of the GADD34 gene (an apoptosis-associated protein) are shown in Figure 2. Of the 17 sites that were sequenced in three individuals, 11 sites were confirmed as common—occurring on at least one chromosome—among the individuals sequenced (Table 3; Genome Research online supplement).
Figure 2.
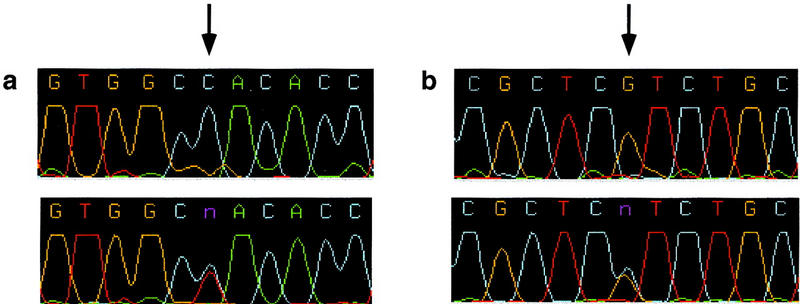
Examples of two SNPs in the GADD34 gene confirmed by DNA sequence analysis. (a) Homozygote C vs. heterozygote C/T; (b) homozygote G vs. heterozygote G/C. Less than 2% of contigs contained more than one SNP. When confirmed the majority of these were in complete linkage disequilibrium with one another.
Table 3.
EST–SNPs Confirmed by Fluorescence-Based Sequencing
| Contiga | EST–SNP sequence contextb | Frequencyc | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | A | G | C | T | G | T | G | G | C | (C/T) | A | C | A | C | C | T | T | C | C | C | 4/2 |
| 1 | C | T | T | C | C | C | G | C | T | C | (G/C) | T | C | T | G | C | T | G | C | T | G | 4/2 |
| 2 | C | C | A | T | A | A | A | T | G | C | (G/A) | C | T | A | A | G | A | T | A | A | A | 5/1 |
| 2 | A | A | T | T | A | A | A | G | A | A | (C/T) | A | A | T | A | A | T | G | T | T | C | 5/1 |
| 3 | T | T | T | T | A | A | C | A | A | A | (G/C) | T | A | A | T | C | T | T | C | A | C | 5/1 |
| 4 | A | T | A | A | G | A | T | T | A | T | (T/A) | A | T | A | A | T | A | T | A | T | A | 5/1 |
| 5 | T | T | C | A | G | T | T | T | T | G | (A/T) | G | G | C | T | C | A | T | G | G | G | 5/1 |
| 6 | C | A | G | T | C | T | G | T | A | T | (C/T) | T | T | C | C | A | A | A | A | A | G | 5/1 |
| 7 | G | A | G | T | C | G | A | A | A | T | (G/C) | T | G | A | T | T | C | T | T | C | A | 4/2 |
| 7 | G | A | A | T | G | C | A | G | T | C | (C/T) | G | T | C | A | T | A | T | G | A | C | 4/2 |
| 7 | T | G | A | C | C | A | C | T | A | A | (C/T) | T | T | G | C | A | T | G | T | G | A | 4/2 |
Summary of SNPs confirmed by fluorescent-based sequencing.
The contig sequences and their corresponding GenBank accession numbers can be found in the NCBI SNP database under the submitter handle debnick SNPS-981209-A and SNPS-981209-B.
More than one SNP per contig was confirmed in contigs 1, 2, and 7.
Context surrounding the substitution noted between parentheses (major allele/minor allele).
Total of six chromosomes sequenced (three individuals).
Because it was necessary to increase the throughput to demonstrate the effectiveness of our strategy, we moved to SNP confirmation by Genetic Bit Analysis (GBA). This method, a solid phase sequencing technique, enabled us to increase the rate of SNP confirmation as well as increase the number of chromosomes assessed for each candidate site. GBA uses primer extension with modified nucleotides to determine the genotype of an amplified DNA sample. The data that results from this typing approach are discrete, allowing for unambiguous identification of homozygous and heterozygous individuals, as well as automatable, high throughput processing and data analysis (Fig. 3).
Figure 3.

