

Abstract
Nrf2 is a major cytoprotective gene and is a key chemopreventive target against cancer and other diseases. Here we show that Nrf2 faces a dilemma in defense against 4-aminobiphenyl (ABP), a major human bladder carcinogen from tobacco smoke and other environmental sources. While Nrf2 protected mouse liver against ABP (which is metabolically activated in liver), the bladder level of N-(deoxyguanosin-8-yl)-4-aminobiphenyl (dG-C8-ABP), the predominant ABP-DNA adduct formed in bladder cells and tissues, was markedly higher in Nrf2+/+ mice than in Nrf2−/− mice after ABP exposure. Notably, Nrf2 protected bladder cells against ABP in vitro. Mechanistic investigations showed that the dichotomous effects of Nrf2 could be explained at least partly by upregulation of UDP-glucuronosyltransferase (UGT). Nrf2 promoted conjugation of ABP with glucuronic acid in the liver, increasing urinary excretion of the conjugate. While glucuronidation of ABP and its metabolites is a detoxification process, these conjugates, which are excreted in urine, are known to be unstable in acidic urine, leading to delivery of the parent compounds to bladder. Hence, while higher liver UGT activity may protect the liver against ABP it increases bladder exposure to ABP. These findings raise concerns of potential bladder toxicity when Nrf2-activating chemopreventive agents are used in humans exposed to ABP, especially in smokers. We further demonstrate that 5,6-dihydrocyclopenta[c][1,2]-dithiole-3(4H)-thione (CPDT) significantly inhibits dG-C8-ABP formation in bladder cells and tissues, but does not appear to significantly modulate ABP-catalyzing UGT in liver. Thus, CPDT exemplifies a counteracting solution to the dilemma posed by Nrf2.
Introduction
NF-E2 related factor 2 (Nrf2) is a ubiquitous transcription factor that stimulates the expression of genes involved in many aspects of cytoprotection, most notably those for Phase 2 enzymes, such as glutathione S-transferase, NAD(P)H:quinone oxidoreductase-1 (NQO1) and UDP-glucuronosyltransferase (UGT). Phase 2 enzymes catalyze detoxification reactions of carcinogens and oxidants and thereby play important roles in prevention of cancer and other diseases. Indeed, Nrf2 knockout mice showed significantly increased susceptibility to cancer (1), neurodegeneration (2), and inflammation (3). Nrf2 works by binding as a heterodimer with Maf or other partners to a cis-acting DNA regulatory element, namely the antioxidant response element (ARE), which is located in the 5′-untranslated region of its target genes, and stimulating gene transcription. Nrf2 is activated itself in response to various stimuli, and chemopreventive and pharmacological agents activate Nrf2 through inhibition of Keap1-mediated Nrf2 ubiquitination, which has been widely advocated as a major strategy for prevention of cancer and other diseases.
4-Aminobiphenyl (ABP) from tobacco smoking and nonsmoking-related environmental and occupational exposure is a well-known cause of human bladder cancer (4, 5). Tobacco smoke is both the main cause of human bladder cancer and the main source of human exposure to ABP (6, 7). In animal studies, ABP causes both bladder and liver cancers (8). While ABP itself is not carcinogenic, it undergoes Phase 1 metabolic activation in the liver to form N-hydroxy-ABP (N-OH-ABP) which is eventually converted to the highly electrophilic arylnitrenium ion that causes DNA damage (9). DNA damage has been shown to be fundamental to ABP-induced bladder tumorigenesis. For example, ABP-DNA adducts were detected in a high percentage of human bladder cancer biopsies(10, 11); higher levels of ABP-DNA adducts in human bladder tumors were associated with more aggressive behavior of the tumors (10); and treatment of animals with ABP caused formation of DNA adducts before tumor formation (8, 12). Hepatic cytochrome P4501A2 was widely suggested to be key to N-OH-ABP formation, but a subsequent study showed that knockout of this gene in mice had no effect on ABP-DNA adduct formation in the bladder (9). However, it is well known that several Phase 2 enzymes, including N-acetyltransferases (NAT1 and NAT2) and UGT (comprising multiple isoforms), attenuate the genotoxicity of ABP by catalyzing the conjugation of ABP or its metabolites with endogenous ligands acetyl-CoA and glucuronic acid, respectively. The conjugates are generally more water-soluble and hence more readily excreted. While there is no evidence that either NAT1 or NAT2 is subjected to Nrf2 regulation, a number of UGT isoforms in both human and animals have been shown to be up-regulated by Nrf2 (13-15).
We recently found that sulforaphane, a naturally occurring Nrf2 activator, inhibited ABP-induced DNA damage in bladder cells and tissues, and that Nrf2 was essential for sulforaphane to inhibit ABP-DNA adduct formation (16). Surprisingly, in the absence of sulforaphane, Nrf2 promoted ABP-DNA adduct formation in the bladder in vivo (16). In the present study, we have further examined the potentiating effect of Nrf2 on ABP-induced DNA damage and investigated the underlying molecular basis of this phenomenon. The data provided in this report has a significant implication for cancer chemoprevention, as they reveal that bladder toxicity may occur if Nrf2-activating chemopreventive agents are used in humans exposed to ABP, such as smokers. Meanwhile, we also demonstrate how the adverse effect of Nrf2 may be mitigated.
