

Abstract
Background
New therapies are necessary to address inadequate asthma control in many patients. This study set out to investigate whether Hypoxia Inducible Factor (HIF) is essential for development of allergic airway inflammation (AAI) and therefore a potential novel target for asthma treatment.
Methods
Mice conditionally knocked out for HIF-1β were examined for their ability to mount an allergic inflammatory response in the lung after intratracheal exposure to ovalbumin. The effects of treating wild-type mice with either ethyl-3,4-dihydroxybenzoate (EDHB) or 2-methoxyestradiol (2ME), which upregulate and downregulate HIF, respectively, were determined. HIF-1α levels were also measured in endobronchial biopsies and bronchial fluid of asthma patients and nasal fluid of rhinitis patients after challenge.
Results
Deletion of HIF-1β resulted in diminished AAI and diminished production of ovalbumin-specific IgE and IgG1. EDHB enhanced the inflammatory response, which was muted upon simultaneous inhibition of Vascular Endothelial Growth Factor (VEGF). EDHB and 2ME antagonized each other with regard to their effects on airway inflammation and mucus production. The levels of HIF-1α and VEGF increased in lung tissue and bronchial fluid of asthma patients and in the nasal fluid of rhinitis patients after challenge.
Conclusions
Our results support the notion that HIF is directly involved in the development of AAI. Most importantly, we demonstrate for the first time that HIF-1α is increased after challenge in asthma and rhinitis patients. Therefore we propose that HIF may be a potential therapeutic target for asthma and possibly for other inflammatory diseases.
Keywords: Allergic Airway Inflammation, Asthma, Hypoxia Inducible Factor, Rhinitis
INTRODUCTION
Asthma is one of the most common chronic diseases in the world. Even with maximal medical therapy many asthmatic patients do not achieve adequate asthma control (1) resulting in a need for additional anti-asthmatic drugs.
The transcriptional response to hypoxia is primarily mediated by the hypoxia-inducible factor (HIF) family of transcription factors. HIFs are heterodimeric proteins containing one α subunit and one β subunit. HIF-1α and HIF-2α are both expressed widely, as is HIF-1β (the Aryl Hydrocarbon Receptor Nuclear Translocator {ARNT}). HIF-2β (ARNT2) has a more limited tissue distribution. The β subunits are constitutively expressed, and regulation of HIFs occurs mainly via effects on the α subunits. Under normoxic conditions, the HIF-α subunits are hydroxylated on key proline residues, thereby allowing for their recognition by the von Hippel-Lindau (pVHL) tumor suppressor protein that targets HIF-α for proteasomal degradation. Hydroxylation of both prolines is catalyzed by a family of three iron-containing prolyl hydroxylases in a reaction requiring O2 and 2-oxoglutarate (2). HIF-1α inhibition also occurs through the hydroxylation of an asparagine residue by an O2, iron, and 2-oxoglutarate-dependent dioxygenase, Factor Inhibiting Hypoxia Inducible Factor-1α , which prevents HIF-1α from interacting with the coactivator p300 under normoxic conditions (3). During hypoxia, HIF-1α and/or HIF-2α dimerize with ARNT and/or ARNT2, and this complex then binds hypoxia response elements (HREs) in the promoters of target genes and up-regulates their transcription (4). HIF target genes include those for several pro-inflammatory cytokines, chemokines, and adhesion molecules, including VEGF. However HIF-1α and HIF-2α regulate unique as well as shared target genes, and play nonredundant roles in the organism (5–6).
It has been proposed that HIF plays a role in human allergic airway diseases (7–11). We previously reported that the non-specific HIF inhibitor 2-methoxyestiadiol (2ME) reduced the allergic pulmonary inflammatory and airway remodelling responses to ovalbumin in a mouse model (9). Guo and coworkers (10) demonstrated that mice heterozygous for a HIF-1α null allele exhibited diminished lung eosinophilia in response to an ovalbumin challenge. However, only limited aspects of the allergic airway response were studied by these last investigators. We therefore decided to thoroughly evaluate the role of HIF in a mouse model of allergic airway response using both genetic and pharmacological perturbations. Our investigations provide compelling evidence for a role of HIF in the development of allergic lung inflammation in the mouse. Importantly, we also demonstrate directly that exposure to allergen leads to upregulation of HIF-1α and VEGF expression in asthmatic and rhinitis patients, as it does in our mouse model of allergic airway inflammation.
