

Abstract
We measured isocyanic acid (HNCO) in laboratory biomass fires at levels up to 600 parts per billion by volume (ppbv), demonstrating that it has a significant source from pyrolysis/combustion of biomass. We also measured HNCO at mixing ratios up to 200 pptv (parts-per-trillion by volume) in ambient air in urban Los Angeles, CA, and in Boulder, CO, during the recent 2010 Fourmile Canyon fire. Further, our measurements of aqueous solubility show that HNCO is highly soluble, as it dissociates at physiological pH. Exposure levels > 1 ppbv provide a direct source of isocyanic acid and cyanate ion (NCO-) to humans at levels that have recognized health effects: atherosclerosis, cataracts, and rheumatoid arthritis, through the mechanism of protein carbamylation. In addition to the wildland fire and urban sources, we observed HNCO in tobacco smoke, HNCO has been reported from the low-temperature combustion of coal, and as a by-product of urea-selective catalytic reduction (SCR) systems that are being phased-in to control on-road diesel NOx emissions in the United States and the European Union. Given the current levels of exposure in populations that burn biomass or use tobacco, the expected growth in biomass burning emissions with warmer, drier regional climates, and planned increase in diesel SCR controls, it is imperative that we understand the extent and effects of this HNCO exposure.
Keywords: troposphere, heterogeneous chemistry
Every day billions of people are exposed to smoke: from tobacco, from biomass, or low-temperature coal combustion used for cooking and heating, and from wildfires (1, 2). Extensive research on the pyrolysis of biomaterials has shown that numerous volatile and semivolatile organic compounds are produced (3–5). It is important to understand the impacts of these emissions from the global scale down to the personal level. Significant human health effects have been associated with smoke exposure, including cataracts, cardiovascular impairment, and chronic diseases such as rheumatoid arthritis (2, 6). A number of studies have shown that protein carbamylation and associated inflammatory response is a common biochemical pathway causing these effects (7–9). In vivo isocyanic acid (HNCO, H─N═C═O) and its aqueous anion, cyanate (NCO-), have been identified as biochemical intermediates in this protein carbamylation (8). In this work we show that smoke from biomass, including tobacco, contains HNCO at concentrations that cause carbamylation at physiologically significant levels. Thus smoke-related HNCO exposure is strongly linked to several classes of major negative health effects.
Isocyanic acid has been known since Liebig and Wöhler (10), but it has not previously been measured in the atmosphere. The compound is moderately acidic (pKa = 3.7) and unstable in pure form as it readily polymerizes (11). However, it is volatile (BP = 23.5 °C estimated) and relatively stable at dilute (several ppmv) concentrations in the gas-phase (12). Isocyanates are toxic at high concentrations, as was demonstrated after the accidental release of methyl isocyanate, CH3NCO, in Bhopal, India, when thousands of people suffered injury and death (13). The work-place exposure to isocyanates is of concern and has been linked to a number of health effects. Consequently, quite low limits for occupational exposure, 0.5 ppbv for methyl isocyanate, and 5 ppbv for total isocyanates have been established in some jurisdictions (14, 15).
We have recently developed a negative-ion proton-transfer chemical ionization mass spectrometer (NI-PT-CIMS) for sensitive [5 pptv (parts-per-trillion by volume) detection limit] and fast response (1 sec) measurement of HNCO and other acids in air (12) (see Materials and Methods below). The NI-PT-CIMS was used to measure HNCO in the emissions from laboratory biomass fires (4, 12, 16), in the Los Angeles (LA) urban area during May and June, 2010 (17), and during a period when Boulder, CO was impacted by emissions from the 2010 Fourmile Canyon wildfire. In addition we measured the Henry’s Law solubility of HNCO at pH = 3, and using the expression for solubility of a weak acid, estimate HNCO to be highly soluble at physiologic conditions, pH = 7.4. Finally we show that the HNCO levels observed in our combustion/pyrolysis measurements, combined with its solubility and reported in vitro biochemical studies, imply that HNCO makes a significant contribution to smoke-related health impacts that are a major societal concern.
Results and Discussion
Sources.
An example emission-time profile from measurements made at the US Forest Service Fire Sciences Laboratory in Missoula, MT (hereafter referred to as the Firelab), Fig. 1A, shows that HNCO and CO are highly correlated during flaming combustion with an HNCO/CO ratio between 0.1% and 0.6%. Smoldering combustion usually produced a second peak of CO emissions, with values of HNCO/CO that were factors of 5–10 lower. The two emission regimes are shown roughly as colored regions in Fig. 1B along with data from LA and the Fourmile Canyon wildfire. HNCO had an ambient “background” concentration, on the order of 10 pptv or less; and ranged up to 100 pptv at the LA site, and up to 200 pptv during the Boulder measurements. The laboratory biomass burning smoke was measured close to the source hence had much higher levels of HNCO and CO with up to 600 ppbv of HNCO. Laboratory biomass burning ratios of HNCO to other well known CN-containing biomass burning marker species ranged from 1 to 1.6 for HNCO/acetonitrile (CH3CN), and from 0.33 to 0.5 for HNCO/hydrogen cyanide (HCN) with one fuel as low as 0.16 (4).
Fig. 1.