Materials and Methods
Chemicals
5,6-Dihydrocyclopenta[c][1,2]-dithiole-3(4H)-thione (CPDT) was synthesized as described previously (17). ABP, N-OH-AABP and rat liver S9 were purchased from Sigma (St Louis, MO), Midwest Research Institute (Kansas City, MO), and Motox (Boone, NC), respectively. An ABP-N-glucuronide (ABP-G) standard was purchased from Toronto Research Chemicals (North York, Ontario).
Cells
RT4 cells were purchased from American Type Culture Collection (ATCC), stored in liquid nitrogen, and passaged in our laboratory for fewer than 6 months. The cell line was characterized by ATCC by antigen expression, DNA profile, cytogenetic profile and isoenzymes. RT4 cells were cultured and treated with CPDT, ABP (plus 6% rat liver S9) or N-OH-AABP as previously reported (16, 18). To silence Nrf2, cells were treated with an Nrf2 siRNA or a control siRNA for 48 h (18).
Mice
Male wild type C57BL/6 mice (NCI, Frederick, MD) and their Nrf2-deficient counterparts (19) (bred at our animal facility), 6-8 weeks of age, were injected intraperitoneally with a single dose of ABP or vehicle (DMSO) and sacrificed 24 h later for quantification of dG-C8-ABP in the liver and bladder tissues (16). For CPDT intervention, the mice were treated with CPDT (in soy oil) or the vehicle by gavage once daily for 5 days; ABP was injected 3 h after the last CPDT dose. To determine the effect of CPDT on Phase 2 enzymes by Western blotting, mice were treated with CPDT by gavage once daily for 5 days and sacrificed 24 h later. Liver and bladder tissues were removed immediately and stored at −80°C before use. To measure urinary levels of ABP-G, mice were given a single dose of ABP i.p. and immediately moved to metabolism cages (2 mice/cage) for 24-h urine collection. The specimens were stored at −80°C before analysis. All animal protocols and procedures were approved by the Roswell Park Cancer Institute Animal Care and Use Committee.
Measurement of N-(deoxyguanosin-8-yl)-4-aminobiphenyl (dG-C8-ABP)
dG-C8-ABP was quantified by capillary liquid chromatography and nanoelectrospray ionization-tandem mass spectrometry as previously described (16, 20).
Western blot analysis
Cellular and tissue levels of Nrf2 and/or Nrf2 target genes were measured by Western blotting as previously described (18). All antibodies were purchased from Santa Cruz Biotechnology, except for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) which was purchased from Millipore.
Measurement of liver UGT activity and urinary levels of ABP-G and creatinine
The assay protocols for measuring liver UGT activity and urinary ABP-G were based on those by Al-Zoughool and Talaska (21) with some modifications. Briefly, livers were homogenized in ice-cold 50 mM potassium phosphate buffer (pH 7.4) in a glass homogenizer. The homogenates were cleared by centrifugation for 20 min at 9,100g at 4°C. UGT activity toward ABP was measured by a biochemical assay coupled with reverse phase high performance liquid chromatography (HPLC). Each liver sample (0.35 mg protein) was first incubated with 17.5 μg alamethicin in 0.171 ml of 50 mM potassium phosphate buffer (pH7.4) containing 8.75 μl methanol on ice for approximately 20 min. The entire mixture was then transferred to a final 0.2 ml reaction solution, containing 50 mM potassium phosphate (pH 7.4), 10 mM magnesium chloride, 5 mM saccharalactone and 0.5 mM ABP (ABP was added in 4 μl methanol). The reaction was initiated by adding 5 mM UDP-glucuronic acid and continued for 30 min in a 37°C water bath. The reaction was stopped by adding 0.2 ml methanol to each reaction solution. A preliminary experiment showed that the formation of ABP-G in the reaction was linear for at least 45 min (data not shown). The solutions were then centrifuged at 16,000g for 3 min at 4°C, and the supernatants were analyzed by HPLC. HPLC analysis of urinary ABP-G was carried out using an Agilent system with a diode-array detector. The mobile phase consisted of 50 mM potassium phosphate (pH 6.8) and methanol. The system was operated at a flow rate of 1.75 ml/min, using a Partisil 10 ODS-2 reverse phase column (4.6 mm × 250 mm, Whatman). A linear gradient gradually increased the methanol concentration: 35% to 55% from 0-7 min, and 55% to 80% from 7-14 min. The ABP-G was monitored at 280 nm and eluted at 5.1 min; the peak area was integrated using Agilent’s ChemStation software. A calibration curve of ABP-G was established using a pure standard (Y = 0.421X + 12.266, r2 = 1.0, where Y is the peak area and X is pmol of ABP-G injected to HPLC). The identity of urinary ABP-G was confirmed by LC-MS/MS (data not shown). Urinary creatinine was measured using a creatinine kit purchased from Caymen, according to the manufacturer’s instruction.
Statistical analysis
All numerical results are expressed as mean ± standard deviation (n=3-6). P<0.05 is considered significant, using unpaired two-tailed Student’s t-test.