METHODS
Breeding and Genotyping of Mice
The original ArntF allele contained a neo cassette, which was excised as described previously (12). The Arntf/f mice, which were of a mixed C57BL/6, 129/Sv and FVB/N genetic background, were back-crossed to homozygous Mx1-Cre+ mice in a C57BL/6 genetic background, and at least ten successive times to the C57BL/6 strain. Genotyping of the ArntF and ArntΔ alleles and the Mx1-Cre transgene was performed by PCR (13). Mx1-Cre+/− heterozygotes could not be distinguished from Mx1-Cre+/+ homozygotes by this procedure, and these genotypes are collectively referred to as Mx1-Cre+.
Treatment of Mice
Deletion of the Arnt allele in Mx1-Cre+: Arntf/f male mice was induced by intraperitoneal (i.p.) injection with 500 μg of polyinosinic-polycitidylic acid (pIpC, Sigma, St.Louis, MO) in PBS on three occasions two days apart. Sensitization was elicited by two i.p. treatments, five days apart, of 10 μg chicken egg ovalbumin (OVA, grade V, Sigma, St. Louis, MO) emulsified in 1 mg of aluminum hydroxide (Pierce Chemical, USA) in a total volume of 100 μL. Mice were challenged via intratracheal (i.t.) administration of 0.75 % OVA on two or three occasions, as indicated. Treatments with ethyl 3,4, dihydroxybenzoate (EDHB) (Sigma, St.Louis, MO), and/or 9 mg/kg (E)- 3-(3,5-Diisopropyl-4-hydroxyphenyl)-2-[(3-phenyl-n-propyl)amino- carbonyl] acrylonitrile (SU1498, Calbiochem, San Deigo, CA), or 30 mg/kg 2- methoxiestradiol (2-ME, Sigma, St. Louis, MO) were performed as indicated. Control mice received vehicle solvent only. Mice were euthanized by inhalation of isofluorane. All experiments were performed in accordance with UCLA and INCMN Mexico City regulations.
Lung histology and morphometric analysis
Calculations of inflammatory area and PAS staining were done as described previously (14.).
Immunocytochemistry
Deparaffinized 4-μm sections were used for immunohistochemical analysis using the LSAB kit (DAKO, Carpinteria, CA, USA) and antibodies against HIF-1α (1:250; Santa Cruz Biotechnology, Santa Cruz, CA), HIF-2α (1:750; Novus Biologicals, Littleton, CO), ARNT, ARNT2 or VEGF (1:500, 1:500, and 1:750, respectively; Santa Cruz). All samples from each group were processed at the same time in a single experiment using a single batch of antibody diluted in PBS with Normal Goat Serum (NGS).
Bronchoalveolar lavage (BAL)
Mice were euthanized by exsanguination. Differential counts were performed on four to six H&E stained cytospin preparations for each experimental group. VEGF in BAL supernatant fluid of rhinitis subjects was determined by ELISA as described previously (15).
OVA-specific IgG1 and IgE
OVA-specific IgE and IgG1 levels were measured in mouse serum samples using ELISA (15).
Human Subjects
Asthma patients
Eleven mild to moderate cat allergic asthmatic subjects (6 female/5 male), ages ranging 18 to 43 (18.5±7.0), with a mean FEV1 of 96±8% were studied at baseline with fiberoptic bronchoscopy in which endobronchial biopsies and bronchoalveolar lavage (BAL) were obtained. Three days later, subjects underwent a naturalistic cat room challenge in which they were exposed to three cats housed in a vivarium room, until their FEV1 dropped by 20% or for an hour, whichever occurred first. The average FELd1 concentration in air samples from the cat room was 17.2±5.8 ng Fel d I/cu meter air Three days later, subjects again underwent repeat fiberoptic bronchoscopy with endobronchial biopsies and BAL. Further details are provided in supplementary materials.
Rhinitis patients
Ten healthy nonsmoking volunteers (1 male and 9 females) ages 20–40 were recruited for the study. Subjects reported a history of cat allergy but no history of perennial allergic rhinitis. Use of antihistamine, intranasal, or immunosuppressive medication was prohibited three days before or at any time during the study. Allergy epicutaneous skin testing was performed with Standardized Cat Allergen Extract - 10,000 BAU units/ml (Hollister Stier, WA) to confirm allergic sensitization. Each subject completed a graded intranasal allergen challenge to determine the cat allergen dose stimulating nasal allergy symptoms. (See supplementary material) The dose stimulating a symptom score of ≥8 was subsequently used for the single allergen dose challenge with nasal lavage samples collected for analysis. Subjects who did not achieve the required symptom score during the graded challenge were excluded from further participation. At least 2 weeks after the graded dose-determination procedures, subjects returned for the single intranasal cat allergen challenge. Subjects performed nasal lavage with 40 ml of sterile saline at baseline (prior to intranasal allergen) and 1 hour after intranasal allergen challenge using previously described methods (15). Nasal lavage samples were spun at 2500 rpm for 30 minutes at 4°C. The cells from the pellet were collected and used for IHC.