(A) Time line for HNCO and CO emissions from a laboratory burn of California sage brush. (B) Measured HNCO vs. CO for the Fire Lab emissions measurements (red circles), CalNex LA ground site (black diamonds), and the Boulder Fourmile Canyon fire (blue triangles), with the general flaming stage and smoldering stage relationships highlighted.
Other laboratory studies have reported HNCO in concentrated emissions from pyrolysis and low-temperature combustion of biomass (18) and coal (19), and pyrolysis of tobacco ingredients (20). HNCO has also been observed as a by-product of urea-selective catalytic reduction systems that are being instituted to control NOx (nitric oxide and nitrogen dioxide) emissions from diesel engines (21). A number of studies show that biomass combustion and pyrolysis produce HNCO from amide or polyamide functionalities (18, 22, 23) {-HN-C(O)-}. Reduced nitrogen coproducts include HCN, CH3CN, and NH3, and the oxidized nitrogen coproducts include N2O, NO, HNO2, NO2, and HNO3. The temperatures involved in natural convection combustion and pyrolysis are low enough that NOx is not formed from N2 and O2, so the emitted nitrogen comes solely from the fuel (24).
Coal is a common fuel used directly for cooking and heating especially in rural areas of developing countries (25). Low-temperature combustion of coal having sufficient nitrogen, has been shown to be a source of HNCO (19). In this case the precursors are likely to be nitrogen heterocyclic compounds, although specific precursors have not been identified. Further work on the combustion of coal char at 600 °C measured the HNCO emission as 12 ± 4.5% of original fuel nitrogen (26).
Tobacco smoking presents an obvious source because, in this case, HNCO is produced from both the plant material, mostly from proteins (polyamides) (18) as well as from pyrolysis of urea, an additive in some cigarettes:
| [1] |
Apparently there are no reports of HNCO measurements in actual tobacco smoke, but a surrogate pyrolysis study found that 93% of added urea (4 mg/g tobacco) was pyrolyzed to HNCO, resulting in an estimated emission rate of 1.9 mg/g tobacco smoked (20). The amount of HNCO delivered to the average smoker, from this source alone, can be estimated using the parameters given in Baker and Bishop (20). If the lower ranges of values are assumed for: cigarette size (0.7 g), fraction of tobacco burned in puffing (0.3), fraction of HNCO transferred to mainstream smoke (0.3), and the fraction transmitted through the filter (0.3), then a smoker consumes 36 μg (filtered) to 108 μg (unfiltered) of HNCO per cigarette. The mixing ratio of HNCO in mainstream cigarette smoke can be estimated from these factors and an average volume of mainstream smoke of 470 mL (27), to be 40 to 140 ppmv. We observed HNCO in cigarette smoke in a brief laboratory test, however the levels were too high for us to quantify with the CIMS instrument configured in the ambient measurement mode. A more detailed study of tobacco smoke was beyond the scope of the current work. We conclude that tobacco-derived HNCO needs to be measured more extensively and potential exposure to it quantified. This measurement is especially important because HNCO is not currently included in the FDA “Proposed Initial List of Harmful/Potentially Harmful Constituents in Tobacco Products, including Tobacco Smoke” (28).
Diesel urea-selective catalytic reduction (SCR) exhaust systems represent a source of emerging interest, because HNCO is a recognized intermediate in this chemistry. These systems work by injecting a small flow (1–3% by volume urea/fuel ratios) of urea solution (32% by weight) into a catalyst system. Conditions and materials are optimized to induce not just reaction [1], but also provide for the complete catalytic hydrolysis of HNCO:
| [2] |
While extensive measurements of HNCO emissions from actual working diesel systems are lacking, there are reports of HNCO emissions from model systems that show that up to 5–10% of injected urea N is measured in exhaust as HNCO, mostly at lower temperatures and with older catalysts (> 1,000 h operation). The observed HNCO mixing ratios in exhaust streams ranged up to 50 ppmv (21). Understanding this source must be given a high priority considering the expected growth in SCR-controlled diesel engines in the European Union (EU) and United States.
Ambient Measurements.
The impact of HNCO emitted by a wildfire on the ambient air of an urban area can be seen in the close correlation of the HNCO and CO levels we measured in Boulder, Colorado during the recent 2010 Fourmile Canyon fire (Fig. 2A). In this case there is essentially minute-by-minute correlation in the levels of the two species. The ratio of HNCO/CO during the three enhancement events is consistent with smoldering in the laboratory biomass fires (Fig. 1B). Concurrent measurements of CH3CN made by gas chromatography/mass spectrometry (GC/MS) (see Materials and Methods below), are also shown in Fig. 2A, and also show close correlation with HNCO and CO, given that the GC/MS sampling time was the first 5 min of each 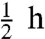 period. The HNCO/CH3CN ratios in these measurements were quite a bit lower than those observed in the laboratory burns described above. This lower ratio implies either very different source ratios for the fuels involved in this fire, or a much shorter lifetime for HNCO.
period. The HNCO/CH3CN ratios in these measurements were quite a bit lower than those observed in the laboratory burns described above. This lower ratio implies either very different source ratios for the fuels involved in this fire, or a much shorter lifetime for HNCO.