Cluster analyses of candidate EST–SNP sites of GBA genotype data from two contigs. The contig number and library source (3′ or 5′) are given. Each GBA experiment assayed 18 genomic DNA samples from three ethnic groups: eight Caucasian DNA samples (+), five African American DNA samples (o), and five Hispanic DNA samples (x), as well as two positive (/) and two negative controls (\ , −). Raw optical density values from the two-color ELISA system were plotted on an xy scatterplot. Allele 1 data (fluorescein–PNPP reactions) were captured by a standard plate reader at 405 nm and were plotted on the x-axis. Allele 2 readings (biotin–TMB reactions, 620 nm) were plotted on the y-axis. Thus, the axes represent the base analog extension signals determined by primer extension. Homozygotes for allele 1 lie on the x-axis, homozygotes for allele 2 lie on the y-axis, and heterozygotes lie on the diagonal. Negative PCR controls (\, reactions controlling for cross-hybridization of the PCR primer and capture–extension primer) and negative GBA controls (−, reactions controlling for self-extension of the capture–extension primer) lie near the plot origins. Synthetic templates (/) were also included as positive controls to monitor hybridization and extension efficiency. Cluster analyses for genotype determination were done automatically by an in-house software program.
Using GBA, 71 additional sites in 60 contigs were tested across three ethnic groups: Caucasian, African-American, and Hispanic. To demonstrate the reliability of the genotype calls, 6 SNP sites of the 71 tested by GBA were also tested and confirmed by fluorescent-based sequencing on three samples (sequence data not shown). Of the 71 sites tested by GBA, 44 sites proved to be polymorphic in one or more of the populations (Table 4; Genome Research online supplement), a frequency that is consistent with that observed in the original 17 fluorescence-based sequenced sites, 62% and 65%, respectively. The average frequency of the major allele from the 71 GBA-tested SNP markers was 0.76. The average heterozygosity was 0.33 and the median heterozygosity was 0.35. Contig 14 (see Table 4) contained multiple SNP sites that do not appear to be in linkage disequilibrium according to the allele frequency data based on 36 chromosomes. Contigs 1, 2, and 7 (see Table 3) also contained multiple SNP sites. The frequencies of the SNP alleles within each of these contigs were identical, but only 6 chromosomes were assayed in these cases. With further analysis, the SNPs within contigs 1 and 2 were found to be in complete linkage disequilibrium; however, the SNPs within contig 7 were not linked (data not shown). Further analyses of contigs 7 and 14 to reveal intron/exon boundaries could provide more information.
Table 4.
Summary of EST SNPs confirmed by GBA
 |
Summary of GBA results. Forty-four candidate EST–SNP sites were confirmed by GBA analysis; a subset of six sites (*) were also confirmed by fluorescent-based sequencing. Multiple SNPs were assayed in contig 14. The sequence context surrounding the base substitution (major allele/minor) is noted. The allele frequency data per ethnic group, and on average, were calculated based on GBA data from a total of 36 chromosomes: 8 individuals (16 chromosomes) for the Caucasian population and 5 individuals (10 chromosomes) for both African American and Hispanic populations. The frequency of the major allele is presented (n/a = not available). Heterozygosity values were calculated based on the average allele frequency for each EST–SNP. The contig sequences and their corresponding GenBank accession numbers can be found in the NCBI SNP database under submitter handle ORCHID SNPS-981210-A.
Overall 63% of the candidate SNPs (55 of 88) tested by either sequence analysis or GBA were confirmed as polymorphic in the population analyzed. In contrast, 29 sites that were clearly high-quality mismatches by visual inspection typed as monomorphic (41% overall). It is possible that a fraction of these sites represent alleles that are population specific or have frequencies less than 3% (1 allele from 18 samples or 36 chromosomes). Interestingly, we identified a small number of the candidate sites (4 of 88) that were heterozygous in all the typed samples. These candidates, although high quality, were likely the result of identifying mismatches between two members of a multigene family.