All subjects signed an informed consent and study activities were approved by the Human Subject Protection Committee of the University of California, Los Angeles.
Statistical analysis
Data were expressed as mean ± standard deviations. Statistical comparisons were performed using one-way analysis of variance followed by Fisher’s exact test, the unpaired Student’s t test or the U Mann Whitney test. For the correlation between HIF-1α and VEGF expression in the human samples, we used Pearson’s correlation analysis.
RESULTS
HIF-1α and HIF-2α are upregulated in mouse lung in a mouse model of allergic airway inflammation
C57BL/6 mice were subjected to a previously described allergenic protocol (14) (Fig. 1A). Lung sections were subjected to hematoxylin-and-eosin (H&E) staining and also stained for either HIF-1α or HIF-2α. Representative lung sections and quantitative analysis of the staining of four mice are presented in Figures 1B, 1C, 1D and 1E. Markedly elevated levels of HIF-1α and HIF-2α were observed in the nuclei of the bronchial epithelial cells of the ovalbumin treated mice. Inflammatory cells in the ovalbumin treated mice also expressed both proteins. Expression was observed in macrophages and lymphocytes.
Figure 1. HIF-1α and HIF-2α are upregulated in mouse lung in a mouse model of allergic airway inflammation.

A. Experimental protocol. B, D. Representative lung sections of C57BL/6 mice treated with OVA (Cre-/OVA) or Sterile Saline (Cre-/SS) and stained for HIF-1α and HIF-2α respectively. C, E. Quantification of the immunostaining for HIF-1α and HIF-2α, respectively, in four mice in each case. † p<0.01.
OVA sensitized/challenged mice conditionally knocked out for Arnt have a reduced allergic response in the lung
We wished to analyze mice deficient in HIF activity. As described above, both HIF-1α and HIF-2α are expressed in mouse lung. However, whereas Arnt is also expressed in the lung, Arnt2 is not (16–18). We therefore studied Arnt-deficient mice. Since mice that are homozygous for an Arnt null allele (like Hif-1α knockout and Hif-2α knockout mice) die in utero (19–20), it was necessary to use conditional knockout mice in which deletion is induced in the Arnt gene in adulthood. To achieve this, we developed Arntf/f:Mx1-Cre+ mice. The Mx1 promoter is silent unless the cell is treated with polyinosinic-polycytidylic acid (pIpC). Thus in Arntf/f:Mx1-Cre+ mice injected intraperitoneally with pIpC, substantial inactivation of the Arnt gene occurs in most tissues of the body, including the lung (12). Immunohistochemical analysis of our mice showed that pIpC treatment led to about an 80% dimunition in the levels of the Arnt protein (Supplementary Figure 1) in lung tissue.
Arntf/f:Mx1-Cre+ and Arntf/f:Mx1-Cre- mice were used. Five mice of each genotype were subjected to the same allergenic protocol as illustrated in Figure 1A except that they were also treated i.p. with 500 ug pIpC (Figure 2A). Cre− and Cre+ mice untreated with OVA exhibited no significant inflammatory infiltration. In contrast, OVA treatment in the Cre− mice elicited a marked perivascular and peribronchiolar inflammatory infiltration comprised mostly of mononuclear cells. The Cre+ mice treated with OVA exhibited a marked reduction in the degree of inflammatory cell infiltration, compared with the similarly treated Cre− mice (Figures 2B and 2C). OVA treatment of Cre− mice increased the levels Arnt and of the HIF target gene, VEGF, in the cytoplasm of cells lining the bronchioles, while there were fewer Arnt and VEGF-positive cells in Cre+ mice treated similarly (Figures 2D to 2G). The increase in VEGF occurring during the inflammatory response is therefore at least partially mediated by Arnt, which is consistent with the upregulation of HIF-1α and HIF-2α that occur in these cells. OVA treatment increased the concentration of total leukocytes in mice of both genotypes. However, there were fewer lymphocytes and eosinophils in Cre+ mice compared with Cre− mice (Figure 2F). Disruption of Arnt was also associated with a decrease in OVA stimulation of OVA-specific IgE and IgG1 production in the sera of the mice (Fig. 2G and 2H). Of note, mice sensitized and not challenged, did not exhibit increase in any of the hallmark paramaters of AAI (data not shown). In conclusion, decreased Arnt expression diminishes manifestations of the allergic response, including lung inflammation with eosinophilia, and allergen-specific IgE and IgG1 production.
Figure 2. OVA sensitized/challenged mice conditionally knocked out for Arnt have a reduced allergic response in the lung.