Fig. 2.

(A) Mixing ratios of HNCO, CO, and acetonitrile from measurements made in Boulder, CO. during the 2010 Fourmile Canyon fire, with HNCO 30-s averages shown in green circles, CO 1-min averages shown by the red line, and CH3CN shown as blue bars representing the 5-min sample time. (B) shows the individual 20-s HNCO measurements (blue dots), and the 5-min averaged HNCO (blue line) and ± 1σ (light blue) vs. time of day for the measurements at the CalNex LA ground site.
There are no other reports of measurements of HNCO in the ambient atmosphere with which to compare, but we can contrast our HNCO observations with those of other reduced nitrogen species known to have strong biomass burning sources: CH3CN and HCN (29). The lowest HNCO values we observe were below 10 pptv, much lower than corresponding background levels of HCN and CH3CN, which are in the range of 50 to 100 pptv. Because biomass burning is a far larger source of trace gases than diesel engines or tobacco smoke (5), it is almost certainly the largest global source of HNCO, thus the disparity in background values implies a shorter lifetime for HNCO relative to the other species.
A clear diurnal variation in HNCO was observed in the LA basin (Fig. 2B) at the Pasadena ground site during the CalNex 2010, a study with combined air quality and climate goals (30). Wildfires were not observed during this period and are therefore not responsible for the observed HNCO. As the diurnal variation of HNCO coincides with those of other photochemical products, such as ozone and oxygenated VOCs, which peaked midday when processed air from the downtown LA area arrived at the site, we suggest that HNCO in LA is also formed photo-chemically, potentially from monomethyl amine, formamide, and acetamide (31, 32) a source that has not been previously considered in urban areas. Moreover, a surface or vehicle source of HNCO would show low midday values when the boundary layer mixing is maximized and higher values during rush hours, particularly in the morning. Our LA measurements give a baseline urban value, in the absence of local wildfires or a possible future increase from vehicle sources.
Atmospheric Removal Processes.
Hydroxyl radicals are generally responsible for removing trace organic species from the atmosphere (33). HNCO is relatively stable against reaction with OH radical [k ≈ 10-15 cm3/molec-s extrapolated from high temperature data (34)], yielding an atmospheric lifetime of several decades for this process. It is possible that there are mechanisms, such as adduct formation, that are important at low temperatures, but we are not aware of any studies of that phenomenon. HNCO is expected to have a very low absorption at near-UV to visible wavelengths that constitute the solar actinic region based on measurements at wavelengths shorter than 280 nm (35, 36). The major dissociation channel (HNCO⇒H + NCO) has a threshold of 261 nm which corresponds to a bond energy Do(H-NCO) = 109.6 ± 0.4 kcal/mole (37), and a channel that forms triplet NH has a 332 nm threshold (38), however the absorption cross-section in that region is quite low. These absorption features result in an HNCO lifetime against photolysis of months.
The major loss processes of HNCO in the lower troposphere are likely the heterogeneous uptake to aerosols or liquid water (fog, clouds, precipitation, and the ocean) and subsequent reactions. HNCO is a moderately weak acid in aqueous solution (pKa = 3.7) (11) and exhibits relatively slow hydrolysis that is pH-dependent (39). The partitioning of HNCO to aqueous solution at low-concentration, i.e., Henry’s Law solubility, H (M/atm), has apparently not been measured previously. We have measured H for isocyanic acid in an aqueous buffer at pH = 3.0 ± 0.1, and room temperature (t = 25 ± 1 °C). The decrease in gas-phase HNCO, exiting a saturated liquid sample, was measured for a range of volume flow-rate to liquid volumes (Fig. 3A). For a system in which the mass transfer between the liquid and gas phases is rapid, the following relationship holds (40):
| [3] |
Where Ct/Co is the relative concentration in the gas phase exiting the reactor, φ is the volumetric flow rate, V is the liquid volume, R is the ideal gas constant, T is temperature, kl is the first order loss rate in solution, and t is the time. The slopes of these exponential curves vs. the flow-rate/liq.-volume yielded the Henry’s Coefficient (Fig. 3B). The resulting value is given in Table 1 and is commensurate with Henry’s Coefficients of some related species (41). HNCO is only slightly soluble at pH = 3, however, it is a weak acid, pKa = 3.7 ± 0.2, hence its effective Henry’s Constant can be calculated as a function of pH with the following relationship (42):
| [4] |
where H* is the intrinsic Henry’s Coefficient independent of any liquid-phase equilibria. This relationship is plotted in Fig. 4 for HNCO using H* derived from the Heff measured at pH = 3, and the known pKa.
Fig. 3.

(A) shows the concentration decay curves data for some of the individual equilibration experiments at the flow rates shown, and (B) shows the resulting fit to Eq. 3 and the points for the individual experiments from (A) along with points from additional experiments (solid dots), which are not shown in (A) for the sake of clarity.
Table 1.
Henry’s Law constants of HNCO and related compounds
| Compound | H*, M/atm | Notes |
| HNCO | 21 | based on Heff at pH3 |
| HONO* | 49 | for the undissociated acid, pKa 3.3 |
| HCN* | 12 | for the undissociated acid, pKa 9.2 |
| CH3CN* | 50 |
*From ref. 41.
