

Abstract
The nucleation event in α-helix formation is a fundamental process in protein folding. However, determining how quickly it takes place based on measurements of the relaxation dynamics of helical peptides is difficult because such relaxations invariably contain contributions from various structural transitions such as, from helical to non-helical states and helical to partial-helical conformations. Herein we measure the temperature-jump (T-jump) relaxation kinetics of three model peptides that fold into a single-turn α-helix, using time-resolved infrared spectroscopy, aiming to provide a direct assessment of the helix nucleation rate. The α-helical structure of these peptides is stabilized by a covalent cross-linker formed between the sidechains of two residues at the i and i+4 positions. If we assume that this cross-linker mimics the structural constraint arising from a strong sidechain-sidechain interaction (e.g., a salt-bridge) in proteins, these peptides would represent good models for studying the nucleation process of an α-helix in a protein environment. Indeed, we find that the T-jump induced relaxation rate of these peptides is approximately (0.6 μs)−1 at room temperature, which is slower than that of commonly studied alanine-based helical peptides but faster than that of a naturally occurring α-helix whose folded state is stabilized by a series of sidechain-sidechain interactions. Taken together, our results put an upper limit of about 1 μs for the helix nucleation time at 20 °C and suggest that the subsequent propagation steps occur with a time constant of about 240 ns.
Keywords: Helix-coil transition, Infrared spectroscopy, Protein folding kinetics, T-jump
INTRODUCTION
The helix-coil transition is a fundamental event in protein folding. As such, numerous studies have been devoted to investigating the dynamics and mechanism of monomeric α-helix folding in solution.1-13 Though apparently one of the simplest folding processes, helix formation proceeds through a complex mechanism thought to consist of, at the very least, a nucleation step followed by a series of propagation events along the remaining length of the chain.14-21 However, previous kinetic studies of the helix-coil transition using temperature-jump (T-jump) methods were performed on α-helices that consisted of several helical turns, making it difficult to dissect the key kinetic events in helix folding, due to the fact that a T-jump induced relaxation process often involves re-equilibration between several conformations, for example, between the helical and non-helical subensembles and between different helical states. Thus, in order to directly assess the nucleation rate in helix formation it is desirable to study the relaxation kinetics of short peptides that fold into a single α-helical turn.
It is well known that the stability of monomeric α-helices depends strongly on the number of consecutively repeating α-helical units.22-28 This can be illustrated by expressing the standard free energy for helix formation in terms of helix length:
| (1) |
Where the penalty of arises from the inability of peptide groups at the end of the helix to form stabilizing i to i+4 hydrogen bonds, requiring a minimum helix length n to compensate and provide an overall stabilizing . As such, very short α-helices can only be stabilized by an external constrain that is positioned to bias the peptide backbone towards the folded state through, for example, cyclization.29 Recently, Fairlie and coworkers30,31 demonstrated such a strategy and showed that a series of pentapeptides can adopt a stable, single-turn helical conformation in water when locked into a 20-membered macrocycle through the formation of a sidechain (i) to sidechain (i+4) linkage (Figure 1). For instance, the pentapeptide Ac-KARAD-NH2 was shown to be highly helical in aqueous solution when the Lys and Asp sidechains are acetamized to form an amide bond31 (the corresponding cyclic peptide is hereafter referred to as cyc-KARAD). Interestingly, the helical structure of cyc-KARAD was found to be stable even in the presence of 8 M guanidinium chloride but unfolds at high temperatures, suggesting a potential desolvation effect of the cross-linker. Thus, cyc-KARAD constitutes an interesting model for studying the kinetics of helix nucleation via temperature perturbation methods. In addition, such cyclic peptides may have an added advantage as they are akin to those containing a photo-switchable linker,32,33 which have been proposed as models of helical segments in proteins because the linker provides constraints likely to be present in globular proteins.21,32-35 Herein, we study the relaxation kinetics of three cyclic peptides (i.e., cyc-KARAD, cyc-RKAAAD, and cyc-RKMAAD) using a T-jump infrared technique6,9 and also perform molecular dynamics (MD) simulations to show such constrained pentapetides indeed adopt an α-helical conformation at low temperatures. Our results show that the T-jump induced relaxation rate of these peptides is slower than that of (unconstrained) alanine-based helical peptides, but is faster than that of a naturally occurring α-helix that is known to be stabilized by sidechain-sidechain salt bridges. We also find that substitution of a bulkier sidechain into the sequence slightly slows down the relaxation kinetics. Additionally, we employ a sequential kinetic model and the relaxation rate of these cyclic peptides to estimate the timescale of helix propagation.
Figure 1.