A. Experimental protocol. B. Representative H&E stained sections of lung tissues were obtained from OVA-sensitized/challenged mice (OVA) and control mice treated with sterile saline (SS), of genotype Arntf/f:Mx1-Cre+ (Cre+) or Arntf/f:Mx-1-Cre – (Cre-). All mice received pIpC. C. The degree of inflammatory infiltration in venules and middle size veins and in bronchioles from five mice from each experimental group was determined as described in “Methods.” Results are expressed as averages and standard deviations of infiltrated area. Empty bars: perivascular infiltrate; solid bars: peribronchiolar infiltrate. D, F. Immunohistochemical staining of VEGF and Arnt in lung tissue of representative Arntf/f:Mx1-Cre+ and Arntf/f:Mx1-Cre- mice treated as described in B. E. G. Quantification of VEGF staining in five mice from each group. H. Total leukocytes and differential cell counts in BAL from OVA-sensitized/challenged and control Arntf/f:Mx1-Cre+ and Arntf/f:Mx1-Cre-mice (cells/ml). I and J. IgE and IgG1 levels in the sera of five mice in each group. *p<0.05, † p < 0.01, Cre+ OVA vs. Cre- OVA (U Mann Whitney).
Increased HIF expression enhances the inflammatory response
Ethyl-3,4-dihydroxybenzoate (EDHB) upregulates HIF-1α (and presumably HIF-2α ) by inhibiting prolyl hydroxylases competitively with regard to two of their cosubstrates, oxoglutarate and ascorbate (23–24), and this compound upregulated HIF-1α in mouse lung (Supplementary Figure 2). We injected Balb/c mice i.p. with EDHB on two occasions, one day before each intratracheal challenge with OVA (Figure 3A). EDHB further enhanced the increase in perivascular and peribronchiolar inflammation elicited by OVA (Figures 3B and 3C).
Figure 3. Increased HIF expression enhances allergic airway inflammation in BALB/c mice.

A. Experimental Protocol. Each group consisted of five mice. B. Representative H&E stained sections of lung tissues showing blood vessels or bronchioles. C. Quantification of the inflammatory response for the five mice from each group. *p<0.05, † p < 0.01, ‡ p < 0.001
EDHB and 2-methoxyestradiol, a non-specific inhibitor of HIF activity, antagonize each other’s effects on airway inflammation and mucus production
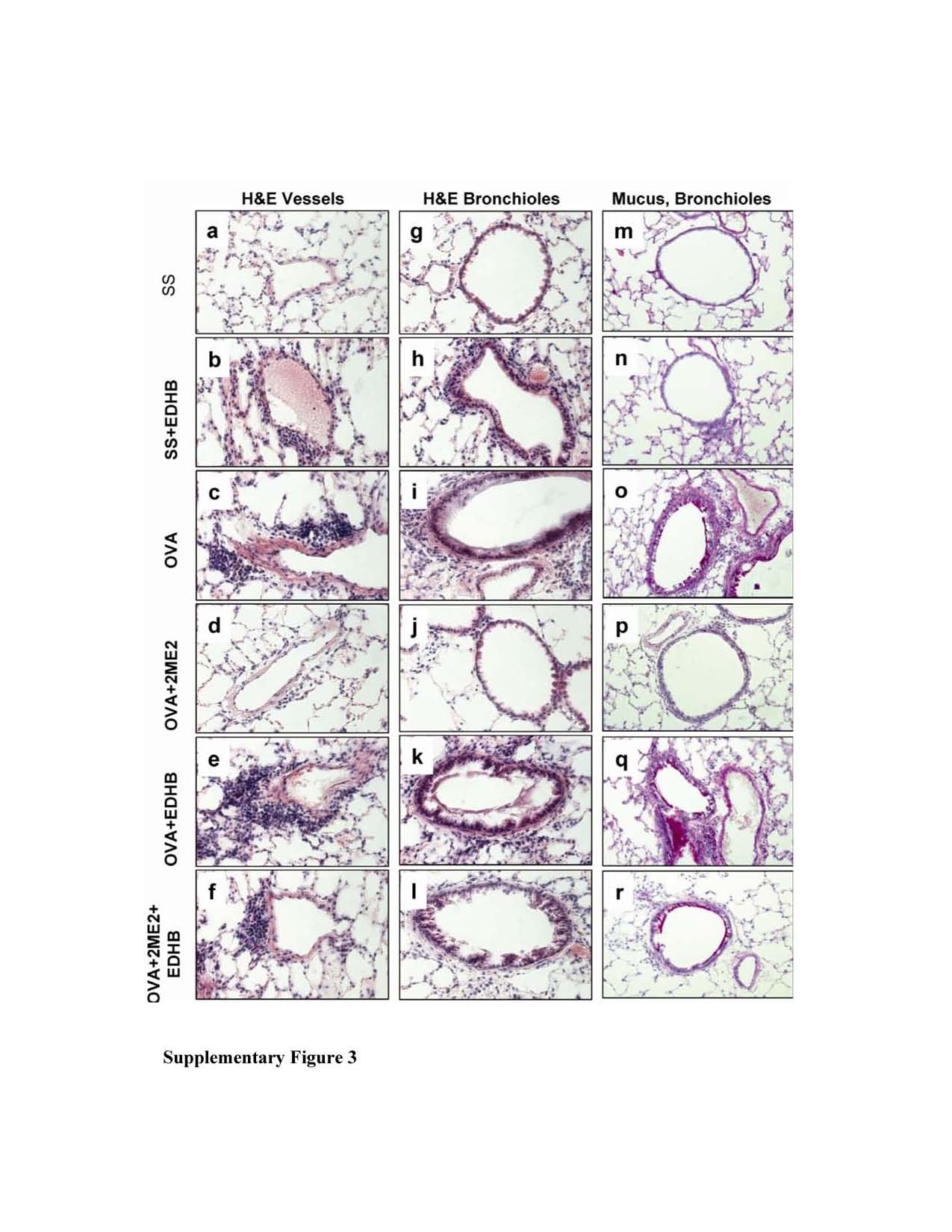
We investigated the effect of using 2ME, a non-specific inhibitor of HIF, which we previously demonstrated to inhibit the AAI response in mice (9), in conjunction with EDHB. However, 2ME destabilizes HIF-1α indirectly, most likely as a consequence of microtubule disruption and/or inhibition of mitochondrial respiration (23-24) and thus has a different molecular target(s) from EDHB. Balb/c mice were treated as outlined in Figure 4A. As expected, OVA treatment led to increases in inflammatory airway inflammation and also in the production of mucin within goblet cells. (Increases in mucin are routinely observed in this mouse model.) (Supplementary Figure 3 and Figures 4B and 4C). EDHB enhanced the inflammatory response to OVA, as before, and also increased mucus production. Interestingly, EDHB, even in the absence of OVA, enhanced inflammation. 