

Abstract
Systemic Lupus Erythematosus (SLE) is a prototypic autoimmune disorder with a complex pathogenesis in which genetic, hormonal and environmental factors play a role. Rare mutations in the TREX1 gene, the major mammalian 3′-5′ exonuclease, have been reported in sporadic SLE cases. Some of these mutations have also been identified in a rare pediatric neurologic condition featuring an inflammatory encephalopathy known as Aicardi-Goutières syndrome (AGS). We sought to investigate the frequency of these mutations in a large multi-ancestral cohort of SLE cases and controls.
Methods
Forty single-nucleotide polymorphisms (SNPs), including both common and rare variants, across the TREX1 gene were evaluated in ∼8370 patients with SLE and ∼7490 control subjects. Stringent quality control procedures were applied and principal components and admixture proportions were calculated to identify outliers for removal from analysis. Population-based case-control association analyses were performed. P values, false discovery rate q values, and odds ratios with 95% confidence intervals were calculated.
Results
The estimated frequency of TREX1 mutations in our lupus cohort was 0.5%. Five heterozygous mutations were detected at the Y305C polymorphism in European lupus cases but none were observed in European controls. Five African cases incurred heterozygous mutations at the E266G polymorphism and, again, none were observed in the African controls. A rare homozygous R114H mutation was identified in one Asian SLE patient whereas all genotypes at this mutation in previous reports for SLE were heterozygous. Analysis of common TREX1 SNPs (MAF >10%) revealed a relatively common risk haplotype in European SLE patients with neurologic manifestations, especially seizures, with a frequency of 58% in lupus cases compared to 45% in normal controls (p=0.0008, OR=1.73, 95% CI=1.25-2.39). Finally, the presence or absence of specific autoantibodies in certain populations produced significant genetic associations. For example, a strong association with anti-nRNP was observed in the European cohort at a coding synonymous variant rs56203834 (p=2.99E-13, OR=5.2, 95% CI=3.18-8.56).
Conclusion
Our data confirm and expand previous reports and provide additional support for the involvement of TREX1 in lupus pathogenesis.
Introduction
Increased expression of interferon regulated genes and disturbance of interferon alpha (IFN-α) homeostasis has a major role in the pathogenesis of the prototypic autoimmune disorder, systemic lupus erythematosus (SLE) (1–3). A perturbation of IFN-α metabolism is also a major pathogenic feature of the inflammatory encephalopathy, Aicardi-Goutières syndrome (AGS) (4). After the discovery of AGS-causing mutations in the TREX1 gene, distinct heterozygous mutations were described in autosomal dominant diseases such as retinal vasculopathy with cerebral leukodystrophy (RVCL) (5), and familial chilblain lupus (FCL) (6). Subsequent studies demonstrate that up to 2% of patients with SLE harbor pathogenic mutations in TREX1 (7). These rare but highly penetrant causative mutations are not detected in genome wide studies and, thereby, may explain part of the missing heritability of lupus as well as provide insights into disease pathogenesis.
The shared TREX1 genetics in clinically distinct human disorders points to a common molecular etiology. Indeed, some AGS individuals also fulfill diagnostic criteria for SLE and have antinuclear antibodies including those with antigenic specificity for ssDNA and dsDNA (8). Likewise, some RVCL patients have autoantibodies to nuclear antigens (unpublished, JPA, PHK).
TREX1 (DNase III) is the major 3′-5′ DNA exonuclease of mammalian cells (9). It has been proposed to have a major role in cell death processes and genomic DNA degradation where it may minimize immune activation by self DNA. It is also a key component of the SET complex, a multitasking protein involved in apoptosis, transcription, nucleosome assembly and histone binding. This complex is normally associated with the endoplasmic reticulum. It is mobilized during the cellular response to oxidative damage and is postulated to participate in the oxidative stress response. A connection between TREX1 and immune activation was initially suggested in the TREX1 null mice that develop an inflammatory myocarditis similar to autoimmune cardiomyopathy and produce type 1 IFN (10, 11). Furthermore, TREX1 deficient cells accumulate single stranded DNA species that may trigger autoimmunity (12).
The TREX1 gene is located on chromosome 3p21.31 and consists of a single exon encoding a 314-amino acid polypeptide. It has three conserved sequence motifs, Exo I, Exo II and Exo III, which form the catalytic site (Fig. 1). TREX1 has a hydrophobic carboxyl-terminal region, predicted to form a transmembrane helix, likely important in defining its intracellular localization (5). In addition, the TREX1 protein contains a proline-rich sequence that is postulated to participate in protein-protein interactions (13) (Fig. 1).
Fig 1.
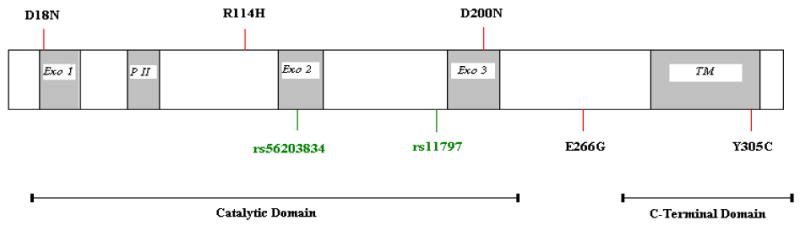
Schematic representation of some important disease associated TREX1 mutations. Coding synonymous SNP (green); coding non-synonymous SNP (red). Exo 1, 2 and 3 denote exonuclease domains, PII denotes polyproline II domain, and TM denotes transmembrane domain.
Mutations throughout the TREX1 gene have been identified in patients with several different human diseases (13). These mutations include null alleles, frameshift mutations and non-synonymous changes in the catalytic domains and the C-terminal region. In AGS, most TREX1 mutations are autosomal recessive and diminish the exonuclease activity of the enzyme, in particular a transition of arginine to histidine at position 114 (R114H). A homozygous change at this locus (R114H) has been found to have a major effect on exonuclease enzyme activity (14,15). The heterozygous R114H mutation has been reported in one individual with SLE (7). Other mutations include D200N and D18N, which have been reported in AGS and FCL, respectively, and are inherited in an autosomal dominant manner (Fig. 1). In lupus, most of the mutations reported thus far are heterozygotes and are usually located outside the catalytic domain in the C-terminal region (7). The functional significance of these mutations is unknown. C-terminal frameshift mutations that retain exonuclease activity are observed in SLE and account for all of the mutations in RVCL (7,13).