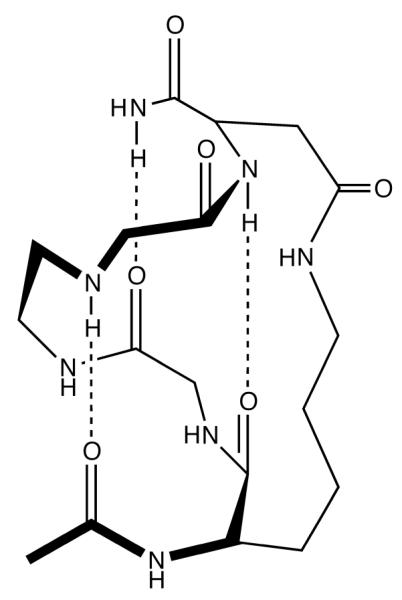
The putative folded structure of cyc-KAAAD, which was constructed based on that proposed by Fairlie and coworkers.31
MATERIALS AND METHODS
Peptide Samples
Peptides (i.e., Ac-KARAD-NH2, Ac-RKAAAD-NH2, and Ac-RKMAAD-NH2) were synthesized using standard Fmoc-protocols on a PS3 automated peptide synthesizer (Protein Technologies, MA). Cyclization of the Lys and Asp sidechains was accomplished using the method of Shepherd et al.31 All peptide products were purified to homogeneity by reverse-phase high performance liquid chromatography and then lyophilized. Multiple rounds of lyophilization were then carried out to remove the residual trifluoroacetic acid (TFA) from peptide synthesis and to ensure amide H/D exchange. Peptide solutions used in both CD and IR measurements were prepared by directly dissolving lyophilized peptide solids in 10 mM (CD) or 20 mM (IR) phosphate buffer (pH* 7) at concentrations of about 0.35 and 11.7 mM, respectively.
Circular Dichroism (CD) and Fourier Transform Infrared (FTIR) Measurements
CD measurements were carried out on an Aviv 62A DS circular dichroism spectrometer (Aviv Associate, NJ) using a 1 mm sample cuvette. FTIR spectra were collected on a Magna-IR 860 spectrometer (Nicolet, WI) using a two-compartment CaF2 sample cell with an optical pathlength of 52 μm. Temperature control and other details of the FTIR setup have been described previously.6
Infrared T-jump Apparatus
The time-resolved T-jump IR apparatus is the same as that described elsewhere9 except that in the current study a continuous wave quantum cascade laser (Daylight Solutions, CA) served as the IR probe. The probing frequency was 1640 cm−1 and the T-jump was in the range of 3–11 °C.
Molecular Dynamics (MD) Simulation
MD simulations were carried out using the NAnoscale Molecular Dynamics (NAMD) program (version 2.5 b1)36 and the CHARMM22 force field.37 The peptide of interest was immersed in 1932 TIP3P water molecules38 in a 40 Å cubic box. Following an initial 1 ns equilibration run at 298 K and 1 atm in the NPT ensemble, a productive run at 298 K in the NVT ensemble was carried out. During the simulation, temperature (298 K) was controlled by Langevin dynamics and the pressure (1 atm) was maintained by the Nosé-Hoover Langevin piston pressure control method. In addition, periodic boundary conditions were used to reduce edge effects. Nonbonded interactions with a cut-off of 12 Å were calculated every time step (2 fs) and full electrostatic interactions were calculated every two time steps. The SHAKE algorithm was employed to constrain all bonds involving the hydrogen atom to their equilibrium values and the full electrostatics were calculated every second step using the particle-mesh Ewald (PME) method.39,40
RESULTS
Equilibrium CD and FTIR measurements
As shown (Figure 2), the far-UV CD spectra of the three cyclic peptides at 2.0 °C exhibit two minima at approximately 206 and 218 nm, respectively. These data are consistent with those reported by Shepherd et al.31 and indicate that these peptides predominantly adopt an α-helical conformation at low temperatures. It is well known that the characteristic CD spectral minima of exceptionally short α-helices are blue-shifted with respect to the typical values (i.e., 208 and 222 nm) observed for longer α-helices.41 In addition, the CD spectra of these peptides measured at 70 °C (Figure 2) are characteristic of disordered or random structures,42 indicating that their α-helical conformations unfold at high temperatures. However, similar to those observed for other monomeric α-helices the CD thermal unfolding transition of these peptides (e.g., that presented in the inset of Figure 1 for cyc-KARAD) is broad, due to a relatively small enthalpy change upon folding for such systems.
Figure 2.
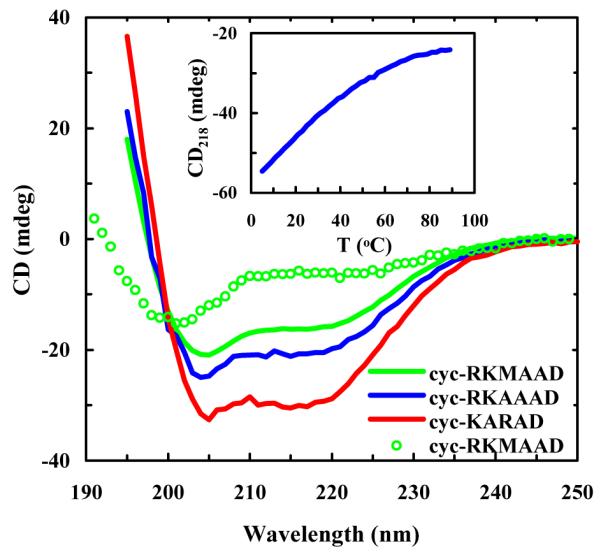
CD spectra of the cyclic peptides in 10 mM phosphate buffer (pH* 7) at 2.0 °C, as indicated. The difference in the amplitudes was due to the concentration difference. Also shown are the CD thermal melting curve of cyc-KARAD measured at 218 nm (inset) and the CD spectrum of cyc-RKMAAD obtained at 70 °C (open cycles).