2ME treatment after OVA challenge reduced the stimulatory effect of OVA on inflammation and mucus production. Importantly, treatment with OVA, 2ME, and EDHB had more of an effect on all the parameters measured than treatment with OVA and 2ME, but had less of an effect on these same parameters than OVA and EDHB. Thus 2ME and EDHB antagonized each other’s effects on these manifestations of allergic airway inflammation, supporting the notion that the effects of each agent on these parameters are indeed mediated by the agents’ effects on HIF, and most importantly, supporting the notion that HIF directly impacts the allergic airway response.
Figure 4. Inhibition of HIF activity attenuates the stimulatory effect of EDHB on airway inflammation and mucus production.

A. Experimental Protocol. B and C. Quantification of the inflammatory response and mucus production, respectively, in five mice from each experimental group. *p < 0.05, † p < 0.01, ‡ p < 0.001.
Attenuation of the allergic inflammatory response after inhibition of VEGF activity
VEGF has been reported to play a role in the airway inflammatory response (25). We applied a VEGF receptor 2 inhibitor, SU1498, after treatment with EDHB (Figure 5A). Treatment with SU1498 reduced inflammation induced by EDHB alone, by OVA alone, and by OVA plus EDHB, indicating that the involvement of HIF in the allergic pulmonary response is at least partly mediated by upregulation of VEGF (Supplementary Figure 4 and Figure 5B).
Figure 5. Attenuation of the allergic inflammatory response after inhibition of VEGF activity.

A. Experimental Protocol. B. Quantification of the inflammatory response in five mice from each experimental group. *p<0.05.
HIF-1α expression is increased in asthmatic patients and rhinitis patients after allergen challenge
We next investigated the potential role of HIF in allergic airway diseases in the human. We measured HIF-1α and VEGF protein levels in cells from BAL and in endobronchial biopsies from eleven asthmatic patients before and after exposure to cats in an enclosed room. Cells from BAL were primarily macrophages (Figure 6A). Both HIF-1α and VEGF levels increased in bronchial lavage cells after allergen challenge (Figs. 6B, C, and D). HIF-1α was predominantly nuclear, although some cytoplasmic staining was observed. VEGF was expressed predominantly in the cytoplasm or in the cell membrane. The levels of both HIF-1α and VEGF in endobronchial biopsies of the eleven patients were also higher after challenge (Figure 6E). (This was also true for HIF-2α; Supplementary Figure 5). In addition, we examined nasal lavage from ten rhinitic patients. We observed increased HIF-1α in nasal lavage cells after cat allergen challenge in the rhinitis patients (Fig. 6F and 6G). The expression was predominantly nuclear, although some cytoplasmic staining was observed. There was a direct correlation between HIF-1α immunostaining and VEGF expression (as measured by ELISA), in the nasal lavage fluid (Fig. 1H) after challenge (*p<0.05, r=0.7930). These results suggest that HIF-1 is likely to play a role in the pathogenesis of allergic disease in the human.