In this study, we evaluated these aforementioned mutations as well as common tagged SNPs in the TREX1 gene in a large, multi-ancestral cohort of SLE cases and controls. This study is the first to investigate TREX1 in populations with a higher prevalence of lupus including African- Americans and Hispanics. Our results confirm and extend the findings in Caucasians and provide additional support for association of SLE with TREX1 in multiple ancestries.
Results
To determine if TREX1 is associated with SLE, we genotyped 40 SNPs in the TREX1 genomic region including both previously reported rare SNPs and more common tag SNPs that capture most of the variation in this region (Table 2). After removing the outliers and correcting for population stratification, 15864 samples (8372 cases and 7492 controls) were included in the analysis (Table 1). All SNPs were in HWE and passed stringent quality control thresholds (see methods).
Table 2.
Selected SNPs in the TREX-1 gene.
| SNP | Position | Minor Allele | Major Allele | Average MAF in European | Average MAF in African | Average MAF in Asian | Average MAF in Hispanic |
|---|---|---|---|---|---|---|---|
| rs922075 | 48464402 | A | G | 0.474 | 0.2975* | 0.3558 | 0.3395 |
| rs6776700 | 48471762 | A | G | 0.4616 | 0.2729 | 0.3566 | 0.2828 |
| rs6442123 | 48475290 | A | G | 0.469 | 0.4512 | 0.3589 | 0.3109 |
| rs34426134 | 48480236 | A | G | 0.0005568 | 0.04316 | 0 | 0.003878 |
| rs62264269 | 48480342 | A | G | 0.00008173 | 0.0003569 | 0 | 0 |
| rs7626978 | 48480835 | C | A | 0.003968 | 0.1098 | 0 | 0.01517 |
| rs3135935 | 48480928 | A | G | 0 | 0.01331 | 0 | 0.0008929 |
| rs2242150 | 48480968 | A | C | 0.4676 | 0.4552 | 0.3558 | 0.311 |
| rs3135936 | 48481222 | 0 | C | 0 | 0 | 0 | 0 |
| rs3135938 | 48481308 | A | G | 0.0002393 | 0.0003521 | 0 | 0.0005984 |
| rs36041404 | 48481452 | A | G | 0.000555 | 0.04846 | 0 | 0.007139 |
| rs12486046 | 48482186 | G | A | 0.03295 | 0.004374 | 0.0001983 | 0.02826 |
| rs3135940 | 48482579 | A | G | 0.001928 | 0.0003521 | 0.0004002 | 0.0009063 |
| rs3135941 | 48482671 | G | A | 0.1787 | 0.03012 | 0.04005 | 0.1778 |
| rs3135942 | 48482826 | 0 | G | 0 | 0 | 0 | 0 |
| D18N | 48483110 | 0 | G | 0 | 0 | 0 | 0 |
| rs55938060 | 48483247 | A | G | 0.001311 | 0.0001779 | 0.0002006 | 0.0003081 |
| rs3135943 | 48483256 | 0 | 0 | NA | NA | NA | NA |
| R114H | 48483399 | A | G | 0.001053 | 0.0003602 | 0.0008094 | 0.0003021 |
| V122A | 48483423 | 0 | A | 0 | 0 | 0 | 0 |
| rs56203834 | 48483454 | A | G | 0.01349 | 0.001092 | 0.001856 | 0.01587 |
| rs3135944 | 48483520 | G | A | 0.0001586 | 0.02172 | 0 | 0.00119 |
| A158V | 48483531 | 0 | G | 0 | 0 | 0 | 0 |
| rs55786737 | 48483544 | A | G | 0.008773 | 0.003002 | 0.003843 | 0.007869 |
| R164X | 48483548 | 0 | G | 0 | 0 | 0 | 0 |
| rs11797 | 48483589 | A | G | 0.4499 | 0.2701 | 0.3499 | 0.279 |
| D200N | 48483656 | 0 | G | 0 | 0 | 0 | 0 |
| V201D | 48483660 | 0 | T | 0 | 0 | 0 | 0 |
| G227S | 48483737 | 0 | 0 | NA | NA | NA | NA |
| R240S | 48483778 | G | C | 0.0000794 | 0.002451 | 0 | 0.0008929 |
| A247P | 48483797 | C | G | 0.00007926 | 0.0014 | 0 | 0 |
| E266G | 48483855 | G | A | 0.002778 | 0.0005248 | 0 | 0.0002976 |
| P290L | 48483927 | 0 | 0 | NA | NA | NA | NA |
| T303P | 48483965 | 0 | A | 0 | 0 | 0 | 0 |
| rs3135945 | 48483970 | A | G | 0.000794 | 0.1282 | 0 | 0.01013 |
| Y305C | 48483972 | G | A | 0.0001587 | 0 | 0 | 0 |
| G306A | 48483975 | 0 | C | 0 | 0 | 0 | 0 |
| rs3135946 | 48484040 | G | A | 0.003578 | 0.1126 | 0 | 0.01492 |
| rs56112131 | 48484061 | A | G | 0 | 0.0001766 | 0 | 0 |
| rs3135947 | 48484132 | 0 | A | 0 | 0 | 0 | 0 |
For this SNP, minor allele for the African populations was allele G.
Table 1.
Demographic distribution of individuals in study.
| European-American Case/Control | Asian Case/Control | African American Case/Control | Gullah* Case/Control | Hispanic (others) Case/Control | |
|---|---|---|---|---|---|
| Total | 3936/3491 | 1265/1260 | 1527/1811 | 152/123 | 1492/807 |
| Male: | 344/1151 | 99/154 | 121/574 | 15/18 | 127/80 |
| Female | 3592/2340 | 1166/1106 | 1406/1237 | 137/105 | 1365/727 |
The Gullah are African-Americans who live in the Low Country of South Carolina and genetically show a much lower admixture rate with non- African populations than other African-Americans.