Fig. 4.

The Henry’s Law constant (blue) from our measurement at pH = 3 and the weak acid equilibrium relationship Eq. 4, first-order loss rate due to hydrolysis from the rate constants measured by Jensen (39) (solid red), and corresponding aqueous phase lifetime (dashed red) of HNCO vs. pH. The open red circle is our measurement of the first-order loss rate at pH = 3 (± 1σ) and the open blue square is our measurement of Heff at pH = 3. Also shown is the Henry’s Law constant for HCN (green). The yellow band indicates the range of pHs most characteristic of ambient aerosol, and the pink band indicates physiological pH, and the error bar at pH = 7.4, is the estimated uncertainty based on the uncertainties in Heff measured at pH = 3, (± 3 M/atm) and, pKa (± 0.2 pH units).
The rate of hydrolysis of HNCO has been measured as a function of pH and found to have several mechanisms, one set that is direct (i.e., first order), and one that is acid-catalyzed (39):
| [5a] |
and
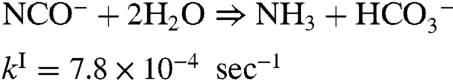 |
[5b] |
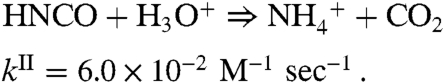 |
[6] |
This hydrolysis rate is also shown in Fig. 4 as a function of pH. A point of comparison is available from the solubility experiment described above, in which 8.7 ± 1.3 × 10-4 sec-1 was measured, a value that is in good agreement with the previous measurement (39). HNCO hydrolyzes slowly enough (t ≈ 20 min) at physiological pHs that there is ample time for carbamylation chemistry, but just fast enough that tradition analytical methods, e.g. aqueous acid/base extractions etc., will underestimate its concentration. While detailed kinetic parameters are lacking, there are a number of studies that show that carbamylation is rapid relative to hydrolysis (8, 43, 44).
Given the above solubility data and the absence of fast gas-phase loss processes, uptake of HNCO on aerosol and cloud droplets, or natural surfaces, will likely be the relevant loss process. The rate of this process can be estimated from knowledge of the Henry’s Law solubility and liquid-phase reaction rate. The loss of a reactive species to a liquid surface of an aerosol particle or droplet can be thought of as a network of resistances, in series and in parallel, that represent diffusion and reaction processes (45). Often one process is limiting, simplifying the representation. The time scale for gas-phase diffusion to particles is on the order of seconds to a few minutes depending on particle size and number. The time scales for loss of HNCO in liquid aerosol or cloud droplets are much longer and can be estimated by the following equation (46);
| [7] |
where Fl is the volume fraction of liquid water, which ranges from 10-12 for an aerosol of total surface area of 200 μm2/cm3, and 0.2 μm mean diameter, and up to 10-6 for fogs or clouds, and khyd is kI. The estimated atmospheric lifetimes for HNCO then range from > 104 years for reaction on an atmospheric aerosol at pH 3, to approximately 0.5 d for reaction in cloud or fog water of pH 5.5 and the above liquid water content. Clearly, the lifetime of HNCO against uptake on aerosols and clouds is limited by liquid-phase reactions.
Slightly different considerations govern the uptake of HNCO on ground or natural water surfaces (lakes, oceans). In these cases it can be assumed that the solubility of HNCO is the dominant factor. Because those surfaces are essentially at neutral pH or above, HNCO should behave similarly to other highly soluble species, such as nitric acid (HNO3). Nitric acid has lifetimes against deposition in the planetary boundary layer in the range of an hour to half a day (33). A comparison of the above lifetimes for HNCO can be made to the other biomass-derived CN compounds, HCN, and CH3CN, which are not very soluble at environmental pHs, and have lifetimes of 5 mo (HCN), and 6.6 mo (CH3CN) against loss, mainly due to deposition to the ocean (29).
Potential Health Impacts.
The chronic and acute health effects of smoke are well documented (2, 6, 8, 47), however detailed causal biochemical pathways are not completely understood and are the subject of current research. The eyes, respiratory, and cardiovascular systems are the areas of the human body that show chronic effects from smoke exposure (2). The potential for health impacts due to HNCO in smoke can be traced to two features of its chemistry: high solubility at physiologic pH as noted above, and the reaction of HNCO with amine, hydroxyl, and sulfhydryl groups, by addition across the N-C bond, to form a carbamyl group, -H2NC(O)-, in a process termed carbamylation (43):
| [8] |
| [9] |
| [10] |
Recent work has shown that carbamylation of proteins is a key step in the inflammatory response that links smoking to cardiovascular disease (8) and rheumatoid arthritis (6), and the link between carbamylation and cataracts has been recognized for some time (7). Wang et al. (8) identified isocyanic acid/cyanate ion as a key intermediate in this reaction. However, the source of cyanate in this chemistry was postulated to be the enzymatic oxidation of thiocyanate (NCS-) by hydrogen peroxide. Our work shows that smoke provides a route to uptake and absorption of HNCO into the blood stream that will drive protein carbamylation directly. The effective Henry’s Law solubility of 105 M/atm at pH 7.4 means that a 1 ppbv (10-9 atm) mixing ratio in inhaled breath will produce an equilibrium aqueous concentration of 100 μM, a concentration that mimics carbamylation in vitro (8). The transport of HNCO into the blood stream and into the sensitive tissues of the eye depends on membrane transport, the precise aspects depending on its pKa and oil/water partition coefficient (48), which is apparently not known for HNCO. However, we would expect HNCO to have similar behavior to formic acid, because of its similar size, polarity, and acidity (pKa = 3.75), which has moderate permeability in lipid bilayer membranes (49). As a result, transport of HNCO should be rapid enough that solution concentrations will be close to those calculated from Henry’s Law equilibrium.