Results
Nrf2 potentiates ABP-induced DNA damage in bladder tissues in vivo, but protects bladder cells against ABP in vitro
Male C57BL/6 mice (6-8 weeks of age, both wild type and Nrf2 knockout) were given a single intraperitoneal dose of ABP at 10 and 50 mg/kg; 24 h later, ABP-DNA adduct levels in bladder tissues were measured. dG-C8-ABP, which accounts for 80% of all ABP-DNA adducts formed in human bladder tissues in vivo (22, 23), was quantified by capillary liquid chromatography and nanoelectrospray ionization-tandem mass spectrometry. ABP caused a marked and dose-dependent increase in dG-C8-ABP formation in the bladder tissues of both wild type mice and Nrf2-deficient mice. However, bladder levels of dG-C8-ABP were 2.4 and 2.7 fold higher in the wild type mice than in their Nrf2-deficient counterparts (Fig. 1A). Thus, Nrf2 significantly potentiated bladder DNA damage caused by ABP in vivo. We wondered if Nrf2 had a similar effect on ABP in bladder cells in vitro. Our recent study showed that ABP as well as its metabolite N-hydroxy-N-acetyl-4-aminobiphenyl (N-OH-AABP) caused significant dG-C8-ABP formation in human bladder RT4 cells and other cells in vitro (16). N-OH-AABP directly caused DNA damage, but ABP required rat liver S9 for metabolic activation, because RT4 cells as well as other bladder cells are apparently deficient in certain ABP-activating enzymes (16). N-OH-AABP rather than N-OH-ABP was used because the latter was known to be highly unstable (24). Nrf2 in RT4 cells was silenced by treatment with a Nrf2-specific siRNA for 48 h (Fig. 1B); these cells and the cells similarly treated with a control siRNA were then exposed to ABP at 0.5 mM (plus 6% rat liver S9) or N-OH-AABP at 0.1 mM for 3 h. Treatment with these carcinogens for 3 h was previously shown to lead to high levels of DNA adduct formation (16). dG-C8-ABP levels in Nrf2-silenced cells were 1.9 fold higher in the case of ABP and 1.8 fold higher in the case of N-OH-AABP than in control cells (Fig. 1C). Thus, Nrf2 provides protection against ABP and N-OH-AABP in bladder cells. However, this finding is in stark contrast to the in vivo effect of Nrf2 described above.
Figure 1.
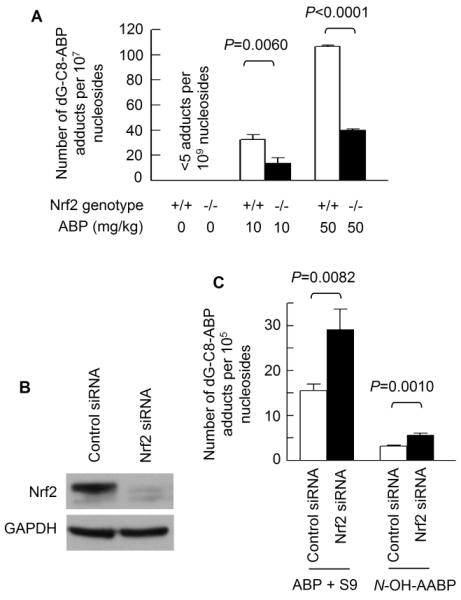
Nrf2 enhances ABP-induced DNA damage in bladder tissues in vivo but inhibits such damage in bladder cells in vitro. (A) Nrf2+/+ mice and Nrf2−/− mice were treated with a single intraperitoneal injection of ABP or vehicle; the bladders were harvested 24 h later for determination of dG-C8-ABP levels by LC/MS/MS. (B) RT4 cells were treated with control siRNA and Nrf2 siRNA for 48 h, followed by Western blotting of Nrf2 in whole cell lysates. GAPDH is a loading control. (C) After 48 h treatment with control siRNA and Nrf2 siRNA, RT4 cells were exposed to ABP (plus S9) or N-OH-AABP for 3 h before dG-C8-ABP measurement.
Nrf2 protects liver against ABP-induced DNA damage and promotes UGT-mediated Phase 2 metabolism and urinary excretion of ABP
While Nrf2 sensitizes bladder to ABP in vivo, hepatic dG-C8-ABP levels were 2.6 fold higher in Nrf2-deficient mice than in wild type mice, after treatment with ABP at 50 mg/kg for 24 h (Fig. 2A). Hence, Nrf2 protects the liver against ABP.
Figure 2.
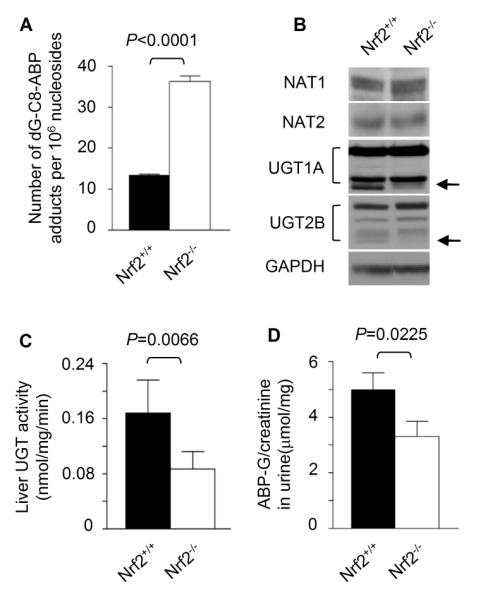
Nrf2 inhibits ABP-induced DNA damage in liver and promotes ABP metabolism. Both Nrf2+/+ mice and Nrf2−/− mice were used in the experiments. (A) Mice were treated with a single intraperitoneal injection of ABP at 50 mg/kg; the livers were harvested 24 h later for determination of dG-C8-ABP levels by LC/MS/MS. (B) Western blotting of liver tissues. GAPDH is a loading control. The arrows indicate the UGT bands (isoforms) that were up-regulated by Nrf2. (C) Microsomal fractions were prepared from liver tissues of untreated mice and measured for UGT activity in catalyzing ABP conjugation with glucuronic acid. (D) Total amounts of glucuronide conjugate of ABP (ABP-G) were measured in 24 h urine collected from mice that were given a single intraperitoneal injection of ABP at 50 mg/kg and were adjusted by urinary creatinine levels. Urinary creatinine concentration (mg/ml) was 0.26 ± 0.10 (mean ± SD).