Observance of rare polymorphisms (MAF<0.01)
Table 3 shows the rare non-synonymous coding SNPs (MAF <0.01) in the TREX1 gene including some detected only in cases. We observed at least three different types of mutations in patients that were not detected in the corresponding controls (Table 3). In Europeans, 5 heterozygous mutations were detected at Y305C in lupus cases but none were observed in the European controls. In African-American and Gullah patients (together), 5 E266G heterozygous mutations were observed but, again, none were observed in the corresponding controls. In the Asians, one homozygous and two heterozygous mutations were observed in lupus cases for the R114H polymorphism, but none in controls. Table 4 describes the ACR criteria fulfilled by these patients and their serologic profiles. Interestingly, two of these 13 SLE cases were males, of which one carried the homozygous R114H mutation and developed SLE at 14 years of age (Table 4).
Table 3.
Frequency of nonsynonymous coding mutations in lupus cases and controls.
| SNP* | Position | Minor allele | Major allele | EA Case (AA/AB/BB) | EA Control (AA/AB/BB) | AA Case | AA Control | Gullah Case | Gullah Control | Asian Case | Asian Control |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D18N | 48483110 | 0 | G | 0/0/3921 | 0/0/3479 | 0/0/1524 | 0/0/1801 | 0/0/152 | 0/0/123 | 0/0/1262 | 0/0/1257 |
| R114H | 48483399 | A | G | 0/9/3856 | 0/5/3389 | 0/1/1511 | 0/1/1734 | 0/0/146 | 0/0/120 | 1/2/1232 | 0/0/1236 |
| V122A | 48483423 | 0 | A | 0/0/3906 | 0/0/3456 | 0/0/1509 | 0/0/1800 | 0/0/152 | 0/0/123 | 0/0/1262 | 0/0/1259 |
| A158V | 48483531 | 0 | G | 0/0/3932 | 0/0/3480 | 0/0/1526 | 0/0/1800 | 0/0/152 | 0/0/123 | 0/0/1263 | 0/0/1257 |
| R164X | 48483548 | 0 | G | 0/0/3928 | 0/0/3487 | 0/0/1527 | 0/0/1807 | 0/0/151 | 0/0/123 | 0/0/1262 | 0/0/1257 |
| D200N | 48483656 | 0 | G | 0/0/3920 | 0/0/3489 | 0/0/1527 | 0/0/1807 | 0/0/151 | 0/0/123 | 0/0/1263 | 0/0/1258 |
| V201D | 48483660 | 0 | T | 0/0/3935 | 0/0/3490 | 0/0/1525 | 0/0/1809 | 0/0/152 | 0/0/123 | 0/0/1264 | 0/0/1260 |
| R240S | 48483778 | G | C | 0/1/3919 | 0/0/3491 | 0/7/1519 | 0/11/1798 | 0/3/149 | 0/1/122 | 0/0/1265 | 0/0/1260 |
| A247P | 48483797 | C | G | 0/0/3936 | 0/1/3489 | 0/3/1524 | 0/7/1803 | 0/0/152 | 0/2/121 | 0/0/1264 | 0/0/1260 |
| E266G | 48483855 | G | A | 0/22/3909 | 0/19/3466 | 0/4/1523 | 0/0/1811 | 0/1/151 | 0/0/123 | 0/0/1265 | 0/0/1260 |
| T303P | 48483965 | 0 | A | 0/0/3918 | 0/0/3458 | 0/0/1511 | 0/0/1789 | 0/0/151 | 0/0/123 | 0/0/1263 | 0/0/1260 |
| Y305C | 48483972 | G | A | 0/5/3924 | 0/0/3489 | 0/0/1527 | 0/0/1811 | 0/0/152 | 0/0/123 | 0/0/1265 | 0/0/1260 |
| G306A | 48483975 | 0 | C | 0/0/3936 | 0/0/3491 | 0/0/1527 | 0/0/1811 | 0/0/152 | 0/0/123 | 0/0/1265 | 0/0/1260 |
Non-Synonymous SNPs with MAF <1%.
Table 4.
Phenotypic characterization of 13 SLE patients with nonsynonymous mutations.
| SLE Cases | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| Detected Mutations | R114H (HZ)* | R114H (HTZ)** | R114H (HTZ) | E266G (HTZ) | E266G (HTZ) | E266G (HTZ) | E266G (HTZ) | E266G (HTZ) | Y305C (HTZ) | Y305C (HTZ) | Y305C (HTZ) | Y305C (HTZ) | Y305C (HTZ) |
| Gender | M | F | F | F | F | F | F | M | F | F | F | F | F |
| Race | Asian | Asian | Asian | African-American | African-American | African-American | African-American | African-Gullah | European | European | European | European | European |
| Age of SLE onset | 14 | 18 | 34 | 16 | 36 | 22 | 50 | 43 | 37 | 27 | N/A | 31 | 37 |
| Malar Rash | + | - | - | - | - | - | - | - | - | + | - | + | + |
| Discoid Rash | + | - | - | - | - | - | + | - | - | + | - | - | - |
| Photosensitivity | + | - | - | + | - | - | - | + | + | + | + | + | - |
| Oral ulcers | - | + | - | + | - | - | - | - | - | + | + | - | - |
| Arthritis | - | - | + | - | + | - | + | + | + | + | + | + | - |
| Serositis | - | + | - | - | + | - | + | + | - | + | + | + | + |
| Nephritis | - | + | + | + | - | + | + | - | - | - | - | - | + |
| Neurologic | - | - | - | - | + | - | - | - | - | + | - | + | - |
| Hematologic | - | - | - | + | - | + | - | - | - | + | + | + | + |
| Immunologic | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Anti-dsDNA | + | - | + | + | - | + | - | + | + | - | N/A | N/A | N/A |
| Anti-Sm | - | - | - | - | + | + | - | - | - | - | N/A | N/A | N/A |
HZ=Homozygous,
HTZ=Heterozygous,
Analyses of polymorphisms with MAF >0.01 and haplotype structure
In addition to these rare SNPs mentioned above, we also evaluated common and tag SNPs that capture most of the variation in this region, as well as SNPs with MAF >1%. TREX1 is a small gene with less than 1000 base pairs and one coding exon (Fig 1). Since it is closely linked to the ATRIP (ATR interacting protein) gene, we selected common SNPs to cover both (shown in Table 2). Figures 2a and 2b demonstrate the haplotype structure of this genomic region in European and African cases and controls using SNPs with MAF>1%. There is a high correlation among all of these common SNPs, especially in the European population (r-squared > 0.9). Also, more SNPs are polymorphic (MAF >0.01) in the African-American population than in other racial groups (see Table 2).