Figure 6. Enhanced HIF-1α and VEGF expression in bronchial lavage and lung tissue from asthmatic patients (A to E) and nasal lavage from rhinitis patients (F to H) after allergen challenge.

A. Representative photomicrographs of HIF-1α and VEGF immunostaining (100X) in epithelial cells from bronchial lavage of asthma patients before and after naturalistic cat challenge. The specificity of the antibodies is shown in staining with the IgG control. B, C. Frequency of HIF-1α and VEGF positive cells in bronchial lavage from 11 asthma patients. D. Representative photomicrographs of HIF-1α and VEGF before and after challenge (40X) in endobronchial biopsies. E. Quantification of the expression of HIF-1α and VEGF, before and after the cat challenge in 11 patients (pre and post challenge data was collected from the same patient). *p < 0.05, † p<0.01 (ANOVA). F. Representative photomicrographs of HIF-1α immunostaining (100X) in rhinitis patients before (a) and after (b) challenge. G. Quantification of HIF-1α positive cells (left), before and after the challenge. The mean and standard deviations were derived from data collected from 10 patients. H. Correlation analysis between HIF-1α immunostaining and VEGF expression as measured by ELISA (right), after the challenge. The analysis was performed from data collected from the 10 patients. Student t-test *p<0.05, † p<0.01 pre vs. post challenge HIF-1α expression. Pearson’s analysis *p<0.05, *r=0.7930 Post challenge HIF-1α vs.VEGF.
DISCUSSION
Our studies on mice confirm and extend previous observations suggesting a role for HIF in allergic airway inflammation (7–10,26). First, we showed that upregulation of HIF-1α and HIF-2α occurs during allergic airway inflammation in mice. Second, we found that the inflammatory response to OVA challenge, including inflammatory cell infiltration, eosinophil recruitment, VEGF expression, and allergen specific-IgE and IgG1 were all attenuated in pIpC-treated Arntfl/fl:Mx1-Cre+ mice, in which Arnt expression is diminished. Arnt can dimerize with certain other members of the bHLHPAS family of proteins, including the aryl hydrocarbon receptor (AHR), single-minded homologs 1 and 2 (Sim 1 and 2), and NXF (27–29). However, it is unlikely that the diminution in the allergic airway response observed in the Arnt deficient mice is due to loss of activity with these alternative dimerization partners, since the Sim proteins function mainly as transcriptional repressors (30), NXF has a very limited tissue-specific expression, excluding the lung (29), and C57BL/6 Ahr-null mice do not exhibit a reduced allergic lung inflammatory response after an OVA challenge protocol very similar to the one we performed here (31).
Third, EDHB markedly enhanced the allergic response to OVA including airway inflammation and mucin production. Fourth, the effects of EDHB and the non-specific HIF-1α inhibitor, 2-ME, on the allergic airway response antagonized each other, thus providing evidence that these agents affect this response via their effects on HIF, and supporting the notion that HIF activity directly affects this response. Interestingly, the effects of EDHB were partially inhibited when followed by treatment with the VEGFR2 inhibitor SU1498, suggesting that the role of HIF in the allergic airway response is at least partly mediated by up-regulation of VEGF.
Interestingly, HIF-1α activity in the alveolar epithelium appeared to protect against the development of an asthma-like inflammatory response to inhaled cobalt in the mouse (32). However, cobalt is a hypoxia mimic and direct inducer of HIF-1α and HIF-2α, and the relevance of this study to ours is unclear.
We show here that allergen exposure leads to upregulation of HIF-1α and VEGF in endobronchial biopsies and bronchial lavage cells of asthmatic patients and in nasal lavage of rhinitis patients. To our knowledge, this is the first time that HIF-1α and VEGF expression have been shown directly to increase in asthmatic and rhinitis patients after allergen challenge. Our clinical observations therefore support a role for HIF in the development of allergic airway diseases.