DISCUSSION
We have demonstrated a strategy for the rapid identification and verification of SNP-based genetic markers using EST data sources. The use of EST sequence data for the identification of SNPs has many advantages that can be exploited to facilitate the development of highly dense genetic maps for the analysis of human populations. One of the main advantages of using EST sources is that markers closely associated with, or directly in the coding region of human genes, can be identified, thus maximizing the density of a map toward gene-associated markers. Approximately 40% of the candidate SNPs we identified were in genes newly identified through cDNA sequencing (Adams et al. 1993; Hillier et al. 1996). In addition to finding variants in new genes, it is also possible that this approach could identify a large number of sequence variants that lead to amino acid substitutions and perhaps lead to functional differences, which could be associated with phenotypic effects that are frequent in the population. A classic example of this is the variation in cytochrome P450 genes. Amino acid variation attributable to several SNP sites in the CYP2D6 gene cause poor metabolism in the debrisoquine oxidation pathway (Sachse et al. 1997).
Another advantage to this EST approach is that it can identify common and perhaps uncommon sequence variations within the human genome. More than half of the nucleotide substitutions we confirmed in this pilot study were common in the population. Although 41% appeared to be monomorphic in the populations we analyzed, the quality of these mismatches indicates that they may represent rarer sequence variations in human genome that may also be population specific (Harding et al. 1997; Nickerson et al. 1998). In this respect, a recent survey of sequence diversity in the human genome suggests that a large proportion of the sequence variants found in the human genome may be population specific or rare in the populations under study. If one considers that the candidates we identified as monomorphic represent rare variants in humans, then the distribution of common to rarer variants is similar to that previously reported (Nickerson et al. 1998). It is important to note that this EST–SNP mining approach could identify some rare variants; however, the majority of these would be missed.
The ability to detect rare polymorphisms, however, should grow as the number of different cDNA libraries increases. As the quality and diversity of the libraries used for sequencing and contig construction improve, it is also likely that errors attributable to sequencing will decrease, whereas the quality of candidate SNPs will increase. In addition, as the quality of 5′ libraries improves, coding sequence variants that change amino acids could potentially be directly screened. To ensure the continued success of this approach it is crucial that the number of cDNA libraries continues to increase.
In this pilot study, we have taken an approach that relied on visual inspection of the traces to enable the development of filters to enrich for high quality mismatches among contigs. Although this approach was clearly effective it was still analysis intensive. One way to further automate this process would be to enrich the data for mismatches based on the quality scores given the two potential alleles by the Phred software tool. Because quality scores in Phred are related to the probability of an error in base-calling, one should be able to select for only those mismatches that had an extremely low probability of error as the result of base-calling. [For example, by selecting mismatches that had a probability of error of 1 in 10,000 (Phred quality 40) for each allele.] We are currently exploring the use of such a filter in identifying new SNP markers in EST data.
One of the obvious benefits of the described approach is its cost effectiveness, as it takes advantage of existing sequence resources generated for gene discovery rather than marker discovery, thus providing a set of useful EST–SNPs with minimal additional requirements. Because the Washington University EST database appears to be doubling every 6 months and is currently growing at a rate of more than 5000 EST sequences per week, the utility of this resource for identifying new genes, as well as new SNPs, will continue to grow, particularly as new libraries representing alternative alleles are also added to this resource. Furthermore, the location of these genes and any new SNP markers will also continue to develop as more ESTs are mapped to chromosomes or chromosome regions.
Finally, as the sequence context of the EST-associated SNPs is already known, it is possible to exploit some of the automated approaches available to genotype human populations on a large scale, such as GBA primer extension, sequencing by hybridization, and hybridization to arrays (Nikiforov et al. 1994a; Chee et al. 1996; Yershov et al. 1996). Because of their potential importance in genetic diseases, as well as their density in the genome and low mutation rate, SNP-based markers will rapidly assume an integral role in large-scale analysis of human genotype–phenotype relationships. As we have shown here, the use of existing resources can rapidly and efficiently lead to high-volume, cost-effective SNP discovery. Given the large number of potential SNPs in the human genome (between 2 and 5 million) and the large amount of sequence data needed to detect them (ultimately between 10 and 20 genomes worth), in silico strategies are essential to rapid, cost-effective polymorphism discovery.