Fig 2.
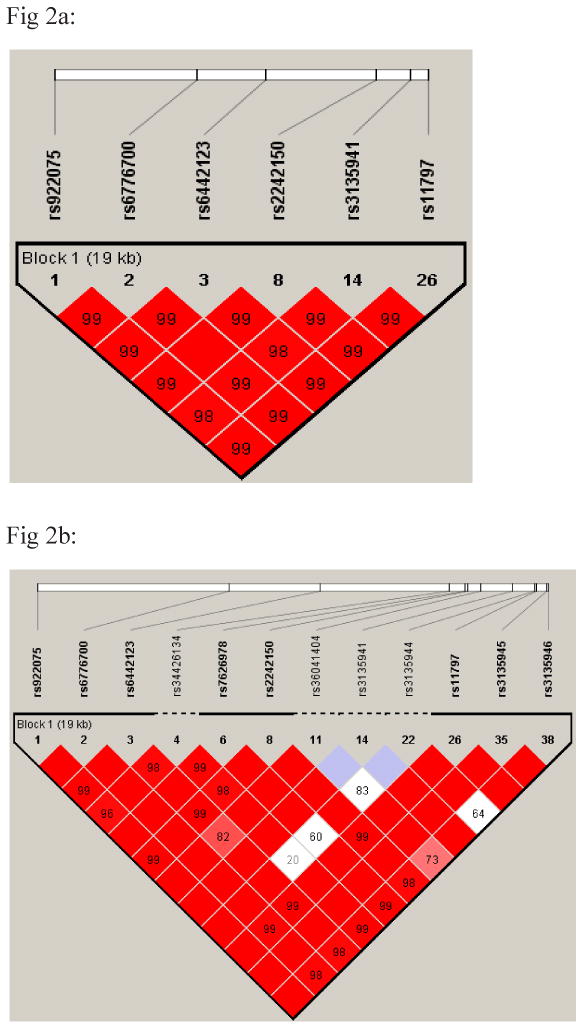
Fig 2a and 2b: European-American (2a) and African-American (2b) haplotype block structure. Blocks connecting SNP pairs are shaded according to the strength of the linkage disequilibrium between the SNPs, from 0.0 (white) to 1.0 (bright red), as measured by the disequilibrium coefficient D′.
Case-control association analysis
In a case control association study, we did not observe significant associations with any of the selected SNPs using the presence of SLE as a phenotype in any of the racial groups. Because of similarities between lupus and neurologic conditions such as AGS, we hypothesized that lupus patients with neurologic manifestations might be enriched for risk alleles in the TREX1 gene. Indeed, in the European population the presence of neurologic manifestations (ACR criteria), especially the presence of seizures (79 European cases), produced significant results at multiple common SNPs when compared with healthy controls (Table 5). The haplotype risk alleles (AAAAAA) (Fig 2a, Table 5) were relatively common in the European population with a frequency of 58% in lupus cases compared to 45% in normal controls (p=0.0008, FDR q=0.007, OR=1.73, 95% CI=1.25-2.39). In addition, in a case only study where these 79 SLE cases with seizure manifestations were compared to 2405 lupus patients with no previous neurologic findings, similar haplotype association results were obtained (p=0.0008, FDR q=0.003, OR=1.75 95% CI=1.25-2.37). Since neurologic manifestations in SLE also correlate with previous cerebrovascular accidents and the presence of antiphospholipid syndrome, we evaluated these sub-groups separately but the results were not significant (data not shown). There was also no evidence of associations with other ACR criteria, gender or age of onset in any population. Of note, all 5 European patients with a mutation at Y305C also had at least one copy of this risk haplotype (2 homozygous and 3 heterozygous patients for the risk allele).
Table 5.
Case control association results in European SLE cases with presence of seizure (79 cases) compared to controls. The bold SNPs represent the common risk haplotype (AAAAAA).
| SNP | BP | Minor Allele | Cases | Controls | Major Allele | CHISQ | P | FDR q | OR | L95 | U95 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs922075 | 48464402 | A | 0.5949 | 0.4718 | G | 9.387 | 0.002185 | 0.007 | 1.644 | 1.192 | 2.267 |
| rs6776700 | 48471762 | A | 0.5921 | 0.4622 | G | 10.09 | 0.001491 | 0.007 | 1.689 | 1.218 | 2.343 |
| rs6442123 | 48475290 | A | 0.6053 | 0.4674 | G | 11.34 | 0.000757 | 0.004 | 1.747 | 1.258 | 2.427 |
| rs2242150 | 48480968 | A | 0.5886 | 0.4662 | C | 9.301 | 0.002291 | 0.007 | 1.638 | 1.189 | 2.257 |
| rs3135941 | 48482671 | G | 0.1429 | 0.1856 | A | 1.83 | 0.1761 | 0.23 | 0.7312 | 0.4637 | 1.153 |
| rs11797 | 48483589 | A | 0.5823 | 0.4485 | G | 11.16 | 0.000835 | 0.005 | 1.714 | 1.245 | 2.36 |
| rs3135945 | 48483970 | A | 0.01266 | 0.000287 | G | 42.15 | 8.46E-11 | 1.69E-09 | 44.65 | 6.25 | 319 |
We also evaluated SLE samples for an association with autoantibodies (anti-Ro, anti-La, anti-RNP, anti-Sm and anti-dsDNA). In the Asian population, 567 SLE patients lacking anti-Ro antibodies were less likely to carry the same common haplotype mentioned above (AAAAAA) compared to 1260 healthy controls (32% in cases versus 36% in controls [p=0.01, FDR q=0.03, OR=0.82 95% CI 0.71-0.96]). Interestingly the same haplotype was observed less frequently among 260 Asian patients with positive anti-nRNP compared with 1260 healthy controls (30% in cases with positive anti-nRNP versus 36% in controls [p=0.003, FDR q=0.008, OR=0.75, 95% CI=0.61-0.92]). In a case only study, these comparisons were not statistically significant, although the frequency of this haplotype was more frequent in Asian cases with positive anti-Ro (335 cases) in comparison to those lupus cases with negative anti-Ro (567 cases) (36% vs. 32% (p=0.22)) and for anti-nRNP autoantibody, less frequent in cases with positive anti-nRNP compared to cases with negative anti-RNP (30% for positives (260 cases) versus 35% for negatives (789 cases) (p=0.05, FDR q= 0.12)).