As shown (Figure 3), the FTIR spectra (in the amide I’ region) of cyc-KARAD and cyc-RKAAAD consist of three resolvable bands. The two minor bands, centered at about 1590 and 1610 cm−1, respectively, can be assigned to νs(CN3H5+) and νas(CN3H5+) of the Arg sidechain,43 whereas the major band arises from the amide carbonyl groups. As shown (Figure 4), the FTIR difference spectra of cyc-KARAD, which were generated by subtracting the spectrum collected at 6.5 °C from those obtained at higher temperatures, exhibit a dominant negative feature centered at 1640 cm−1 and a positive feature centered at about 1668 cm−1. Because the latter is widely accepted as an indication of the formation of disordered peptide conformations,44 these temperature-induced spectral changes, which are consistent with the CD results, indicate that the α-helical population decreases with increasing temperature and can be used to probe the conformational relaxation kinetics of the peptide. Moreover, since fully solvated α-helices typically yield difference IR spectra with a negative band centered at about 1630 cm−1 upon thermal denaturation,44 this result suggests that the helical amides of these cyclic peptides are less solvated presumably due to the shielding effect of the sidechain-sidechain cross-linker, similar to the desolvation effect of long charged sidechains (e.g., Arg and Lys).45,46 In addition, desolvation has also been shown to increase the stability of α-helices.47,48 Another possibility is, as has been suggested by transition dipole coupling theories,43,49,50 that this relative blue-shift is due to the very short helical length of the cyclic peptide.
Figure 3.
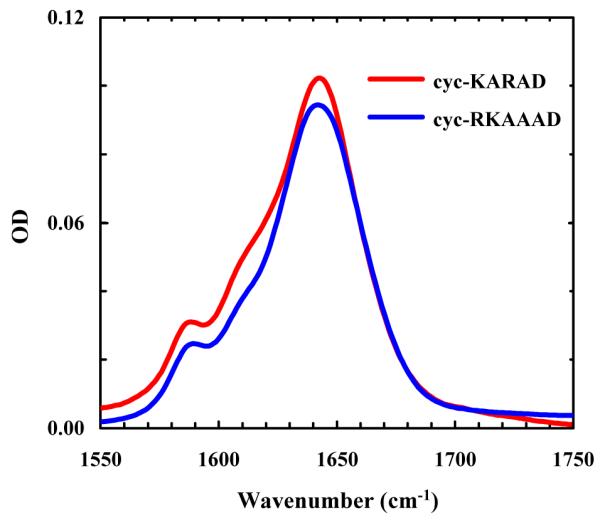
FTIR spectra (in the amide I’ region) of cyc-KARAD and cyc-RKAAAD at 6.5 and 5.4 °C, respectively.
Figure 4.
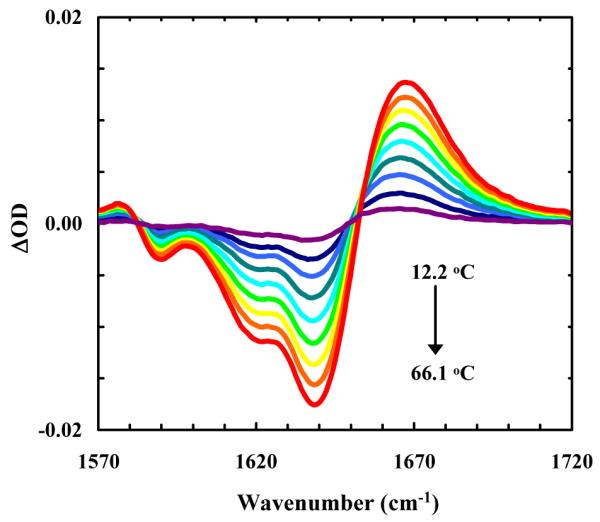
Difference FTIR spectra of cyc-KARAD, which were generated by subtracting the spectrum collected at 6.5 °C from those collected at higher temperatures in a step of 6–7 °C.
T-jump induced relaxation kinetics
Based on the FTIR results, the T-jump induced relaxation kinetics for these peptides were probed at 1640 cm−1, where the folded helical conformation absorbs. As shown (Figure 5), the T-jump induced relaxation proceeds in two distinct and well-separated phases, in agreement with our previous studies on alanine-based peptides.7,9-11 The fast phase is fully developed within the response time of the IR detector and thus is not time resolved. According to our earlier interpretation, this unresolved kinetic phase likely arises from a temperature-induced shift of the amide I’ band.51 The slow phase, which is well-resolved below approximately 45 °C, is attributed to a conformational redistribution process between helical and non-helical conformations in response to the T-jump. While non-exponential relaxation kinetics have been observed for alanine-based peptides9 and photo-switchable peptides,33,35 in the current case the T-jump-induced conformational relaxation can be modeled satisfactorily with a single-exponential function convoluted with a 20 ns instrument response function (Figure 5). However, similar to those observed for other helical peptides,11 the relaxation rate constants obtained here exhibit Arrhenius temperature dependence with an apparent activation energy of about 12.4 ± 0.5, 10.9 ± 0.7, and 10.6 ± 0.3 kcal/mol for cyc-RKAAAD, cyc-KARAD, and cyc-RKMAAD, respectively (Figure 6).