Conclusions.
Our work shows that there is potential for significant exposure of humans to HNCO as a consequence of biomass burning, biofuel usage, cooking, and tobacco usage, and perhaps by the use of new diesel SCR emission control systems. Studies of indoor CO in rural areas of China, where biomass or coal is burned in open indoor fires for cooking and heating, report average concentrations in the range 4–12 ppmv (25). If our data from Fig. 1 are typical of biomass cooking fuels, then HNCO levels up to 10 ppbv or higher could exist in those homes and result in blood NCO- levels far in excess of those shown to produce protein carbamylation. The tobacco source of HNCO has apparently not been quantified directly, but pyrolysis of urea, (a major tobacco additive) produces HNCO directly (20), implying that this source could be considerable. HNCO mixing ratios in the 1 ppbv range were not measured in urban areas, but the possibility that new diesel SCR sources could increase ambient HNCO is a real concern. Wildfire impacts on populated areas and firefighters could also be significant based on our Firelab measurements (Fig. 1B).
These results suggest several issues that require further study. Accurate HNCO emission factors are needed for the different biofuels and coals used for cooking, and the fuels that burn in wildfires and tropical deforestation. Emission ratios for diesel SCR vehicles need to be determined. Human exposure to HNCO needs to be studied in depth, including lung and membrane transport, and their relation to blood and tissue levels of NCO-. Increased biomass burning emissions are expected with warmer, drier regional climates (50), and diesel SCR controls are being phased-in by the EU and some states in the United States. Extensive source characterization and targeted toxicological and exposure studies are needed to better understand and mitigate this potentially harmful HNCO exposure.
Materials and Methods
Acid CIMS Instrument.
Our HNCO method is described by Veres et al. (51) and Roberts et al. (12) and involves [1] selective ionization of HNCO by reaction with acetate ion via proton transfer in the gas phase and [2] detection of the NCO- ion at 42 amu by a quadrupole mass spectrometer. Contributions to mass 42 from other compounds such as HN3 and other H C N O isomers, were considered and rejected for reasons discussed by Roberts et al. (12). The possibility that mass 42 is 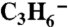 produced by electron transfer from
produced by electron transfer from  can be ruled out by the fact that
can be ruled out by the fact that  had at most a 0.1% abundance relative to acetate ions in our Firelab and ambient studies, so even if the reaction occurred at the gas kinetic limit, sensitivity to propene would be about 0.3%, because acetate-acid reactions occur at about 1/3 the gas kinetic limit (16). The detection limit of this measurement is 5 pptv for a 1 sec measurement, and the uncertainties are ± (25%+5 pptv) for ambient measurements. Biomass burning emissions measurements were conducted at the US Department of Agriculture, Fire Science Laboratory in Missoula, MT (4). The LA measurements were made at the CalNex-LA Pasadena ground site (34.140582 N, 118.122455 W) (30), and the Boulder measurements were made right outside the NOAA/ESRL laboratory (39.991431 N 105.261032 W). The inlet time constant was a few seconds in the case of LA and Firelab measurements and was estimated to be 10 to 15 s during the Boulder Fourmile Canyon measurements.
had at most a 0.1% abundance relative to acetate ions in our Firelab and ambient studies, so even if the reaction occurred at the gas kinetic limit, sensitivity to propene would be about 0.3%, because acetate-acid reactions occur at about 1/3 the gas kinetic limit (16). The detection limit of this measurement is 5 pptv for a 1 sec measurement, and the uncertainties are ± (25%+5 pptv) for ambient measurements. Biomass burning emissions measurements were conducted at the US Department of Agriculture, Fire Science Laboratory in Missoula, MT (4). The LA measurements were made at the CalNex-LA Pasadena ground site (34.140582 N, 118.122455 W) (30), and the Boulder measurements were made right outside the NOAA/ESRL laboratory (39.991431 N 105.261032 W). The inlet time constant was a few seconds in the case of LA and Firelab measurements and was estimated to be 10 to 15 s during the Boulder Fourmile Canyon measurements.
HNCO Calibration.
Calibration of the HNCO signal was accomplished by FTIR measurement of a gas-phase diffusion source of HNCO. A gas stream of HNCO at mixing ratios in the range of 1–2 ppmv in 50 sccm (standard cubic centimeters per minute) zero air was produced by the thermal decomposition (210°–230 °C) of cyanuric acid, the trimer of HNCO. Limiting the HNCO mixing ratio to a few ppmv was found to be essential to avoid polymerization as the gas stream cooled to room temperature down stream of the source. The concentration of HNCO in the diffusion source was measured by FTIR in a room pressure (620 Torr) multipass cell (4.8 m length). The standard HNCO absorption cross-section from the database described by Sharpe et al. (52) and Johnson et al. (53) was used to calculate mixing ratios. The HNCO source was diluted in several stages by larger flows of zero air or nitrogen to the ranges appropriate for the Firelab study and the LA study.