In addition, the presence of anti-nRNP in the European population was strongly associated with another coding synonymous SNP rs56203834 that was extremely rare in the Asian population. In this subgroup, 269 European cases positive for anti-nRNP were compared with European controls. The MAF for this SNP was only 1% in controls (Table 2) but 5% in European cases with positive anti-nRNP (p=2.99E-13, FDR q=5.97E-12, OR=5.2, 95% CI=3.18-8.56). Furthermore, in case only study, if these 269 cases were compared with 1413 SLE patients with negative anti-nRNP again similar results were obtained (p= 2.74E-06, FDR q= 2.73E-05, OR=3.33 95% CI 1.95-5.65). In the Hispanic population suggestive results were also obtained but the number of samples for subset analysis was small.
Discussion
To explore the frequency of mutations in the TREX1 gene and their relationship to SLE, we evaluated 40 SNPs, including rare variants, previously reported to be associated with several human diseases in large multi-ethnic sample populations. The large sample size of cases and controls (>15800) provide enough power to evaluate rare variations in TREX1. One important mutation described in AGS (R114H) is on the dimer interface of the protein (14,15). Homozygous mutations at this position (i.e TREX1 R114H/R114H) degrade dsDNA 300-fold less efficiently than TREX1 wild type (16). Although AGS cases are associated with homozygous R114H mutations, their parents, who are heterozygous carriers of the mutation, have no abnormal phenotypes reported (17). A heterozygous mutation at this location has been reported in one European SLE case (7). In our European population, we identified 9 SLE cases with heterozygous mutations at this locus and 5 heterozygotes in the European healthy controls (Table 3). In the Asian population, on the other hand, we identified one homozygous SLE case. In addition, two heterozygous SLE cases were detected in the same population, but none in Asian controls. The homozygous case was a male lupus patient with early onset lupus, positive anti-dsDNA antibody and predominant skin manifestations, but no neurological manifestations. The latter is intriguing since all previously reported homozygous R114H mutations have been in AGS patients, who invariably have had central nervous system disease in early childhood. (Table 4).
We identified 5 European lupus cases with heterozygous mutations at another variant (Y305C) but none in controls. This is a missense (coding) mutation located outside of the catalytic domain of TREX1. This polymorphism was previously reported in one European lupus patient (7). None of these 5 patients were carriers for R114H mutant alleles while all of them were carriers for the common risk haplotype (AAAAAA) (2 homozygotes and 3 heterozygotes). This suggests a correlation of this common risk haplotype with coding mutation at Y305C that could be functionally important.
In the African population, we also identified 5 lupus patients with the E266G mutation, which was absent in African controls (Fig 1). This mutation was detected in Europeans but as previously reported in European cohorts, there was no significant difference in cases and controls (Table 3) (7). This association has not been previously reported in the African population. Overall the coding mutation frequency in our SLE cases was ∼0.5%.
AGS, an autosomal disease (usually recessive but rare dominant case) characterized by progressive encephalopathy of early onset, basal ganglia calcifications and chronic cerebrospinal fluid (CSF) lymphocytosis, is also associated with increased levels of IFNα in the CSF. SLE patients likewise may have high level of IFNα and neurologic manifestations similar to AGS, including seizures. Indeed, neuroimaging in patients with SLE may show calcifications, white matter changes, and atrophy, as typically observed in patients with AGS (18-20). Given these overlapping phenotypes, one may speculate that a common pathogenic mechanism underlies the neurologic phenotype of AGS and cerebral lupus.
In a recent study, up to 60% of patients with AGS presented with clinical findings such as skin rash, arthritis, oral ulcer as well as laboratory findings commonly seen in patients with lupus including ANAs, reduced complement levels, thrombocytopenia, leukocytopenia. Furthermore, if seizures are taken into account, up to 75% of those patients with AGS showed manifestations of lupus (21). Because of these similarities, Vries et al. sequenced genomic DNA of 60 European lupus patients with neurologic manifestations for exonic TREX1 mutations and identified a novel R128H mutation in one of these SLE patients (22). Brain magnetic resonance imaging (MRI) of this patient showed generalized atrophy, extensive symmetric cerebral white matter hyperintensities and cerebellar infarcts, without evidence for ischemia. This rare mutation is located within the highly conserved second exonuclease domain (ExoII) of the TREX1 gene.
In our study we also tested this hypothesis and identified a relatively common risk haplotype in the TREX1 gene among European lupus patients with seizure (58% in SLE cases with seizure compare to 45% in normal controls). The frequency of this haplotype in normal European controls was consistent with the HapMap data for the CEU study population (45%). This haplotype spans 19 kb and covers both TREX1 and ATRIP genes. These two genes are closely linked and some mRNAs encode ATRIP and TREX1 in different reading frames. In fact, these two genes previously were considered a single gene (NCBI-35). ATRIP is an essential component of the DNA damage checkpoint, and binds to ssDNA and interacts with proteins such as ataxia telangiectasia, Rad3 related protein, and breast-ovarian cancer susceptibility 1 (BRCA1) (23). It has central role in checkpoint activation in response to DNA damage and is important for chromosomal stability. Because of the high LD between SNPs in this risk haplotype in Europeans (Fig 2a), conditional analyses were not conclusive; however, analyses on this haplotype suggest that rs11797, a common synonymous SNP located in the exonic region of TREX1, can better explain the whole association in this haplotype and therefore the effect likely originates from the TREX1 gene.