Our human and animal studies data suggest that HIF levels may serve as a useful biomarker for poor asthma control and a clinical therapeutic target. With regard to the latter notion, several compounds have been identified that downregulate HIF and there is a major effort underway in both academia and the pharmaceutical industry to develop specific small molecule inhibitors of HIF (33–34). Our studies suggest that such molecules will represent potential novel treatments for asthma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Supported by a Collaborative Research Grant from the University of California Institute for Mexico and the United States (UC MEXUS-CONACYT) (S.H.Y and O.H), Mexico Federal Funds Grant HIM/2008/034 (S.H.-Y., G.B.-G.), National Institutes of Health grants R01 CA28868 (O.H.) and R01 HL080343 (E.K. and M.Z.), and the National Cancer Institute Intramural Research Program (F.J.G.), and the US Environmental Protection Agency (D.D-S.).
We thank Kelly Joiner and Stephen Hop for helping prepare the manuscript for submission and Xiaomeng Wu for technical assistance.
Footnotes
STATEMENT OF CONTRIBUTION: S.H.-Y. designed and performed experiments, analyzed data, helped write the paper and helped coordinate the research. G.B.-G., R.H.-P. and M.V performed the animal experimentas and obtained the corresponding histochemistry and immunohistochemistry data, R.H.-P. assisted in the design of the animal experiments and assisted in interpretation of the immunohistochemical data. M.V. assisted in experimental design. I.G.B. assisted in performing the immunohistochemistry experiments and helped write the paper. D.D.-S. and M.R. designed and conducted the human rhinitis studies. MR. also provided immunoassay data and wrote portions of the paper. E.K, D.P.T, and M.Z, participated in the design of the human studies involving the naturalistic cat allergen exposure, the collection, analysis and interpretation of the data from these studies and the review and editing of the manuscript. F.J.G. provided materials and advice and reviewed the manuscript, B.B. assisted in experimental design and in writing the paper, C.L. assisted in interpretation of the immunohistochemical data and helped write portions of the paper. O.H. designed and interpreted the mouse experiments, wrote the paper and coordinated the research.
STATEMENT OF CONFLICT OF INTEREST: On behalf of all the authors, there are no conflicts of interest.
References
- 1.Bateman ED, Boushey HA, Bousquet J, Busse WW, Clark TJ, Pauwels RA, et al. Can guideline-defined asthma control be achieved? The Gaining Optimal Asthma ControL study. Am J Respir Crit Care Med. 2004;170:836–44. doi: 10.1164/rccm.200401-033OC. [DOI] [PubMed] [Google Scholar]
- 2.Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621–7. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 3.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent KA, Feron O, Kelly RA. Harnessing the response to tissue hypoxia: HIF-1 alpha and therapeutic angiogenesis. Trends Cardiovasc Med. 2002;12:362–7. doi: 10.1016/s1050-1738(02)00186-x. [DOI] [PubMed] [Google Scholar]
- 5.Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18:4528–42. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW. Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer. 2007;96:1284–92. doi: 10.1038/sj.bjc.6603675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SY, Chung SM. Neovastat (AE-941) inhibits the airway inflammation via VEGF and HIF-2 alpha suppression. Vascul Pharmacol. 2007;47:313–8. doi: 10.1016/j.vph.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Lee SY, Kwon S, Kim KH, Moon HS, Song JS, Park SH, et al. Expression of vascular endothelial growth factor and hypoxia-inducible factor in the airway of asthmatic patients. Ann Allergy Asthma Immunol. 2006;97:794–9. doi: 10.1016/S1081-1206(10)60971-4. [DOI] [PubMed] [Google Scholar]
- 9.Huerta-Yepez S, Baay-Guzman GJ, Garcia-Zepeda R, Hernandez-Pando R, Vega MI, Gonzalez-Bonilla C, et al. 2-Methoxyestradiol (2-ME) reduces the airway inflammation and remodeling in an experimental mouse model. Clin Immunol. 2008;129:313–24. doi: 10.1016/j.clim.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 10.Guo J, Lu W, Shimoda LA, Semenza GL, Georas SN. Enhanced interferon-gamma gene expression in T Cells and reduced ovalbumin-dependent lung eosinophilia in hypoxia-inducible factor-1-alpha-deficient mice. Int Arch Allergy Immunol. 2009;149:98–102. doi: 10.1159/000189191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fajardo I, Svensson L, Bucht A, Pejler G. Increased levels of hypoxia-sensitive proteins in allergic airway inflammation. Am J Respir Crit Care Med. 2004;170:477–84. doi: 10.1164/rccm.200402-178OC. [DOI] [PubMed] [Google Scholar]
- 12.Tomita S, Sinal CJ, Yim SH, Gonzalez FJ. Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1alpha. Mol Endocrinol. 2000;14:1674–81. doi: 10.1210/mend.14.10.0533. [DOI] [PubMed] [Google Scholar]