Acetonitrile Measurement.
The gas chromatographic/mass spectrometric method used for the measurement of acetonitrile in Boulder during the Fourmile Canyon fire, was described by Gilman et al. (54). The instrument is a custom built two channel GC coupled to an Agilent 5973 quadrupole mass spectrometer. Air was sampled from the west side of the laboratory building at 7 L/ min through a short (several meter) Teflon PFA sample tube. A small subflow of 70 sccm was sampled off of this flow for 5 min through a series of traps designed to reduce water, carbon dioxide, and O3 interferences (55). Acetonitrile was separated on a semipolar (Restek-MXT-624) capillary column [20 m × 0.18 mmID (inner diameter)] and detected at m/z 41. The GC separation required 25 min, permitting a 5 min sample to be analyzed every 30 min. Calibrations were accomplished by dynamic dilution of gravimetrically prepared gas-phase standards.
Henry’s Constant Measurement.
Henry’s Law describes the equilibration between a gas-phase chemical species and a liquid-phase, at infinite dilution. At equilibrium, the partial pressure of a species in the gas phase is proportional to its concentration in the liquid phase:
| [11] |
where the Henry’s Law constant, H is typically given in units of M/atm. Measurement of H is relatively simple if the compound of interest is stable in both phases. However, HNCO hydrolyzes at appreciable rates in a pH-dependent way. Therefore a dynamic method was used in this work, following the description by Kames and Schurath (40), and utilizing the apparatus described by Roberts (56), and the HNCO sources described by Roberts et al. (12). The apparatus consisted of two fritted bubblers (Aldrich 250 mL Gas Reducing Flask) placed in series: the first bubbler contained de-ionized (DI) water to humidify the gas stream, and the second contained the sample of DI water buffered to pH = 3 ± 0.1 with a citric acid/NaOH/NaCl buffer (Fluka Chemicals), through which a flow of zero air, in the flow range 100 to 1,000 sccm, was directed. The HNCO source was connected to the gas stream in between the two bubblers by means of a 3-way valve, so that the source could be directed into the bubbler stream, or to vent, without perturbing the main flow. The outlet of the bubbler stream was teed into the inlet of the NI-PT-CIMS so that the pressure at the outlet remained at room pressure. Equilibration experiments were conducted by placing a sample of pH = 3.0 solution (25 ± 0.25 mL) into the bubbler and measuring the HNCO at the outlet at a series of flow rates, as the HNCO source was switched in-line and equilibrated with the solution, and then switched out of line and observed to decay due to a combination of hydrolysis and loss to the gas phase. The exponential decays were then fit to the relationship given in Eq. 3, as shown in Fig. 3. Measurement of H at physiologic pH is not possible with this method as it is too high to yield decay curves on a meaningful time scale, rather we rely on the well demonstrated relationship in Eq. 4 to calculate H at pH = 7.4.
Acknowledgments.
We thank the USDA Fire Lab, Missoula, Montana, for the use of their facility, and the California Institute of Technology for hosting the CalNex 2010 ground site. We acknowledge useful discussions with Paul Wennberg, Robert Harley, and Armin Wisthaler, and John Holloway for CO measurements in the Pasadena studies. This work was supported by the NOAA’s Health of the Atmosphere Program and NOAA’s Climate Goal, the NOAA-ISET Program, National Science Foundation (NSF) Grant number ATM 1542457, the Cooperative Institute for Research in the Environmental Sciences (CIRES) Innovative Research Program, and Department of Defense (DoD) Strategic Environmental Research and Development Program (SERDP) Grant Numbers SI-1648 and SI-1649.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Crutzen PJ, Andreae MO. Biomass burning in the tropics: impact on atmospheric chemistry and biogeochemical cycles. Science. 1990;250:1669–1678. doi: 10.1126/science.250.4988.1669. [DOI] [PubMed] [Google Scholar]
- 2.Fullerton DG, Bruce N, Gordon SB. Indoor air pollution from biomass fuel smoke is a major health concern in the developing world. Trans Roy Soc Trop Med H. 2008;102:841–952. doi: 10.1016/j.trstmh.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreae MO, Merlet P. Emission of trace gases and aerosols from biomass burning. Global Biogeochem Cy. 2001;15:955–966. [Google Scholar]
- 4.Burling IR, et al. Laboratory measurements of trace gas emissions from biomass burning of fuel types from the southeastern and southwestern United States. Atmos Chem Phys. 2010;10:11115–11130. [Google Scholar]
- 5.Akagi SK, et al. Emission factors for open and domestic biomass burning for use in atmospheric models. Atmospheric Chemistry Physics Discussions. 2010;10:27523–27602. [Google Scholar]
- 6.Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 7.Beswick HT, Harding JJ. Conformational changes induced in bovine lens α-crystallin by carbamylation, relevance to cataract. Biochem J. 1984;223:221–227. doi: 10.1042/bj2230221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, et al. Protein carbamylation links infammation, smoking, uremia, and atherogenesis. Nat Med. 2007;13:1176–1184. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 9.Mydel P, et al. Carbamylation-dependent activation of T cells: a novel mechanism in the pathogensis of autoimmune arthritis. J Immunol. 2010;184:6882–6890. doi: 10.4049/jimmunol.1000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liebig J, Wöhler F. Investigations of cyanic acid (German) Ann Phys. 1830;20:396–400. [Google Scholar]
- 11.Belson DJ, Strachan AN. Preparation and properties of isocyanic acid. Chem Soc Rev. 1982;11:41–56. [Google Scholar]
- 12.Roberts JM, et al. Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions. Atmospheric Measurement Technology. 2010;3:981–990. [Google Scholar]
- 13.Mishra PK, et al. Bhopal gas tragedy: review of clinical and experimental findings after 25 years. Int J Occup Med Env. 2009;22:193–202. doi: 10.2478/v10001-009-0028-1. [DOI] [PubMed] [Google Scholar]
- 14.State of California. Determination of noncancer chronic reference exposure levels Batch 2B December 2001, chronic toxicity summary, methyl isocyanate. 2001. oehha.ca.gov/air/chronic_rels/pdf/methyliso.pdf.