In an attempt to further understand the dichotomy of Nrf2, we examined enzymes that are known to be involved in ABP metabolism. Aromatic amine acetyltransferases, NAT1 and NAT2, participate in ABP metabolism. Liver NAT1 and NAT2 are thought to contribute to ABP detoxification by catalyzing acetylation reactions of ABP or its N-hydroxylated metabolite (N-OH-ABP). However, Western blot analysis of liver tissues of wild type mice and Nrf2-deficient mice showed that Nrf2 had no effect on either NAT1 or NAT2 (Fig. 2B). UGT is also known to detoxify ABP by catalyzing the conjugation of ABP and N-OH-ABP with glucuronic acid in liver. The glucuronides, which are excreted in urine, however, are known to dissociate in acidic urine and thereby deliver the carcinogenic parent compounds to bladder tissue (25-27). UGT represents a supergene family, which is divided mainly into two subfamilies: UGT1A and UGT2B, each of which is further divided into multiple isoforms (28). Several Western blot bands (isoforms) of both UGT1A and UGT2B were detected in the livers of Nrf2+/+ mice and Nrf2−/− mice, but at least one isoform in each family showed a profound decrease in the latter (Fig. 2B). Experiments to identify the isoforms were not pursued for reasons provided in the Discussion. In accordance with the Western blot results, liver enzymatic activity in catalyzing the conjugation of ABP with glucuronic acid was 1.94 fold higher in the wild type mice than in the Nrf2-deficient mice (Fig. 2C). Likewise, the 24-h urinary level of ABP-G (adjusted by urinary creatinine) was 34% higher in the wild type mice than in the Nrf2-deficient mice, after treatment with ABP at 50 mg/kg (Fig. 2D). Urinary levels of the glucuronic acid conjugate of N-OH-ABP could not be measured due to high instability (26).
Taken together, our study has shown that Nrf2 protects the liver against DNA damage caused by ABP, but renders bladder more susceptible to this carcinogen. Furthermore, our results also strongly suggest that UGT is responsible at least in part for the dichotomous effects of Nrf2 on ABP in bladder and liver.
CPDT inhibits ABP-induced DNA damage and activates the Nrf2 cytoprotective signaling system in cultured bladder cells
We have recently shown that CPDT is a highly effective Nrf2 activator and Phase 2 enzyme inducer in cultured bladder cells, and that its induction of Phase 2 enzymes depends on Nrf2 (18). In a rat study in vivo, CPDT was particularly effective in the bladder, and more significantly, Nrf2 activation and induction of Phase 2 enzymes in the bladder by CPDT occurred exclusively in the epithelium, which is the principal site of bladder cancer development (18). To find out if CPDT is capable of inhibiting ABP-induced DNA damage in bladder cells, RT4 cells were first treated with CPDT at 12.5 and 25 μM for 48 h; cells were then incubated in fresh medium containing 0.5 mM ABP (plus 6% rat liver S9) or 0.1 mM N-OH-AABP for 3 h. dG-C8-ABP levels in these cells were then measured. The CPDT concentrations as well as the treatment conditions for ABP and N-OH-AABP were based on previous findings (16, 18). CPDT treatment led to significant and dose-dependent inhibition of dG-C8-ABP formation, up to 59% inhibition in the case of ABP and up to 84% inhibition in the case of N-OH-AABP (Fig. 3A). Since CPDT was not present in the culture medium during carcinogen exposure, a direct antagonistic effect of CPDT on ABP or N-OH-ABP was unlikely.
Figure 3.
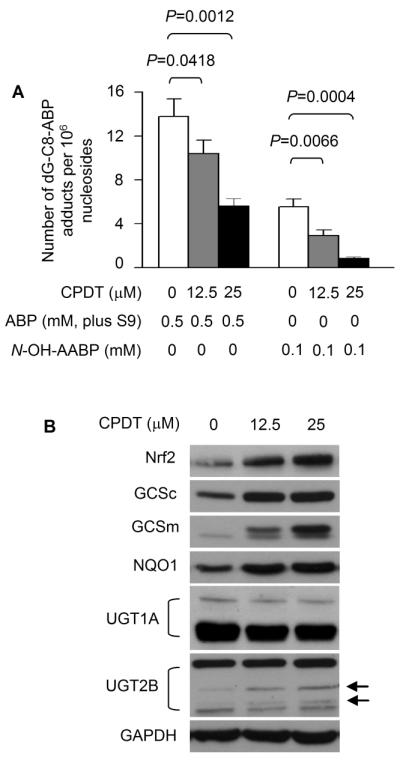
CPDT inhibits ABP-induced DNA damage, elevates Nrf2, and induces Phase 2 enzymes in bladder cells in vitro. (A) RT4 cells were treated with CPDT or solvent for 24 h and then exposed to ABP (with S9) or N-OH-AABP for 3 h, followed by dG-C8-ABP analysis by LC/MS/MS. (B) RT4 cells were treated with CPDT for 24 h. Nrf2 and Nrf2 target genes in whole cell lysates were measured by Western blotting. GAPDH is a loading control. The arrows indicate two UGT2B bands (isoforms) induced by CPDT.