- 13.Takagi S, Tojo H, Tomita S, Sano S, Itami S, Hara M, et al. Alteration of the 4-sphingenine scaffolds of ceramides in keratinocyte-specific Arnt-deficient mice affects skin barrier function. J Clin Invest. 2003;112:1372–82. doi: 10.1172/JCI18513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yepez SH, Pando RH, Argumedo LS, Paredes MV, Cueto AH, Isibasi A, et al. Therapeutic efficacy of an E coli strain carrying an ovalbumin allergenic peptide as a fused protein to OMPC in a murine model of allergic airway inflammation. Vaccine. 2003;21:566–78. doi: 10.1016/s0264-410x(02)00244-x. [DOI] [PubMed] [Google Scholar]
- 15.Macy E, Kemeny M, Saxon A. Enhanced ELISA: how to measure less than 10 picograms of a specific protein (immunoglobulin) in less than 8 hours. FASEB J. 1988;2:3003–9. doi: 10.1096/fasebj.2.14.3263291. [DOI] [PubMed] [Google Scholar]
- 16.Aitola MH, Pelto-Huikko MT. Expression of Arnt and Arnt2 mRNA in developing murine tissues. J Histochem Cytochem. 2003;51:41–54. doi: 10.1177/002215540305100106. [DOI] [PubMed] [Google Scholar]
- 17.Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, et al. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt) Mol Cell Biol. 1996;16:1706–13. doi: 10.1128/mcb.16.4.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain S, Maltepe E, Lu MM, Simon C, Bradfield CA. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech Dev. 1998;73:117–23. doi: 10.1016/s0925-4773(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 19.Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Dev Biol. 1997;191:297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- 20.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–7. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 21.Majamaa K, Gunzler V, Hanauske-Abel HM, Myllyla R, Kivirikko KI. Partial identity of the 2-oxoglutarate and ascorbate binding sites of prolyl 4-hydroxylase. J Biol Chem. 1986;261:7819–23. [PubMed] [Google Scholar]
- 22.Wang J, Buss JL, Chen G, Ponka P, Pantopoulos K. The prolyl 4-hydroxylase inhibitor ethyl-3,4-dihydroxybenzoate generates effective iron deficiency in cultured cells. FEBS Lett. 2002;529:309–12. doi: 10.1016/s0014-5793(02)03389-6. [DOI] [PubMed] [Google Scholar]
- 23.Hagen T, D'Amico G, Quintero M, Palacios-Callender M, Hollis V, Lam F, et al. Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochem Biophys Res Commun. 2004;322:923–9. doi: 10.1016/j.bbrc.2004.07.204. [DOI] [PubMed] [Google Scholar]
- 24.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–75. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 25.Lee YC, Kwak YG, Song CH. Contribution of vascular endothelial growth factor to airway hyperresponsiveness and inflammation in a murine model of toluene diisocyanate-induced asthma. J Immunol. 2002;168:3595–600. doi: 10.4049/jimmunol.168.7.3595. [DOI] [PubMed] [Google Scholar]
- 26.Lee KS, Kim SR, Park HS, Park SJ, Min KH, Lee KY, et al. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-kappaB and hypoxia-inducible factor-1alpha. Exp Mol Med. 2007;39:756–68. doi: 10.1038/emm.2007.82. [DOI] [PubMed] [Google Scholar]
- 27.Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science. 1992;256:1193–5. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 28.Probst MR, Fan CM, Tessier-Lavigne M, Hankinson O. Two murine homologs of the Drosophila single-minded protein that interact with the mouse aryl hydrocarbon receptor nuclear translocator protein. J Biol Chem. 1997;272:4451–7. doi: 10.1074/jbc.272.7.4451. [DOI] [PubMed] [Google Scholar]
- 29.Ooe N, Saito K, Mikami N, Nakatuka I, Kaneko H. Identification of a novel basic helix-loop-helix-PAS factor, NXF, reveals a Sim2 competitive, positive regulatory role in dendritic-cytoskeleton modulator drebrin gene expression. Mol Cell Biol. 2004;24:608–16. doi: 10.1128/MCB.24.2.608-616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woods SL, Whitelaw ML. Differential activities of murine single minded 1 (SIM1) and SIM2 on a hypoxic response element. Cross-talk between basic helix-loop-helix/per-Arnt-Sim homology transcription factors. J Biol Chem. 2002;277:10236–43. doi: 10.1074/jbc.M110752200. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence BP, Denison MS, Novak H, Vorderstrasse BA, Harrer N, Neruda W, et al. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low-molecular-weight compound. Blood. 2008;112:1158–65. doi: 10.1182/blood-2007-08-109645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saini Y, Kim KY, Lewandowski R, Bramble LA, Harkema JR, Lapres JJ. Role of hypoxia-inducible factor 1{alpha} in modulating cobalt-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L139–47. doi: 10.1152/ajplung.00252.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–34. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009;13:2780–6. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.