- 15.Swedish Work Environment Authority. 2005. Occupational exposure limit values and measures against air contaminants. http://www.av.se/dokument/inenglish/legislations/eng0517.pdf. [Google Scholar]
- 16.Veres P, et al. Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) J Geophys Res-Atmos. 2010;115:D23302. doi: 10.1029/2010JD014033. [Google Scholar]
- 17.NOAA Earth System Research Laboratories, Chemical Sciences Division. CalNex 2010, research at the nexus of air quality and climate change. 2010. http://www.esrl.noaa.gov/csd/calnex/
- 18.Hansson K-M, Samuelsson J, Tullin C, Amand L-E. Formation of HNCO, HCN, and NH3 from the pyrolysis of bark and nitrogen-containing model compounds. Combust Flame. 2004;137:265–277. [Google Scholar]
- 19.Nelson PF, Li C-Z, Ledesma E. Formation of HNCO from the rapid pyrolysis of coals. Energ Fuels. 1996;10:264–265. [Google Scholar]
- 20.Baker RR, Bishop LJ. The pyrolysis of tobacco ingredients. J Anal Appl Pyrol. 2004;71:223–311. [Google Scholar]
- 21.Kröcher O, Elsener M, Koebel M. An ammonia and isocyanic acid measuring method for soot containing exhaust gases. Analytica Chemica Acta. 2005;573:393–400. [Google Scholar]
- 22.Ren QQ, et al. Formation of NOx precursors during wheat straw pyrolysis and gasification with O2 and CO2. Fuel. 2010;89:1064–1069. [Google Scholar]
- 23.Wang XB, et al. Nitrogen, sulfur, and chlorine transformations during the pyrolysis of straw. Energy Fuel. 2010;24:5215–5221. [Google Scholar]
- 24.Lobert JM, Scharffe DH, Hao WM, Crutzen PJ. Importance of biomass burning in the atmospheric budgets of nitrogen-containing gases. Nature. 1990;346:552–554. [Google Scholar]
- 25.Wang S, Wei W, Li D, Aunan K, Hao J. Air pollutants in rural homes in Guizhou China—concentrations, speciation and size distribution. Atmos Environ. 2010;44:4575–4581. [Google Scholar]
- 26.Nicholls P, Nelson PF. Detection of HNCO during the low-temperature combustion of coal chars. Energy Fuel. 2000;14:943–944. [Google Scholar]
- 27.Shopland DR, editor. FTC Cigarette Test Method for Determining Tar, Nicotine, and Carbon Monoxide Yields of U.S. Cigarettes, Report of the NCI Expert Committee. Darby, PA: Diane Publishing Co.; 1996. p. 275. [Google Scholar]
- 28.U.S. FDA. Draft proposed initial list of harmful/potentially harmful constituents in tobacco products, including tobacco smoke. 2010. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/TobaccoProductsScientificAdvisoryCommittee/ucm221804.htm.
- 29.Singh HB, et al. In situ measurements of HCN and CH3CN over the Pacific Ocean: Sources, sinks, and budgets. J Geophys Res-Atmos. 2003;108:8795. doi: 10.1029/2002JD003006. [Google Scholar]
- 30.CalNex-LA. 2010. http://cires.colorado.edu/jimenez-group/wiki/index.php/CalNex-LA.