Figure 5.
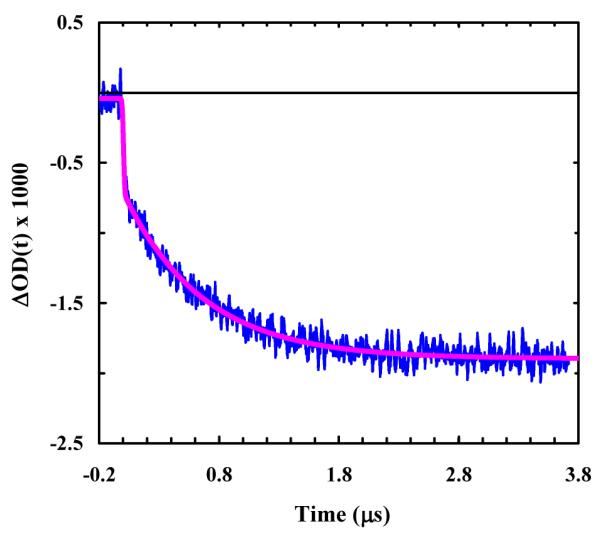
A representative relaxation kinetic trace of cyc-RKAAAD in response to a T-jump of 3.7 °C, from 14.2 to 17.9 °C. The blue line is a convolution of the instrument response function with the following exponential function: ΔOD(t) = A · [1− B · exp(−t /τ)], with A = −1.9, B = 0.64 and τ = 0.71 μs.
Figure 6.
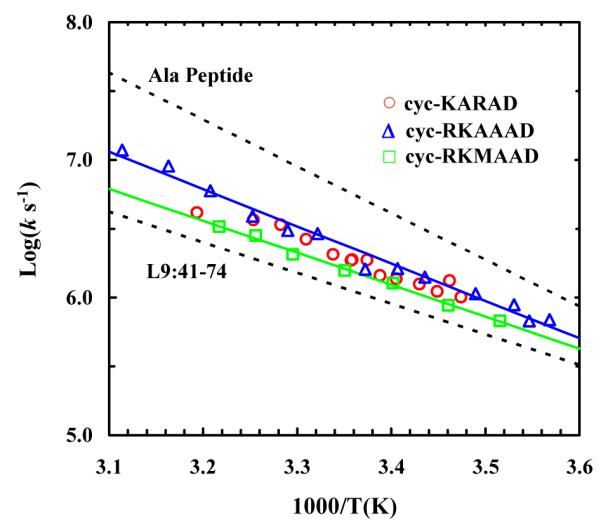
Arrhenius plot of the T-jump induced conformational relaxation rates of the cyclic peptides studied, as indicated. The corresponding smooth lines are the respective linear regressions of these data. Also shown for comparison are the relaxation rates of an alanine-based peptide6 and a naturally occurring helical peptide L9:41-74,44 as indicated.
MD simulations
To further confirm that these cyclic peptides fold into an α-helical conformation at low temperatures, we performed two sets of MD simulations (A and B) of Ac-AARAA-NH2 with a starting structure having the typical backbone dihedral angles of an α-helix (i.e., φ = −58° and ψ = −47°).52 Simulation A was carried out at 298 K for 60 ns, whereas simulation B was run at 298 K first for 20 ns and then at 308 K for 10 ns followed by a production run 298 K for 30 ns. In addition, following the work of Paoli et al.,53 in simulation B a 3.0 kcal/(mol Å2) harmonic constraint was added between the β–carbons of the Ala1 and Ala5 residues to mimic the sidechain (i) to sidechain (i+4) linkage. As shown (Figure 7), the unconstrained model unfolded within the first 5 ns of the simulation (i.e., simulation A), whereas the one with the harmonic constraint did not unfold during the first 40 ns of the simulation (i.e., simulation B). While at a later time the constrained peptide briefly adopted an unfolded structure with an RMSD close to that of the unfolded structure obtained in simulation A, this result is not surprising since the simulation was carried out at a temperature where both folded and unfolded conformations are expected to be significantly populated (Figure 2). After returning to the folded basin, the dihedral angles sampled by the peptide in the last 10 ns of the simulation were found to be distributed in the helical region (i.e., φ and ψ ranging from −93° to −52° and −70° to −25°, respectively). Thus, these simulation results support the notion that the folded state of the cyclic peptides studied here is α-helical and their unfolded state has a random-like structure.52
Figure 7.
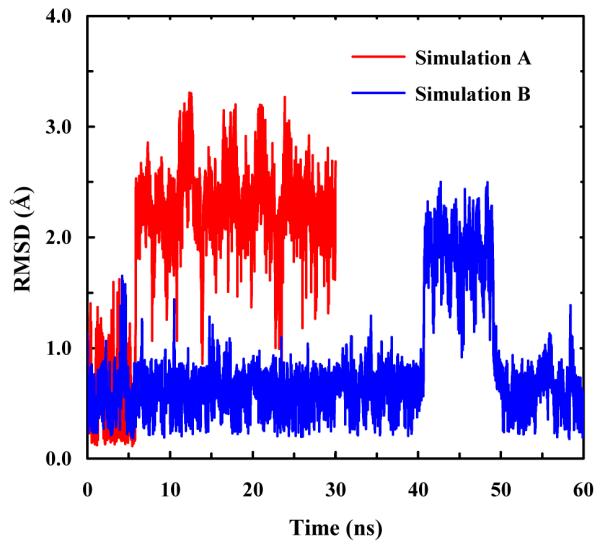
The peptide backbone RMSD obtained from simulations A and B, as indicated.