There was also evidence for association of certain SNPs with the presence of autoantibodies in SLE. In particular, there was a positive association with the presence of anti-nRNP in Europeans for SNP rs56203834 (p=2.99E-13, FDR q=5.97E-012, OR = 5.2, 95% CI=3.18-8.56). This is another synonymous SNP located in the TREX1 exon and is less than 60 base pairs away from R114H (Fig. 1). Because of low MAF in this SNP, no clear correlation between this SNP and the common risk haplotype for neurologic manifestation in European can be detected, and in fact all available European cases with seizure manifestations were homozygous for the major wild type allele for this SNP, suggesting that these two effects might be independent. This SNP was extremely rare in the Asian and African populations while in the Hispanic population the result was suggestive.
In regard to the Asian population, the common haplotype that was associated with neurologic manifestations in Europeans was also more frequently seen in Asian patients with this phenotype (42% in cases compare to 38% in controls, p=NS); although it was not significant. As described in the Results, in the Asian population, this haplotype was less frequently observed with the presence of anti-nRNP or in patients with absence of anti-Ro antibodies. In fact this negative correlation with anti-Ro in TREX1 has been previously reported in the same population (24). The reason for this opposite association between anti-Ro and anti-nRNP in Asian with the same haplotype is not clear, and requires additional confirmation. Since SLE is an extremely heterogenous disease, this could be partly related to different disease manifestations. In complex diseases such as SLE, many subtle inherited elements could directly or indirectly affect these autoantibodies with subsequent clinical sequel. Some autoantibodies associated with lupus tend to cluster together and usually result in specific clinical manifestations. For example, the association of anti-Ro with secondary Sjogren's syndrome or leukopenia, anti-RNP with Raynaud's phenomenon, and anti-dsDNA with nephritis have been noted and replicated in many studies. In addition, anti-Ro antibodies have been reported as one of the independent predictors of neurologic damage in lupus (25). This correlation could explain our results in regard to the risk haplotype and anti-Ro antibodies.
Overall, our results with TREX1 indicate a complex relationship between genetic loci, SLE sub-phenotypes and different population ancestry that demands further studies of this gene. Our data, combined with the findings in Trex1-deficient mice which develop lethal autoimmunity accompanied with a high production of type I IFN, suggest that TREX1 is involved in lupus pathogenesis and probably essential for the prevention of autoimmunity.
Materials and Methods
Recruitment and Biological Sample Collection
The participants were enrolled in the Lupus Family Registry and Repository and Lupus Genetics Studies at the Oklahoma Medical Research Foundation (OMRF) as described (26) and by collaborators (27-30). A total of 15864 study participants were used in the current study (Table 1). Protocols were approved by the Institutional Review Boards at each respective institution. Patients met four of the 11 revised 1997 ACR criteria for the classification of SLE (31). Ethnicity was self-reported and verified by principal component and admixture proportion calculations.
Genotyping
This genotyping project was part of the Lupus Large Association Study (LLAS) that involved different investigators and more than 32,000 SNPs. Data were generated using the Illumina iSelect technology at the OMRF. Genotype calls were made using all samples to maximize the accuracy of the cluster plots. Following genotype scoring, SNP clusters were evaluated electronically using the Illumina BEADSTUDIO(r) software package (http://www.illumina.com). Ambiguous SNP clusters were evaluated manually and SNPs with poor cluster characteristics were flagged.
Genotypic data were only used from samples with a call rate >90% (average sample call rate=99.1%) and from SNPs with a call frequency >90% (average SNP call rage=99.0%). Initial QC analyses were performed by plate, by lot of reagents, and by date genotyped to be certain that systematic error did not find its way into our data. A sample report was generated for every sample attempted in the project, including sample barcode, ethnicity, gender, pedigree information, no calls, calls, call rate, genotype frequency and Gencall Score. Any sample with previous genotype data was analyzed for concordance. A summary SNP report was also generated containing chromosome and location, call rate, genotype frequency, and Gencall score.
Statistical Analyses
Testing for association was completed using PLINK (32). Haploview version 4.0 (33) was used to estimate the linkage disequilibrium (LD) between markers and haplotypes in the different racial groups. Conditional haplotype analyses were conducted using the WHAP program version 2.09 (34). To correct for multiple testing, false discovery rate (FDR) methods were used and q values were calculated using PLINK (32). Q values correspond to the proportion of false positives among the results. Thus, q values < 0.05 signify less than a 5% false positive rate and are taken as a measure of significance. For each SNP, missing data proportions for cases and controls, minor allele frequencies, ORs, 95% CI intervals, P values and exact tests for departures from Hardy-Weinberg expectations were calculated. SNPs needed to pass stringent quality control criteria that included: Hardy-Weinberg proportions (HWP) with a p>0.01 in the controls and >0.0001 for cases, total proportion missing <5%, and p>0.05 for differential missingness between cases and controls. Samples with a <90% call rate or increased heterozygosity (>5 std. dev. around the mean) were excluded from the analysis. The remaining samples were then evaluated for duplicates or related individuals. Genetic outliers were removed from further analysis as determined by principal components analysis (35) and admixture proportions calculated using ADMIXMAP. Principal components were calculated using all SNPs and admixture proportions were calculated using 347 AIMs.