- 31.Schade GW, Crutzen PJ. Emission of aliphatic amines from animal husbandry and their reactions: potential source of N2O and HCN. J Atmos Chem. 1995;22:319–346. [Google Scholar]
- 32.Barnes I, Solignac G, Mellouki A, Becker KH. Aspects of the atmospheric chemistry of amides. Chem Phys Chem. 2010;11:3844–3857. doi: 10.1002/cphc.201000374. [DOI] [PubMed] [Google Scholar]
- 33.Finlayson-Pitts BJ, Pitts JN., Jr. Chemistry of the Upper and Lower Atmosphere. San Diego: Academic Press; 2000. [Google Scholar]
- 34.Tsang W. Chemical kinetic data base for propellant combustion II: reactions involving CH, NCO, and HNCO. J Phys Chem Ref Data. 1992;21:753–791. [Google Scholar]
- 35.Dixon RN, Kirby GH. Ultra-violet absorption spectrum of isocyanic acid. Transactions of the Faraday Society. 1968;64:2002–2012. [Google Scholar]
- 36.Brownsword RA, Laurent T, Vatsa RK, Volpp H-R, Wolfrum J. Photodissociation dynamics of HNCO at 248 nm. Chem Phys Lett. 1996;258:164–170. [Google Scholar]
- 37.Brown SS, Berghout HL, Crim FF. The HNCO heat of formation and the N-H and C-N bond enthalpies from initial state selected photodissociation. J Chem Phys. 1996;105:8103–8110. [Google Scholar]
-
38.Berghout HL, Brown SS, Delgado R, Crim FF. Nonadiabatic effects in the photodissociation of vibrationally excited HNCO: The branching between singlet (a1Δ) and triplet (
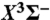 ) NH. J Chem Phys. 1998;109:2257–2263. [Google Scholar]
) NH. J Chem Phys. 1998;109:2257–2263. [Google Scholar] - 39.Jensen MB. On the kinetics of the decomposition of cyanic acid. Acta Chem Scand. 1958;12:1657–1670. [Google Scholar]
- 40.Kames J, Schurath U. Henry’s Law and hydrolysis-rate constants for peroxyacyl nitrates (PANs) using a homogeneous gas-phase source. J Atmos Chem. 1995;21:151–164. [Google Scholar]
- 41.Sander R. Compilation of Henry's Law constants for inorganic and organic species of potential importance in environmental chemistry (Version 3) 1999. http://www.henrys-law.org.
- 42.Sander R. Modeling atmospheric chemistry: interactions between gas-phase species and liquid cloud/aerosol particles. Surv Geophys. 1999;20:1–31. [Google Scholar]
- 43.Stark GR, Stein WH, Moore S. Reactions of cyanate present in aqueous urea with amino acids and proteins. J Biol Chem. 1960;235:3177–3181. [Google Scholar]
- 44.Lee CK, Manning JM. Kinetics of the carbamylation of the amino groups of sickle cell hemoglobin by cyanate. J BioChem. 1973;248:5861–5865. [PubMed] [Google Scholar]
- 45.Davidovits P, Kolb CE, Williams LR, Jayne JT, Worsnop DR. Mass accommodation and chemical reactions at gas-liquid interfaces. Chem Rev. 2006;106:1323–1354. doi: 10.1021/cr040366k. [DOI] [PubMed] [Google Scholar]
- 46.Molina MJ, Molina LT, Kolb CE. Gas-phase and heterogeneous chemical kinetics of the troposphere and stratosphere. Ann Rev Phys Chem. 1996;47:327–367. [Google Scholar]
- 47.Dherani M, et al. Indoor air pollution from unprocessed solid fuel use and pneumonia risk in children aged under five years: a systematic review and meta-analysis, B World Health Organ. 2008;86:390–398. doi: 10.2471/BLT.07.044529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Missner A, Pohl P. 110 Years of the Meyer—Overton rule: predicting membrane permeability of gases and other small compounds. Chem Phys Chem. 2009;10:1405–1414. doi: 10.1002/cphc.200900270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walter A, Hastings D, Gutknecht J. Weak acid permeability through lipid bilayer membranes. J Gen Physiol. 1982;79:917–933. doi: 10.1085/jgp.79.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parry ML, Canziani OF, Palutikof JP, van der Linden PJ, Hanson CE, editors. Climate Change 2007: Impacts, Adaptation and Vulnerability. Cambridge, United Kingdom: Cambridge University Press; 2007. p. 976. [Google Scholar]
- 51.Veres P, et al. Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for the measurement of gas-phase organic acids in the atmosphere. Int J Mass Spectrom. 2008;274:48–55. [Google Scholar]
- 52.Sharpe SW, et al. Gas-phase databases for quantitative infrared spectroscopy. Appl Spectrosc. 2004;58:1452–1459. doi: 10.1366/0003702042641281. [DOI] [PubMed] [Google Scholar]
- 53.Johnson TJ, Profeta LTM, Sams RL, Griffith DWT, Yokelson RJ. An infrared spectral database for detection of gases emitted by biomass burning. Vib Spectrosc. 2010;53:97–102. [Google Scholar]
- 54.Gilman JB, et al. Ozone variability and halogen oxidation within the Arctic and sub-Arctic springtime boundary layer. Atmos Chem Phys. 2010;10:10223–10236. [Google Scholar]
- 55.Goldan PD, et al. Nonmethane hydrocarbon and oxy hydrocarbon measurements during the 2002 New England Air Quality Study. J Geophys Res-Atmos. 2004;109:D21309. doi: 10.1029/2003JD004455. [Google Scholar]
- 56.Roberts JM. Measurement of the Henry's Law coefficient and the first order loss rate of PAN in n-octanol. Geophys Res Lett. 2005;31:L08803. doi: 10.1029/2004GL022327. [Google Scholar]