METHODS
EST Sequence Data
The sequence data used in this study were obtained through the Washington University Genome Center web server (http://genome.wustl.edu/gsc/gschmpg.html) in a FASTA format and were generated by a collaborative effort between the Washington University Genome Center in St. Louis and Merck Pharmaceuticals (Hillier et al. 1996). The libraries analyzed here were obtained from a collection of normalized cDNA libraries generated at Columbia University by Bento Soares and included 19 libraries (both 5′ and 3′).The library descriptions were as follows: Library 1 Soares_adult_brain_N2b4HB55Y, male 55 years; Library 2 Soares_adult_brain_N2b5HB55Y, male 55 years; Library 3 Soares_breast_2NbHBst, female adult; Library 4 Soares_breast_3NbHBst, female adult; Library 5 Soares_fetal_heart_NbHH19W 19 weeks; Library 6 Soares_fetal_liver_spleen_1NFLS, male 20 weeks; Library 7 Soares_fetal_liver_spleen_1NFLS_S1; Library 8 Soares_fetal_lung_NbHL19W, 19 weeks; Library 9 Soares_infant_brain_1NIB, 73 days; Library10 Soares_melanocyte_2NbHM, male; Library 11 Soares_multiple_sclerosis_2NbHMSP, male 46 years; Library 12 Soares_ovary_tumor_NbHOT; Library 13 Soares_parathyroid_tumor_NbHPA, adult; Library 14 Soares_pineal_gland_N3HPG; Library 15 Soares_placenta_1NHP; Library 16 Soares_placenta_8to9weeks_2NbHP8to9W; Library 17 Soares_retina_N2b4HR, male 55 years; Library 18 Soares_retina_N2b5HR, male 55 years; Library 19 Soares_senescent_fibroblasts_NbHSF.
EST Assembly
Sequences from the 3′ and 5′ ends of the cDNA clones were assembled in two separate groups using the Phrap sequence assembler [http://genome.washington.edu (Ewing and Green 1998; Ewing et al. 1998)]. In general, the library files were not screened before contig assembly and alignment. One exception to this rule were the immunoglobin sequences associated with the Soares_fetal_liver_spleen_1NFLS library (both 3′ and 5′). Sequences from this large multigene family were abundant in this library and without a prescreen, several thousand of them aligned into a single contig, requiring >1 gB of computer memory allocation to complete. Masking these immunoglobulin sequences was necessary to curtail memory usage during the assembly procedure.
Viewing Software
Consed (Gordon et al. 1998) was used for the comparison of multiple sequences trace files that were processed by Phred (Ewing and Green 1998; Ewing et al. 1998) and aligned by Phrap (http://genome.washington.edu). This software was used to visually inspect the ABI trace files of candidate EST–SNP selections by the filtering software.
DNA Samples and Amplification
DNA samples from randomly selected individuals representing three ethnic groups (Caucasian, African American, and Hispanic) were used in this analysis. Blood samples were prepared according to standard phenol/chloroform extraction and ethanol precipitation procedures (Sambrook et al. 1989) or by chelex isolations (Walsh et al. 1991). The DNA isolates were assembled in 96-well plates for amplification by PCR as in Reynolds et al. (1997).
Genotyping Methods
Primer Designs
Primers for PCR and GBA were designed by an in-house software program, GBA-Primer 1.1, that was based on the 1991 PRIMER program developed by Mark J. Daly, Steve E. Lincoln, and Eric S. Lander (http://www-genome.wi.mit.edu/ftp/pub/software/primer.0.5). GBA–Primer was used to evaluate primer melting temperature, annealing temperature, and the likelihood of oligonucleotide self-priming. Primers were selected to perform under the same conditions (annealing temperature of 55°C). GBA primers were designed to capture single-stranded PCR products by hybridization (Reynolds et al. 1997). The primers were designed such that their 3′ ends were immediately adjacent to the polymorphism of interest and were ∼25 bp in length. Because either the coding or noncoding DNA strand may be typed, selection of the target strand and GBA primer were based on evaluations of PCR product and GBA primer secondary structure stabilities (measured in kcal/mole; data not shown).