Acknowledgments
The cooperation of patients and normal control individuals involved in this study as well as physicians that provide samples is gratefully acknowledged. At the Oklahoma Medical Research Foundation, the work was supported by the NIH (AR042460, RR015577, AR048940, RR020143, AI24717, AR62277), Lupus Foundation of America, the Alliance for Lupus Research, the U.S. Department of Veterans Affairs. At the Washington University School of Medicine (St. Louis, MO) the work was supported by grants (HL083822 [PHK], and NS062069 [JPA]), and the Cerebral Retinal Vasculopathy Fund. At the University of Alabama at Birmingham, the work was supported by grants AR33062 (RPK), AR49084 (RPK), and 5UL1RR02777. At the Johns Hopkins University School of Medicine the work was supported by The Hopkins Lupus Cohort (NIH AR43727), and UL1RR025005 from the National Center for Research Resources (NCRR). At the University of Colorado, Denver School of Medicine, the work was supported by NIH grant R21AI070304. At the Medical University of South Carolina, Charleston, the project was supported by award number UL1RR029882 from the National Center for Research Resources. (The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health). At the Rosalind Russell Medical Research Center for Arthritis, University of California, San Francisco, the work was supported by Kirkland Scholar Award, U.S. Public Health Service National Center for Research Resources, M01 RR-00079 and NIH grants AR044804, AR02175, and AR052300. At the Northwestern University, Feinberg School of Medicine, Chicago, IL, the work was supported by NIH grants K24 AR 002138, P60 2 AR30692, PO1 AR49084, UL1RR025741. At the Wake Forest University Health Sciences, Department of Biochemistry, the work was supported by R01GM069962 and Alliance for Lupus Research (67692) awards. The authors acknowledge grants from the European Science Foundation for the BIOLUPUS network, the Swedish Research Council, Swedish Association against Rheumatism, the Swedish International Development Agency (SIDA) and Gustaf Vth-80th-jubilee Foundation to MEAR and the NIH grant CA141700-01. For GENLES: Argentina: Hugo R. Scherbarth MD, Pilar C. Marino MD, Estela L. Motta MD Servicio de Reumatología, Hospital Interzonal General de Agudos “Dr. Oscar Alende”, Mar del Plata, Argentina; Susana Gamron MD, Cristina Drenkard MD, Emilia Menso MD Servicio de Reumatología de la UHMI 1, Hospital Nacional de Clínicas, Universidad Nacional de Córdoba, Córdoba, Argentina; Alberto Allievi MD, Guillermo A. Tate MD Organización Médica de Investigación, Buenos Aires, Argentina; Jose L. Presas MD Hospital General de Agudos Dr. Juán A. Fernandez, Buenos Aires, Argentina; Simon A. Palatnik MD, Marcelo Abdala MD, Mariela Bearzotti PhD Facultad de Ciencias Medicas, Universidad Nacional de Rosario y Hospital Provincial del Centenario, Rosario, Argentina; Alejandro Alvarellos MD, Francisco Caeiro MD, Ana Bertoli MD Servicio de Reumatología, Hospital Privado, Centro Medico de Córdoba, Córdoba, Argentina; Sergio Paira MD, Susana Roverano MD, Hospital José M. Cullen, Santa Fe, Argentina; Cesar E. Graf MD, Estela Bertero PhD Hospital San Martín, Paraná; Cesar Caprarulo MD, Griselda Buchanan PhD Hospital Felipe Heras, Concordia, Entre Ríos, Argentina; Carolina Guillerón MD, Sebastian Grimaudo PhD, Jorge Manni MD Departamento de Inmunología, Instituto de Investigaciones Médicas “Alfredo Lanari”, Buenos Aires, Argentina; Luis J. Catoggio MD, Enrique R. Soriano MD, Carlos D. Santos MD Sección Reumatología, Servicio de Clínica Medica, Hospital Italiano de Buenos Aires y Fundación Dr. Pedro M. Catoggio para el Progreso de la Reumatología, Buenos Aires, Argentina; Cristina Prigione MD, Fernando A. Ramos MD, Sandra M. Navarro MD Servicio de Reumatología, Hospital Provincial de Rosario, Rosario, Argentina; Guillermo A. Berbotto MD, Marisa Jorfen MD, Elisa J. Romero PhD Servicio de Reumatología Hospital Escuela Eva Perón. Granadero Baigorria, Rosario, Argentina; Mercedes A. Garcia MD, Juan C Marcos MD, Ana I. Marcos MD Servicio de Reumatología, Hospital Interzonal General de Agudos General San Martín, La Plata; Carlos E. Perandones MD, Alicia Eimon MD Centro de Educación Médica e Investigaciones Clínicas (CEMIC), Buenos Aires, Argentina; Cristina G. Battagliotti MD Hospital de Niños Dr. Orlando Alassia, Santa Fe, Argentina. Perú is represented by: Dr Eduardo Acevedo and Dr Mariano Cucho, Hospital Nacional Guillermo Almenara Irigoyen, Lima, Perú. Mexico: Dr Ignacio García de la Torre, Hospital General de Occidente, Zapopan, Jalisco; Dr Mario Cardiel Ríos, Hospital General Dr Miguel Silva, Morelia, Michoacán; Dr. José Francisco Moctezuma, Hospital General de México, México, D.F.; Dr Marco Maradiaga Ceceña, Hospital General de Culiacán, Culiacán, Sinaloa. The members of the BIOLUPUS network are: Sandra D'Alfonso, Department of Medical Sciences and IRCAD, University of Eastern Piedmont, 28100, Novara, Italy; Bernard R. Lauwerys, Cliniques Universitaires Saint-Luc, Université catholique de Louvain, B-1200, Bruxells, Belgium; Emoke Endreffy, Department of Pediatrics and Health Center, University of Szeged, Szeged, Hungary, László Kovács, Department of Rheumatology, Albert Szent-Györgyi Clinical Centre, University of Szeged, Szeged, Hungary;, Carlos Vasconcelos, Hospital Santo Antonio and ICBAS, 4099, Porto, Portugal, Berta Martins da Silva, UMIB/ICBAS, Immunogenetics laboratory, Universidado do Porto, Porto, Portugal.; Iñigo Rúa Figueroa, Hospital Doctor Negrin, Las Palmas de Gran Canaria, Spain. Sandra D'Alfonso, Nadia Barizzone (Department of Medical Sciences and IRCAD, University of Eastern Piedmont, Novara, Italy), Gian Domenico Sebastiani (Azienda Ospedaliera San Camillo-Forlanini, Roma, Italy), Mauro Galeazzi, (Siena University, Siena, Italy) Maria Giovanna Danieli (Clinica Medica di Scienze Mediche e Chirurgiche, Universitá Politecnica delle Marche, Ancona, Italy), Giovanni La Montagna (Rheumatology Unit Second University of Naples, Naples, Italy), Maria Grazia Sabbadini (Vita-Salute San Raffaele University – Milan, Italy).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Stetson DB, Medzhitov R. Antiviral defense: interferons and beyond. J Exp Med. 2006;203:1837–1841. doi: 10.1084/jem.20061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crow MK. Type I interferon in systemic lupus erythematosus. Curr Top Microbiol Immunol. 2007;316:359–386. doi: 10.1007/978-3-540-71329-6_17. [DOI] [PubMed] [Google Scholar]