DISCUSSION
The folding times of model alanine-based peptides have been well established to be on the nanosecond timescale.7,50 In contrast, several photoswitchably-constrained α-helices33,35 and a naturally occurring α-helical peptide,51 whose folded state is stabilized by a series of sidechain-sidechain salt-bridges and/or hydrogen bonds, have been shown to fold on the microsecond timescale. Because in proteins α-helices are stabilized by inter- and/or intra-strand sidechain-sidechain interactions, the folding of such conformationally biased or constrained α-helical peptides may be a more realistic representation of the process of α-helix formation in proteins.54 Herein, we further investigate the conformational relaxation kinetics of three exceptionally short peptides, each of which folds into a single-turn α-helical structure due to a cross-linker formed between the sidechains of two residue at the i and i+4 positions, and aim to provide a direct assessment of the nucleation time of α-helix formation.
Our results show that the T-jump induced relaxation kinetics of these cyclic peptides can be fit to a single-exponential function and are independent of the T-jump amplitude, suggesting that the underlying conformational transition occurs between well defined helical and non-helical states. This notion is consistent with the study of Fairlie and coworkers,31 which showed that at least three backbone hydrogen bonds are simultaneously required to maintain the folded α-helical conformation. On the other hand, Hamm and coworkers have shown that the conformational relaxation kinetics of a longer helical peptide with an azobenzene cross-linker placed between residues i and i+11 exhibit stretched-exponential behavior with relaxation times ranging from 0.4 to 3.3 μs at 19 °C, depending on the position of the spectroscopic probe. They attributed this distribution of relaxation times to conformational heterogeneity in the unfolded ensemble.33,55 Thus, we believe that the conformational simplicity of these cyclic penta-peptides makes them better candidates for estimating the fundamental folding rate of a single-helical turn or the helix nucleation rate, especially for α-helices that are stabilized by specific sidechain-sidechain interactions between residues i and i+4. As shown (Figure 6), this view is supported by the fact that over the temperature range studied the conformational relaxation of these peptides is slower than that of alanine-based helical peptides,6 but faster than that of a naturally occurring peptide, L9:41-74, whose helical conformation is stabilized by a series of sidechain-sidechain interactions.51 For example, at room temperature the relaxation time constant of cyc-RKAAAD is about 0.6 μs, while those of alanine-based peptides11 and the L9:41-74 peptide44 are about 0.2 and 1.2 μs, respectively.
The folding and unfolding rates (kf and ku) of a two-state folder can be deduced from the experimentally determined relaxation rate (kR) if the free energy change (ΔG) of the underlying conformational transition is known (because kR = kf + ku and Keq = kf/ku). However, the CD thermal unfolding transitions of these cyclic peptides are rather broad (Figure 2), owing to the small enthalpy change (ΔH) associated with the folding of a single-turn helix. This makes accurate determination of ΔG(T) or Keq(T) difficult due to the intrinsic uncertainty in determining the folded and unfolded CD baselines. Nevertheless, in order to estimate the folding times of cyc-RKAAAD at different temperatures for comparison with other studies, we fit its CD thermal unfolding curve to a two-state model56 assuming temperature independent CD baselines. We found that the fit is satisfactory and that the resultant thermodynamic parameters for unfolding are: ΔH = 7.7 kcal mol−1 and ΔS = 26 cal K−1 mol−1. Based on these values, we further estimated the folding times of cyc-RKAAAD to be, for example, 1.2 and 2.6 μs, at 20 and 5 °C, respectively. Recently, Kiefhaber and coworkers57 have studied the local folding-unfolding dynamics of a 23-resdue alanine-based helical peptide using a triplet-triplet energy transfer technique. They found that the local helix formation time (i.e., the formation time of a helical segment between residues i and i+6) is approximately 0.4 μs at 5 °C. Thus, these findings suggest that the steric constraint arising from a cross-linker or a specific sidechain-sidechain interaction (e.g., a salt-bridge) in a helical peptide can effectively reduce the rate of folding, consistent with our previous study.51 The question then arises as to why such constraints make the peptide fold slower.