GBA Methodology
The hybridization conditions, extension reactions, and colorimetric detection of incorporated labeled nucleotides were followed as in Reynolds et al. (1997). Single-stranded PCR templates were generated by selective digestion with T7 gene 6, 5′ → 3′ exonuclease (Nikiforov et al. 1994b) and hybridized to GBA primers in a 96-well plate format at room temperature. After hybridization of the template strands, GBA primers were extended by one base at the polymorphic site of interest, complimentary to the PCR template strands. The extension mixes contained two labeled dideoxynucleotides (one fluorescein, one biotin) and two unlabeled dideoxynucleotides. Extension reactions were performed at room temperature for 15 min using the Klenow fragment of DNA polymerase I (exonuclease-free) as described in Reynolds et al. (1997). Detection of the extended primers was done by standard enzyme-linked immunofluorescent techniques (ELISA). Antifluorescein–alkaline phosphatase (Boehringer Mannheim, Indianapolis, IN) was used with the substrate p-nitrophenyl phosphate (PNPP; Moss, Pasadena, MD) to detect fluoresceinated nucleotides (405 nm wavelength), representing allele 1. An antibiotin–horseradish peroxidase conjugate (Zymed, San Francisco, CA) followed by the substrate tetramethylbenzidine (TMB; Moss, Pasadena, MD) was then used to detect biotinylated nucleotides (620 nm), representing allele 2, for each EST–SNP.
GBA Genotype Determinations
The raw OD data from the ELISA detection were captured by a standard plate reader (ICN, Costa Mesa, CA) and analyzed by an in-house software program, GenoMatic, which uses cluster analyses of the raw OD signals to determine sample genotypes. Each genotype call was automatically assigned a confidence measure according to the most likely or probable cluster in which a data point was located. Automated genotype calls were corroborated by visual inspection of the data.
Sequencing Methods
Twenty-six percent of the candidate SNPs were confirmed by sequence analysis. PCR products were sequenced by standard dye terminator methods using AmpliTaq DNA polymerase, FS as previously described in detail (Nickerson et al. 1997). In some cases, the PCR primers were also used as the sequencing primers.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lpn@orchidbio.com; FAX (410) 558-5910.
REFERENCES
- Adams MD, Soares MB, Kerlavage AR, Fields C, Venter JC. Rapid cDNA sequencing (expressed sequence tags) from a directionally cloned human infant brain cDNA library. Nat Genet. 1993;4:373–380. doi: 10.1038/ng0893-373. [DOI] [PubMed] [Google Scholar]
- Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, Trent JM. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci. 1998;95:9979–9984. doi: 10.1073/pnas.95.17.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. Comprehensive human genetic maps: Individual and sex-specific variation in recombination. Am J Hum Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D, Winkler J, Lockhart DJ, Morris MS, Fodor SP. Accessing genetic information with high-density DNA arrays. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- Cl’ement K, Vaisse C, Lahlow N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- Collins FS. Positional cloning moves from perditional to traditional. Nat Genet. 1995;9:347–350. doi: 10.1038/ng0495-347. [DOI] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Charkravarti A. Variations on a theme: Cataloging human DNA sequence variation. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M. Human gene mutation. UK: Coronet Books, Bios Scientific Publishers, Ltd.; 1995. [Google Scholar]
- Cooper DN, Smith BA, Cooke H, Niemann S, Schmidtke J. An estimate of unique sequence heterozygosity in the human genome. Hum Genet. 1985;69:201–205. doi: 10.1007/BF00293024. [DOI] [PubMed] [Google Scholar]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazen J, Seboun E, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:A1–A138. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Hacia JG, Brody LC, Chee MS, Fodor SP, Collins FS. Detection of heterozygous mutations in BRCA1 using high density oligonucleotide arrays and two-colour fluorescence analysis. Nat Genet. 1996;14:441–447. doi: 10.1038/ng1296-441. [DOI] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB. Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E. Linkage disequilibrium mapping in isolated founder populations: Diastrophic dysplasia in Finland. Nat Genet. 1992;2:204–211. doi: 10.1038/ng1192-204. [DOI] [PubMed] [Google Scholar]