As expected, the protective effects of CPDT against ABP and N-OH-AABP in RT4 cells were associated with significant stimulation of the Nrf2 signaling system. RT4 cells after treatment with CPDT at 12.5 and 25 μM for 48 h showed marked increases in protein levels of Nrf2 and several well-known Nrf2 target genes, including Phase 2 gene NQO1 and both the catalytic and the regulatory subunits of glutamate cysteine ligase (GCSc and GCSm) (Fig. 3B). These genes were assessed as biomarkers of Nrf2 transactivation activity and may not necessarily mediate the inhibitory effect of CPDT. Two UGT1A bands and four UGT2B bands were detected in RT4 cells, but CPDT had no effect on UGT1A and slightly elevated two UGT2B isoforms (Fig. 3B, indicated with the arrows). The identities of these isoforms have not been determined.
In vivo, CPDT inhibits ABP-induced DNA damage in the bladder but not in the liver
Our present study reveals that liver UGT assumes dual roles in response to ABP – protecting the liver against ABP, but increasing bladder exposure to ABP through increased synthesis and urinary excretion of glucuronides of ABP and its metabolites. Since in RT4 cells CPDT strongly inhibited DNA damage caused by ABP and N-OH-AABP and also activated Nrf2 cytoprotective signaling, and our recent study showed that CPDT activated Nrf2 signaling in the rat bladder in vivo (18), we predicted that CPDT would inhibit ABP-induced DNA damage in the mouse bladder in vivo. However, it was difficult to tell if CPDT would protect liver against ABP or if CPDT would modulate liver UGT in vivo. The experiments to address these questions were carried out in both wild type mice and Nrf2-deficient mice, so that the potential involvement of Nrf2 could also be assessed. The mice were treated with CPDT at 0, 5, 20 and 80 mg/kg by gavage once daily for 5 days; 3 h after the last CPDT dose, each mouse was given ABP at 50 mg/kg i.p.; the mice were killed 24 h after the ABP injection, and the bladders and livers were harvested for measurement of tissue dG-C8-ABP levels. In a parallel experiment, mice were treated with only CPDT and were killed 24 h after the last CPDT dose for measurement of induction of UGT and other Phase 2 enzymes.
CPDT inhibited ABP-induced DNA damage in the bladders of the wild type mice in a dose-dependent manner; dG-C8-ABP level was reduced by up to 68% (Fig. 4A). However, CPDT was much less effective in the Nrf2-deficient mice, as bladder dG-C8-ABP levels in mice treated with CPDT were reduced by only 9-33% (Fig. 4A). Hence, Nrf2 plays an important role in the ability of CPDT to protect bladder against ABP-induced DNA damage in vivo. In contrast, CPDT treatment had virtually no effect on ABP-induced dG-C8-ABP formation in the livers of the wild type mice, and was ineffective in the livers of Nrf2-deficient mice, except at the lowest CPDT dose where hepatic dG-C8-ABP levels in Nrf2-deficient mice were 30% lower than in the control (P<0.05) (Fig. 4B). The reason for this outlier, which is reproducible, is unknown.
Figure 4.

CPDT protects against ABP in bladder but not in liver in vivo. (A) Nrf2+/+ mice and Nrf2−/− mice were treated with CPDT or vehicle by gavage once daily for 5 days; 3 h after the last CPDT dose, each mouse was given ABP i.p.; 24 h later, animals were killed. dG-C8-ABP levels in the bladders and livers were quantified by LC/MS/MS. (B) Nrf2+/+ mice and Nrf2−/− mice were treated with CPDT or vehicle by gavage once daily for 5 days; 24 h after the last CPDT dose, animals were killed. Nrf2 and Nrf2 target genes in the bladders and livers were measured by Western blotting. GAPDH is a loading control. The arrows indicate the UGT bands (isoforms) that were regulated in a Nrf2-dependent manner.
The potential impact of CPDT on UGT and other Nrf2 target genes in the bladder and liver tissues was examined by Western blotting. Besides UGT1A and UGT2B, other genes, including GCSc, GCSm and NQO1, were assessed as indicators of Nrf2 activation by CPDT. In agreement with our previous data in rat (18), CPDT was more effective in the bladder than in the liver in activating Nrf2 signaling, as GCSc, GCSm and NQO1 were all significantly elevated by CPDT in the bladders of Nrf2+/+ mice (Fig. 4C), but only NQO1 was elevated in the livers of these animals (Fig. 4D). CPDT was unable to significantly induce these enzymes in the bladders and livers of Nrf2−/− mice. Interestingly, while both GCSc and GCSm were largely undetectable in the bladders of Nrf2−/− mice, significant levels of these enzymes were detected in their liver tissues, implying that these genes are subjected to regulation by Nrf2-independent mechanisms in the liver. Multiple bands of both UGT1A and UGT2B were detected in the bladder and liver tissues (Fig. 4C and 4D), reflecting their multi-isomeric nature. Three UGT1A bands were detected in both bladder and liver, two of which were apparently not regulated by Nrf2, whereas the third band (indicated by an arrow) was slightly up regulated by Nrf2 and CPDT. Many more UGT2B isoforms seem to exist in the liver than in the bladder, some of which were expressed in a Nrf2-independent manner, but one isoform in both bladder and liver was clearly up regulated by Nrf2 and stimulated by CPDT (indicated by an arrow). However, since CPDT showed no protective effect on ABP-induced DNA damage in the liver as described above, it is unlikely that the limited induction of UGT by CPDT in the liver is functionally important with regard to ABP detoxification (glucuronidation). It is also possible that ABP is not the substrate of the isoforms that were slightly induced by CPDT. Indeed, liver activities of ABP glucuronidation were similar between the control Nrf2+/+ mice and Nrf2+/+ mice treated with the highest dose of CPDT (0.20 ± 0.05 nmol/min/mg vs 0.18 ± 0.03 nmol/ming/mg).