In a related study, Caflisch and coworkers53 compared the folding rates of a 16-residue alanine-based peptide constrained either with an azobenzene cross-linker or with a simple harmonic force constraint (i.e., an implicit cross-linker that does not impair sidechain motions) via molecular dynamics simulations. Their results indicated that the azobenzene cross-linker slows down folding (about 20 times compared to that obtained with the simple harmonic restraint) due to steric hindrance rather than to a restraining effect on the two ends of the helical segment.53 Interestingly, our results show that over the entire temperature range studied the T-jump induced relaxation rate of cyc-KARAD is indistinguishable within our experimental uncertainty from that of cyc-RKAAAD (Figure 6), indicating that the long Arg sidechain in this case does not impair folding. This finding does not necessarily contradict the results of Caflisch and coworkers,53 but rather suggests that in the present case the cross-linker has little, if any, hindrance effect on the motion of Arg. This is consistent with the proposed folded structure of the cyc-KAAAD (Figure 1), which shows that the third sidechain points away from the linker group.31 On the other hand, the relaxation rates of cyc-RKAAAD and cyc-RKMAAD show a small but measureable difference (Figure 6). For example, at 25 °C the relaxation time of cyc-RKAAAD is 0.42 μs, while at the same temperature cyc-RKMAAD relaxes in 0.63 μs. These results indicate that a bulky sidechain at the i+1 position (where i represents one end of the linker position) slows down the rate of conformation relaxation, due to the proximity of the bulkier Met sidechain to the cross-linker. This is in agreement with the finding of Caflisch and coworkers53 that steric hindrance exerted by the cross-linker on the motion of a sidechain to its native state, which effectively increases the internal friction along the folding coordinate, can decrease the folding rate.
Interestingly, the relaxation rate of these cyclic peptides is more similar to that obtained for an α-helix (YGG-3V) undergoing cold denaturation below room temperature (in 10% hexafluoroisopropanol (HFIP)). Werner et al.8 showed that the relaxation time of YGG-3V in response to a T-jump from 1.2 to 23.2 °C is 0.63 μs, comparable to the relaxation time of 0.62 μs measured for cyc-RKAAAD at 23.4 °C. Since the fractional helicity of the YGG-3V peptide changes from ~36% to 62% with a T-jump from 1.2 to 23.2 °C, these authors argued that the measured relaxation time directly reports the rate of α-helix nucleation.
Thus, results of this and other relevant studies support the notion that the folding rate of these cyclic peptides provides a realistic approximation of the α-helix nucleation rate, especially for α-helices that are stabilized by strong sidechain-sidechain interactions. (It is noted that for alanine-based peptides the helix nucleation rate could be faster.58) In order to further substantiate this claim, we simulated the relaxation kinetics of the L9:41-74 peptide using the folding rate of cyc-RKMAAD as the helix nucleation rate. To simplify the calculation, we employed a sequential kinetic model59 wherein the helix formation was assumed to proceed unidirectionally from a single nucleation site with a site-independent propagation rate constant, kp. In addition, we assumed that all of the microscopic unfolding rate constants (ku) have the same value. Specifically, the population relaxation kinetics of the L9:41-74 peptide were simulated by numerically solving the following kinetic master equation,
| (2) |
with
| (3) |
where l is the peptide length and was set to 33, while the nucleation rate constant, knuc, was set to (1.2 μs)−1, and ku was set to (1.2 μs)−1, both of which were estimated based on the relaxation rate of cyc-RKMAAD at 20 °C. As shown (Figure 8), we found that the normalized kinetic signal, S(t), which was determined from the fractional helicity of the peptide as a function of time, i.e.,
| (4) |
where h is a proportionality constant, becomes almost indistinguishable from the experimentally measured relaxation kinetics of L9:41-74 when kp = (240 ns)−1. As expected, this value is considerably slower than the propagation rate constant of (51 ns)−1 obtained by Kiefhaber and coworkers for an alanine-based peptide.57 Thus, this simple calculation provides further evidence supporting the aforementioned conclusions and also an estimate of the rate of helix propagation for the formation of α-helices that are stabilized by sidechain-sidechain interactions.
Figure 8.
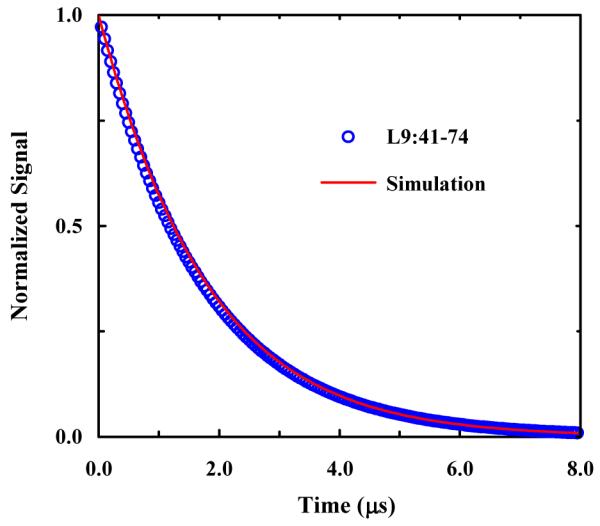
Comparison of the (normalized) relaxation kinetics of the L9:41-74 peptide and the simulated relaxation signal obtained with kp = (240 ns)−1. The L9:41-74 relaxation trace was determined based on the kinetic parameter of Mukherjee et. al.44
CONCLUSIONS
The helix-coil transition kinetics have been extensively studied by many laboratories using various relaxation methods and peptide sequences. However, the α-helix nucleation rate could only be inferred from these studies because they invariably employed α-helices composed of several turns. Thus, the resulting relaxation kinetics do not directly report on the folding rate of a single helical turn or the nucleation rate. In order to provide a direct assessment of this key elementary kinetic event in protein folding, we measure the T-jump induced relaxation kinetics of three cyclic peptides (i.e., cyc-KARAD, cyc-RKAAAD and cyc-RKMAAD) that adopt a folded α–helical conformation consisting of only one helical turn. We believe, as others have previously proposed,33 that the folding kinetics of such structurally constrained helical peptides may better represent the kinetics of helical structure formation in proteins in which similar constraining effects arise from sidechain-sidechain interactions. Our results show that the relaxation rate of cyc-RKAAAD over the temperature range studied is indistinguishable from that of cyc-KARAD, but is slightly faster than that of cyc-RKMAAD. This is due to the proximity of the bulkier Met sidechain to the amide cross-linker compared to the Ala sidechain, further corroborating the idea that sidechain-sidechain interactions between nearby groups can slow down the kinetics of helix folding. A two-state analysis further indicates that the folding time of cyc-RKAAAD is about 1.2 μs at 20 °C, which we believe represents an upper limit of the helix nucleation time in proteins. In addition, kinetic simulations involving a sequential kinetic model further suggest that the helix propagation time is about 240 ns.