- Head SR, Rogers Y-H, Parikh K, Lan G, Anderson S, Goelet P, Boyce-Jacino MT. Nested genetic bit analysis (N-GBA) for mutation detection in the p53 tumor suppressor gene. Nucleic Acids Res. 1997;25:5065–5071. doi: 10.1093/nar/25.24.5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier L, Lennon G, Becker M, Bonaldo MF, Chiapelli B, Chissoe S, Dietrich N, DuBuque T, Favello A, Gish W, et al. Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 1996;6:807–828. doi: 10.1101/gr.6.9.807. [DOI] [PubMed] [Google Scholar]
- Koop BF, Rowan L, Chen WQ, Deshpande P, Lee H, Hood L. Sequence length and error analysis of Sequenase and automated Taq cycle sequencing methods. BioTechniques. 1993;14:442–447. [PubMed] [Google Scholar]
- Kruglyak L. The use of a genetic map of biallelic markers in linkage studies. Nat Genet. 1997;17:21–24. doi: 10.1038/ng0997-21. [DOI] [PubMed] [Google Scholar]
- Kwok PY, Carlson C, Yager TD, Ankener W, Nickerson DA. Comparative analysis of human DNA variations by fluorescence-based sequencing of PCR products. 1994. Genomics. 1994;23:138–144. doi: 10.1006/geno.1994.1469. [DOI] [PubMed] [Google Scholar]
- Li WH, Sadler LA. Low nucleotide diversity in man. Genetics. 1991;129:513–523. doi: 10.1093/genetics/129.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall B, Tay G, Marley J, Abraham LJ, Dawkins RL. Analysis of MHC genomic structure and gene content between HLA-B and TNF using yeast artificial chromosomes. Genomics. 1993;17:435–441. doi: 10.1006/geno.1993.1344. [DOI] [PubMed] [Google Scholar]
- Mein CA, Esposito L, Dunn MG, Johnson GC, Timms AE, Goy JV, Smith AN, Sebag-Montefiore L, Merriman ME, Wilson AJ, et al. A search for type 1 diabetes susceptibility genes in families from the United Kingdom. Nat Genet. 1998;19:213–214. doi: 10.1038/991. [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavitigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. PolyPhred: Automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson DA, Taylor SL, Weiss KM, Clark AG, Hutchinson RG, Stengard J, Salomaa V, Vartiainen E, Boerwinkle E, Sing CF. DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat Genet. 1998;19:233–240. doi: 10.1038/907. [DOI] [PubMed] [Google Scholar]
- Nikiforov TT, Rendle RB, Goelet P, Rogers Y-H, Kotewicz ML, Anderson S, Trainor GL, Knapp M. Genetic bit analysis: A solid phase method for typing single nucleotide polymorphisms. Nucleic Acids Res. 1994a;22:4167–4175. doi: 10.1093/nar/22.20.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov TT, Rendle RB, Kotewicz ML, Rogers Y-H. The use of phosphorothioate primers and exonuclease hydrolysis for the preparation of single-stranded PCR products and their detection by solid-phase hybridization. PCR Methods Applic. 1994b;3:285–291. doi: 10.1101/gr.3.5.285. [DOI] [PubMed] [Google Scholar]
- Reynolds JE, Head SR, McIntosh TC, Picoult-Vrolijk L, Boyce-Jacino MT. Genetic bit analysisTM : A solid-phase method for genotyping single nucleotide polymorphisms. In: Caetano-Anolles G, editor. DNA markers: Protocols, applications, and overviews. New York, NY: Wiley-Liss; 1997. pp. 199–211. [Google Scholar]
- Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: Allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60:284–295. [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schork NJ. Genetically complex cardiovascular traits. Origins, problems, and potential solutions. Hypertension. 1997;29:145–149. doi: 10.1161/01.hyp.29.1.145. [DOI] [PubMed] [Google Scholar]
- Yershov G, Barsky V, Belgovskiy A, Kirillov E, Kreindlin E, Ivanov I, Parinov S, Guschin D, Drobishev A, Dubiley S, Mirzabekov A. DNA analysis and diagnostics on oligonucleotide microchips. Proc Natl Acad Sci. 1996;93:4913–4918. doi: 10.1073/pnas.93.10.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. BioTechniques. 1991;10:506–513. [PubMed] [Google Scholar]
- Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]