Discussion
ABP is a major human bladder carcinogen from tobacco smoke and other environmental sources, and is known to be metabolically activated in liver. dG-C8-ABP is the predominant DNA adduct formed in human bladder cells and tissues exposed to ABP and is closely associated with human bladder cancer development and prognosis. While numerous studies have shown that Nrf2 plays a critical role in cellular defense against chemical carcinogens and oxidants, our present study has revealed a serious dilemma that Nrf2 faces in defense against ABP. On one hand, Nrf2 protects liver against ABP-induced DNA damage, as Nrf2 knockout increased liver dG-C8-ABP level by 2.7 fold in mice exposed to ABP (Fig. 2A). On the other hand, Nrf2 renders bladder significantly more susceptible to the genotoxicity of ABP, as Nrf2 knockout decreased bladder dG-C8-ABP level by 2.7 fold in mice exposed to the same dose of ABP (Fig. 1A). However, in vitro, Nrf2 protects bladder cells against ABP and its metabolite (N-OH-AABP), as dG-C8-ABP levels in Nrf2-silenced cells increased 1.8-1.9 fold (Fig. 1C). This suggested that Nrf2 acted at a site remote from the bladder in potentiating ABP-induced DNA damage in this organ in vivo. Indeed, further studies in wild type mice and Nrf2-deficient mice showed that a) levels of certain UGT1A and UGT2B isoforms were significantly higher in Nrf2+/+ liver than in Nrf2−/− liver (Fig. 2B); b) Nrf2 knockout eliminated 49% of liver UGT activity in catalyzing ABP glucuronidation (Fig. 2C); c) Nrf2 knockout also led to a 34% decrease in the urinary level of ABP-G in mice dosed with ABP (Fig. 2D). Given that it has been well documented that the glucuronide conjugates of ABP and its N-hydroxylated metabolites, which are produced in the liver and disposed via urinary excretion, are unstable in acidic urine and deliver the parent carcinogenic compounds to the bladder, our results strongly suggest that liver UGT is responsible at least in part for the dichotomous effects of Nrf2 on ABP in bladder and liver. NAT1 and NAT2 are also involved in ABP metabolism, but Nrf2 had no effect on these enzymes in the liver (Fig. 2B).
UGT is a supergene family, consisting of UGT1A and UGT2B subfamilies, each of which is further divided into multiple isoforms (29). A recent study showed that multiple UGT isoforms might be up-regulated by Nrf2 in mouse liver, including UGT1A6, UGT1A7, UGT2B1, UGT2B35 and UGT2B36 (1, 13). It is not known at the present time which of these UGT isoform(s) may catalyze the glucuronidation of ABP and its metabolites. In our experiments, only one band of UGT1A and UGT2B was clearly up-regulated by Nrf2 and CPDT, but this may be due to antibody specificity and sensitivity. We did not attempt to measure the catalytic activity of specific mouse UGT isoforms toward ABP or to determine the identities of Nrf2-regulated UGT isoforms, because many UGT isoforms that are expressed in mice are pseudogenes in humans, or vice versa (30, 31), and a different set of UGT isoforms appear to be up-regulated by Nrf2 in humans, including UGT1A1, UGT1A8 and UGT1A10 (15, 32). In view of the potentially important role of UGT in bladder cancer, however, it is important in future studies to identify the human UGT isoform(s) that is both regulated by Nrf2 and catalyzes ABP glucuronidation.
Nrf2 plays an important role in bladder cell defense against ABP locally (Fig. 1C). However, given that certain UGT isoforms that metabolize ABP are up-regulated by Nrf2, that increased liver UGT activity likely increases bladder exposure to ABP, and that ABP is a major human bladder carcinogen, it seems important to identify Nrf2-activating cancer chemopreventive agents that do not significantly induce liver UGT that promotes glucuronidation of ABP and its metabolites, if such agents are to be developed for use in humans exposed to ABP, particularly in smokers. Our recent study showed that sulforaphane, a naturally occurring chemopreventive agent, activated Nrf2 and protected against ABP-induced DNA damage in the bladder in mice, but sulforaphane also activated Nrf2 in the liver (16), although it has not been determined to what extent sulforaphane induces liver UGT. In the present study, in cultured bladder cells, CPDT strongly activated Nrf2 and Nrf2-regulated cytoprotective genes and inhibited DNA damage caused by ABP and N-OH-AABP (Fig. 3). In the wild type mice In vivo, it also strongly inhibited bladder DNA damage in mice exposed to ABP and activated Nrf2 signaling in that tissue (Fig. 4). In the liver, however, CPDT had little effect on ABP-induced DNA damage and did not induce two of the three Nrf2 genes examined. Its inducing effect on UGT in the liver was also limited and did not modulate liver activity of ABP glucuronidation. Thus, the chemopreventive activity of CPDT is relatively bladder-specific. Our data also show that the chemopreventive activity of CPDT was significantly attenuated but not completely eliminated in Nrf2-deficient mice (Fig. 4), indicating that its activity is mediated through both Nrf2-dependent and Nrf2-independent pathways.