ACKNOWLEDGMENT
We gratefully acknowledge financial support from the National Institutes of Health (GM-065978 and RR-01348).
References
- (1).Williams S, Causgrove TP, Gilmanshin R, Fang KS, Callender RH, Woodruff WH, Dyer RB. Biochemistry. 1996;35:691–697. doi: 10.1021/bi952217p. [DOI] [PubMed] [Google Scholar]
- (2).Thompson PA, Eaton WA, Hofrichter J. Biochemistry. 1997;36:9200–9210. doi: 10.1021/bi9704764. [DOI] [PubMed] [Google Scholar]
- (3).Volk M, Kholodenko Y, Lu HSM, Gooding EA, DeGrado WF, Hochstrasser RM. J. Phys. Chem. B. 1997;101:8607–8616. [Google Scholar]
- (4).Lednev IK, Karnoup AS, Sparrow MC, Asher SA. J. Am. Chem. Soc. 1999;121:8074–8086. doi: 10.1021/ja003381p. [DOI] [PubMed] [Google Scholar]
- (5).Jas GS, Eaton WA, Hofrichter J. J. Phys. Chem. B. 2001;105:261–272. [Google Scholar]
- (6).Huang CY, Klemke JW, Getahun Z, DeGrado WF, Gai F. J. Am. Chem. Soc. 2001;123:9235–9238. doi: 10.1021/ja0158814. [DOI] [PubMed] [Google Scholar]
- (7).Huang CY, Getahun Z, Wang T, DeGrado WF, Gai F. J. Am. Chem. Soc. 2001;123:12111–12112. doi: 10.1021/ja016631q. [DOI] [PubMed] [Google Scholar]
- (8).Werner JH, Dyer RB, Fesinmeyer RM, Andersen NH. J. Phys. Chem. B. 2002;106:487–494. [Google Scholar]
- (9).Huang CY, Getahun Z, Zhu YJ, Klemke JW, DeGrado WF, Gai F. Proc. Natl. Acad. Sci. U. S. A. 2002;99:2788–2793. doi: 10.1073/pnas.052700099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang T, Du DG, Gai F. Chem. Phys. Lett. 2003;370:842–848. [Google Scholar]
- (11).Wang T, Zhu YJ, Getahun Z, Du DG, Huang CY, DeGrado WF, Gai F. J. Phys. Chem. B. 2004;108:15301–15310. doi: 10.1021/jp037272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gooding EA, Ramajo AP, Wang JW, Palmer C, Fouts E, Volk M. Chem. Commun. 2005:5985–5987. doi: 10.1039/b511072f. [DOI] [PubMed] [Google Scholar]
- (13).Balakrishnan G, Hu Y, Bender GM, Getahun Z, DeGrado WF, Spiro TG. J. Am. Chem. Soc. 2007;129:12801–12808. doi: 10.1021/ja073366l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schwarz G, Seelig J. Biopolymers. 1968;6:1263. doi: 10.1002/bip.1968.360060904. &. [DOI] [PubMed] [Google Scholar]
- (15).Zana R. Biopolymers. 1975;14:2425–2428. [Google Scholar]
- (16).Bose SK, Chernovitz PA, Emptage MR. J. Chem. Phys. 1980;73:1368–1375. [Google Scholar]
- (17).Fujita S, Blaistenbarojas E, Torres M, Godoy SV. J. Chem. Phys. 1981;75:3097–3102. [Google Scholar]
- (18).Dill KA, Fiebig KM, Chan HS. Proc. Natl. Acad. Sci. U. S. A. 1993;90:1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Buchete NV, Straub JE. J. Phys. Chem. B. 2001;105:6684–6697. [Google Scholar]
- (20).Morozov AN, Lin SH. J. Phys. Chem. B. 2006;110:20555–20561. doi: 10.1021/jp061781e. [DOI] [PubMed] [Google Scholar]
- (21).Vorov OK, Livesay DR, Jacobs DJ. Biophys. J. 2009;97:3000–3009. doi: 10.1016/j.bpj.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zimm BH, Bragg JK. J. Chem. Phys. 1959;31:526–535. [Google Scholar]
- (23).Marqusee S, Robbins VH, Baldwin RL. Proc. Natl. Acad. Sci. U. S. A. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Scholtz JM, York EJ, Stewart JM, Baldwin RL. Hong Q, editor. Biopolymers. 1991;31:1463–1470. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- (25).Hong Q, Schellman JA. J. Phys. Chem. 1992;96:3987–3994. [Google Scholar]
- (26).Scholtz JM, Baldwin RL. Annu. Rev. Bioph. Biom. 1992;21:95–118. doi: 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]
- (27).Doig AJ. Biophys. Chem. 2002;101:281–293. doi: 10.1016/s0301-4622(02)00170-9. [DOI] [PubMed] [Google Scholar]
- (28).Ohkubo YZ, Brooks CL. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13916–13921. doi: 10.1073/pnas.2334257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ghadiri MR, Choi C. J. Am. Chem. Soc. 1990;112:1630–1632. [Google Scholar]
- (30).Shepherd NE, Abbenante G, Fairlie DP. Angew. Chem. Int. Edit. 2004;43:2687–2690. doi: 10.1002/anie.200352659. [DOI] [PubMed] [Google Scholar]