- 3.Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dussaix E, Lebon P, Ponsot G, Huault G, Tardieu M. Intrathecal synthesis of different alpha-interferons in patients with various neurological diseases. Acta Neurol Scand. 1985;71:504–509. doi: 10.1111/j.1600-0404.1985.tb03235.x. [DOI] [PubMed] [Google Scholar]
- 5.Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- 6.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 8.De Laet C, Goyens P, Christophe C, Ferster A, Mascart F, Dan B. Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutières syndrome. Neuropediatrics. 2005;36:399–402. doi: 10.1055/s-2005-873058. [DOI] [PubMed] [Google Scholar]
- 9.Mazur DJ, Perrino FW. Structure and expression of the TREX1 and TREX2 3′-5′ exonuclease genes. J Biol Chem. 2001;276:14718–14727. doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 10.Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, et al. Gene-targeted mice lacking the TREX1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stetson DB, Ko JS, Heidmann T, Medzhitov R. TREX1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Kavanagh D, Spitzer D, Kothari PH, Shaikh A, Liszewski MK, Richards A, et al. New roles for the major human 3′–5′ exonuclease TREX1 in human disease. Cell Cycle. 2008;7:1718–1725. doi: 10.4161/cc.7.12.6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, Hollis T. The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a polyproline II helix for protein partnering. J Biol Chem. 2007;282:10537–43. doi: 10.1074/jbc.M700039200. [DOI] [PubMed] [Google Scholar]
- 16.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 Double-stranded DNA Degradation Activity Is Defective in Dominant Mutations Associated with Autoimmune Disease. J Biol Chem. 2008;283:31649–56. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 18.Raymond AA, Zariah AA, Samad SA, Chin CN, Kong NC. Brain calcification in patients with cerebral lupus. Lupus. 1996;5:123–128. doi: 10.1177/096120339600500207. [DOI] [PubMed] [Google Scholar]
- 19.Appenzeller S, Vasconcelos FA, Li LM, Costallat LT, Cendes F. Quantitative magnetic resonance imaging analyses and clinical significance of hyperintense white matter lesions in systemic lupus erythematosus patients. Ann Neurol. 2008;64:635–43. doi: 10.1002/ana.21483. [DOI] [PubMed] [Google Scholar]
- 20.Sibbitt WL, Brooks WM, Kornfeld M, Hart BL, Bankhurst AD, Roldan CA. Magnetic resonance imaging and brain histopathology in neuropsychiatric systemic lupus erythematosus. Semin Arthritis Rheum. 2010;40:32–52. doi: 10.1016/j.semarthrit.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 2010;62:1469–1477. doi: 10.1002/art.27367. [DOI] [PubMed] [Google Scholar]
- 22.De Vries B, Steup-Beekman GM, Haan J, Bollen EL, Luyendijk J, Frants RR, et al. TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann Rheum Dis. 2010 Apr 13; doi: 10.1136/ard.2009.114157. [DOI] [PubMed] [Google Scholar]
- 23.Venere M, Snyder A, Zgheib O, Halazonetis TD. Phosphorylation of ATR-interacting protein on Ser239 mediates an interaction with breast-ovarian cancer susceptibility 1 and checkpoint function. Cancer Res. 2007;67:6100–6105. doi: 10.1158/0008-5472.CAN-07-0369. [DOI] [PubMed] [Google Scholar]
- 24.Hur JW, Sung YK, Shin HD, Park BL, Cheong HS, Bae SC. TREX1 polymorphisms associated with autoantibodies in patients with systemic lupus erythematosus. Rheumatol Int. 2008;28:783–789. doi: 10.1007/s00296-007-0509-0. [DOI] [PubMed] [Google Scholar]
- 25.Mikdashi J, Handwerger B. Predictors of neuropsychiatric damage in systemic lupus erythematosus: data from the Maryland lupus cohort. Rheumatology (Oxford) 2004;43:1555–1560. doi: 10.1093/rheumatology/keh384. [DOI] [PubMed] [Google Scholar]
- 26.Kaufman KM, Kelly JA, Herring BJ, Adler AJ, Glenn SB, Namjou B, et al. Evaluation of the genetic association of the PTPN22 R620W polymorphism in familial and sporadic systemic lupus erythematosus. Arthritis Rheum. 2006;54:2533–2540. doi: 10.1002/art.21963. [DOI] [PubMed] [Google Scholar]
- 27.International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN) Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacob CO, Reiff A, Armstrong DL, Myones BL, Silverman E, Klein-Gitelman M, et al. Identification of novel susceptibility genes in childhood-onset systemic lupus erythematosus using a uniquely designed candidate gene pathway platform. Arthritis Rheum. 2007;56:4164–4173. doi: 10.1002/art.23060. [DOI] [PubMed] [Google Scholar]
- 29.Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Cohen PL, et al. Differences by race, sex and age in the clinical and immunologic features of recently diagnosed systemic lupus erythematosus patients in the southeastern United States. Lupus. 2002;3:161–167. doi: 10.1191/0961203302lu161oa. [DOI] [PubMed] [Google Scholar]
- 30.Alarcón GS, McGwin G, Jr, Petri M, Ramsey-Goldman R, Fessler BJ, Vilá LM, et al. PROFILE Study Group Time to renal disease and end-stage renal disease in PROFILE: a multiethnic lupus cohort. PLoS Med. 2006;3:e396. doi: 10.1371/journal.pmed.0030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 32.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. American Journal of Human Genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S, Daly MJ, Sham PC. WHAP: haplotype-based association analysis. Bioinformatics. 2007;23:255–256. doi: 10.1093/bioinformatics/btl580. [DOI] [PubMed] [Google Scholar]
- 35.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]