In summary, while Nrf2 protects bladder tissues against ABP locally, it counters such protective effect by stimulating UGT and promoting UGT-catalyzed glucuronidation of ABP and its metabolites in liver and increasing urinary excretion of such conjugates, which causes increased exposure of bladder tissue to ABP. It is possible that Nrf2 may act in a similar manner toward other arylamine bladder carcinogens. Indeed, Williams and coworkers previously showed that feeding rats with butylated hydroxytoluene, a Nrf2 activator, reduced the formation of liver tumors following treatment with N-2-fluorenylacetaminde (FAA), but led to an increase in bladder tumors – tumors not seen in rats treated with FAA alone (33). However, such a dichotomous effect of Nrf2 was not detected in bladder carcinogenesis induced by the nitrosamine carcinogen N-nitrosobutyl(4-hydrobutyl)amine in mice (1). CPDT is a promising bladder cancer chemopreventive agent, as it strongly protects bladder tissues and cells against ABP, and activates Nrf2 in the bladder, but does not stimulate ABP glucuronidation in the liver. Neither Nrf2 nor CPDT have been assessed for their effects on ABP-induced bladder and liver cancer formation. These experiments remain difficult, as the only available animal protocol shows low tumor incidence, high mortality and long (2-year) tumor induction time by ABP (34).
Acknowledgements
We thank Thomas W. Kensler (University of Pittsburgh) for providing Nrf2−/− mouse breeders and Yun Li for valuable technical assistance. This work was supported by National Institutes of Health Grants R01CA69390, R01CA112231 and R01CA120533.
References
- 1.Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64:6424–31. doi: 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, et al. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann N Y Acad Sci. 2008;1147:61–9. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vineis P. Epidemiology of cancer from exposure to arylamines. Environ Health Perspect. 1994;102(Suppl 6):7–10. doi: 10.1289/ehp.94102s67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skipper PL, Tannenbaum SR, Ross RK, Yu MC. Nonsmoking-related arylamine exposure and bladder cancer risk. Cancer Epidemiol Biomarkers Prev. 2003;12:503–7. [PubMed] [Google Scholar]
- 6.Negri E, La Vecchia C. Epidemiology and prevention of bladder cancer. Eur J Cancer Prev. 2001;10:7–14. doi: 10.1097/00008469-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Smith CJ, Perfetti TA, Garg R, Hansch C. IARC carcinogens reported in cigarette mainstream smoke and their calculated log P values. Food Chem Toxicol. 2003;41:807–17. doi: 10.1016/s0278-6915(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 8.Poirier MC, Beland FA. DNA adduct measurements and tumor incidence during chronic carcinogen exposure in rodents. Environ Health Perspect. 1994;102(Suppl 6):161–5. doi: 10.1289/ehp.94102s6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuneoka Y, Dalton TP, Miller ML, Clay CD, Shertzer HG, Talaska G, et al. 4-aminobiphenyl-induced liver and urinary bladder DNA adduct formation in Cyp1a2(−/−) and Cyp1a2(+/+) mice. J Natl Cancer Inst. 2003;95:1227–37. doi: 10.1093/jnci/djg025. [DOI] [PubMed] [Google Scholar]
- 10.Airoldi L, Orsi F, Magagnotti C, Coda R, Randone D, Casetta G, et al. Determinants of 4-aminobiphenyl-DNA adducts in bladder cancer biopsies. Carcinogenesis. 2002;23:861–6. doi: 10.1093/carcin/23.5.861. [DOI] [PubMed] [Google Scholar]
- 11.Curigliano G, Zhang YJ, Wang LY, Flamini G, Alcini A, Ratto C, et al. Immunohistochemical quantitation of 4-aminobiphenyl-DNA adducts and p53 nuclear overexpression in T1 bladder cancer of smokers and nonsmokers. Carcinogenesis. 1996;17:911–6. doi: 10.1093/carcin/17.5.911. [DOI] [PubMed] [Google Scholar]
- 12.Block NL, Sigel MM, Lynne CM, Ng AB, Grosberg RA. The initiation, progress, and diagnosis of dog bladder cancer induced by 4-aminobiphenyl. Invest Urol. 1978;16:50–4. [PubMed] [Google Scholar]
- 13.Reisman SA, Yeager RL, Yamamoto M, Klaassen CD. Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol Sci. 2009;108:35–47. doi: 10.1093/toxsci/kfn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeager RL, Reisman SA, Aleksunes LM, Klaassen CD. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol Sci. 2009;111:238–46. doi: 10.1093/toxsci/kfp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalthoff S, Ehmer U, Freiberg N, Manns MP, Strassburg CP. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyltransferase 1A10. J Biol Chem. 2010;285:5993–6002. doi: 10.1074/jbc.M109.075770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Y, Paonessa JD, Randall KL, Argoti D, Chen L, Vouros P, et al. Sulforaphane inhibits 4-aminobiphenyl-induced DNA damage in bladder cells and tissues. Carcinogenesis. 2010;31:1999–2003. doi: 10.1093/carcin/bgq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legrand L, Mollier Y, Lozac’h N. Sulfuration des composes organiques (IV). Dithiole-1,2-thiones-3 comportant des substituants hydrocarbones ou des noyaux condenses. Bull Soc Chim Fr. 1953:327–31. [Google Scholar]