- (31).Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. J. Am. Chem. Soc. 2005;127:2974–2983. doi: 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]
- (32).Ihalainen JA, Bredenbeck J, Pfister R, Helbing J, Chi L, van Stokkum IHM, Woolley GA, Hamm P. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5383–5388. doi: 10.1073/pnas.0607748104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ihalainen JA, Paoli B, Muff S, Backus EHG, Bredenbeck J, Woolley GA, Caflisch A, Hamm P. Proc. Natl. Acad. Sci. U. S. A. 2008;105:9588–9593. doi: 10.1073/pnas.0712099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Paoli B, Seeber M, Backus EHG, Ihalainen JA, Hamm P, Caflisch A. J. Phys. Chem. B. 2009;113:4435–4442. doi: 10.1021/jp810431s. [DOI] [PubMed] [Google Scholar]
- (35).Backus EHG, Bloem R, Donaldson PM, Ihalainen JA, Pfister R, Paoli B, Caflisch A, Hamm P. J. Phys. Chem. B. 2010;114:3735–3740. doi: 10.1021/jp911849n. [DOI] [PubMed] [Google Scholar]
- (36).Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- (37).MacKerell AD, Bashford D, Jr., Bellott M, Dunbrack RL, Evanseck JD, Jr., Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, III, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- (38).Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- (39).Darden T, York D, Pederson L. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- (40).Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pederson LG. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- (41).Chin DH, Woody RW, Rohl CA, Baldwin RL. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15416–15421. doi: 10.1073/pnas.232591399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Greenfield NJ. Methods Enzymol. 2004;383:282–317. doi: 10.1016/S0076-6879(04)83012-X. [DOI] [PubMed] [Google Scholar]
- (43).Barth A, Zscherp C. Q. Rev. Biophys. 2002;35:369–430. doi: 10.1017/s0033583502003815. [DOI] [PubMed] [Google Scholar]
- (44).Dyer RB, Gai F, Woodruff WH. Acc. Chem. Res. 1998;31:709–716. [Google Scholar]
- (45).Garcia AE, Sanbonmatsu KY. Proc. Natl. Acad. Sci. U. S. A. 2002;99:2782–2787. doi: 10.1073/pnas.042496899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ghosh T, Garde S, Garcia AE. Biophys. J. 2003;85:3187–3193. doi: 10.1016/S0006-3495(03)74736-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Levy Y, Jortner J, Becker OM. Proc. Natl. Acad. Sci. U. S. A. 2001;98:2188–2193. doi: 10.1073/pnas.041611998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Peng Y, Hansmann UHE, Alves NA. J. Chem. Phys. 2003;118:2374–2380. [Google Scholar]
- (49).Nevskaya NA, Chirgadze YN. Biopolymers. 1976;15:637–648. doi: 10.1002/bip.1976.360150404. [DOI] [PubMed] [Google Scholar]
- (50).Thompson PA, Munoz V, Jas GS, Henry ER, Eaton WA, Hofrichter J. J. Phys. Chem. B. 2000;104:378–389. [Google Scholar]
- (51).Mukherjee S, Chowdhury P, Bunagan MR, Gai F. J. Phys. Chem. B. 2008;112:9146–9150. doi: 10.1021/jp801721p. [DOI] [PubMed] [Google Scholar]
- (52).Creighton T. Proteins. Freeman and Company; 1993. pp. 171–199. [Google Scholar]
- (53).Paoli B, Pellarin R, Caflisch A. J. Phys. Chem. B. 2010;114:2023–2027. doi: 10.1021/jp910216j. [DOI] [PubMed] [Google Scholar]
- (54).Lyu PC, Liff MI, Marky LA, Kallenbach NR. Science. 1990;250:669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- (55).Hamm P, Helbing J, Bredenbeck J. Chem. Phys. 2006;323:54–65. [Google Scholar]
- (56).Xu Y, Bunagan MR, Tang J, Gai F. Biochemistry. 2008;47:2064–2070. doi: 10.1021/bi702195c. [DOI] [PubMed] [Google Scholar]
- (57).Fierz B, Reiner A, Kiefhaber T. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1057–1062. doi: 10.1073/pnas.0808581106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sancho DD, Best RB. J. Am. Chem. Soc. 2011;133:6809–6816. doi: 10.1021/ja200834s. [DOI] [PubMed] [Google Scholar]
- (59).Brooks CL. J. Phys. Chem. 1996;100:2546–2549. [Google Scholar]