- 18.Paonessa JD, Munday CM, Mhawech-Fauceglia P, Munday R, Zhang Y. 5,6-Dihydrocyclopenta[c][1,2]-dithiole-3(4H)-thione is a promising cancer chemopreventive agent in the urinary bladder. Chem Biol Interact. 2009;180:119–26. doi: 10.1016/j.cbi.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S, et al. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur J Pharmacol. 2009;620:138–44. doi: 10.1016/j.ejphar.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Randall KL, Argoti D, Paonessa JD, Ding Y, Oaks Z, Zhang Y, et al. An improved liquid chromatography-tandem mass spectrometry method for the quantification of 4-aminobiphenyl DNA adducts in urinary bladder cells and tissues. J Chromatogr A. 2010;1217:4135–43. doi: 10.1016/j.chroma.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Zoughool M, Talaska G. 4-Aminobiphenyl N-glucuronidation by liver microsomes: optimization of the reaction conditions and characterization of the UDP-glucuronosyltransferase isoforms. J Appl Toxicol. 2006;26:524–32. doi: 10.1002/jat.1172. [DOI] [PubMed] [Google Scholar]
- 22.Talaska G, al-Juburi AZ, Kadlubar FF. Smoking related carcinogen-DNA adducts in biopsy samples of human urinary bladder: identification of N-(deoxyguanosin-8-yl)-4-aminobiphenyl as a major adduct. Proc Natl Acad Sci U S A. 1991;88:5350–4. doi: 10.1073/pnas.88.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beland FA, Beranek DT, Dooley KL, Heflich RH, Kadlubar FF. Arylamine-DNA adducts in vitro and in vivo: their role in bacterial mutagenesis and urinary bladder carcinogenesis. Environ Health Perspect. 1983;49:125–34. doi: 10.1289/ehp.8349125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Z, Hu W, Rom WN, Beland FA, Tang MS. 4-aminobiphenyl is a major etiological agent of human bladder cancer: evidence from its DNA binding spectrum in human p53 gene. Carcinogenesis. 2002;23:1721–7. doi: 10.1093/carcin/23.10.1721. [DOI] [PubMed] [Google Scholar]
- 25.Al-Zoughool M, Succop P, Desai P, Vietas J, Talaska G. Effects of N-glucuronidation on urinary bladder genotoxicity of 4-aminobiphenyl in male and female mice. Environmental Toxicology and Pharmacology. 2006;22:153–9. doi: 10.1016/j.etap.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Radomski JL, Hearn WL, Radomski T, Moreno H, Scott WE. Isolation of the glucuronic acid conjugate of n-hydroxy-4-aminobiphenyl from dog urine and its mutagenic activity. Cancer Res. 1977;37:1757–62. [PubMed] [Google Scholar]
- 27.Babu SR, Lakshmi VM, Huang GP, Zenser TV, Davis BB. Glucuronide conjugates of 4-aminobiphenyl and its N-hydroxy metabolites. pH stability and synthesis by human and dog liver. Biochem Pharmacol. 1996;51:1679–85. doi: 10.1016/0006-2952(96)00165-7. [DOI] [PubMed] [Google Scholar]
- 28.King CD, Rios GR, Green MD, Tephly TR. UDP-glucuronosyltransferases. Curr Drug Metab. 2000;1:143–61. doi: 10.2174/1389200003339171. [DOI] [PubMed] [Google Scholar]
- 29.Mackenzie PI, Bock KW, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, et al. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 2005;15:677–85. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- 30.Buckley DB, Klaassen CD. Tissue- and gender-specific mRNA expression of UDP-glucuronosyltransferases (UGTs) in mice. Drug Metab Dispos. 2007;35:121–7. doi: 10.1124/dmd.106.012070. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura A, Nakajima M, Yamanaka H, Fujiwara R, Yokoi T. Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab Dispos. 2008;36:1461–4. doi: 10.1124/dmd.108.021428. [DOI] [PubMed] [Google Scholar]
- 32.Yueh MF, Tukey RH. Nrf2-Keap1 signaling pathway regulates human UGT1A1 expression in vitro and in transgenic UGT1 mice. J Biol Chem. 2007;282:8749–58. doi: 10.1074/jbc.M610790200. [DOI] [PubMed] [Google Scholar]
- 33.Maeura Y, Weisburger JH, Williams GM. Dose-dependent reduction of N-2-fluorenylacetamide-induced liver cancer and enhancement of bladder cancer in rats by butylated hydroxytoluene. Cancer Res. 1984;44:1604–10. [PubMed] [Google Scholar]
- 34.Schieferstein GJ, Littlefield NA, Gaylor DW, Sheldon WG, Burger GT. Carcinogenesis of 4-aminobiphenyl in BALB/cStCrlfC3Hf/Nctr mice. Eur J Cancer Clin Oncol. 1985;21:865–73. doi: 10.1016/0277-5379(85)90227-5. [DOI] [PubMed] [Google Scholar]