

Abstract
Signal Transducer and Activator of Transcription 3 (STAT3) is persistently activated and contributes to malignant progression in various cancers. Janus kinases (JAKs) phosphorylate STAT3 in response to stimulation with cytokines or growth factors. The STAT3 signaling pathway has been validated as a promising target for development of anti-cancer therapeutics. Small-molecule inhibitors of JAK/STAT3 signaling represent potential molecular-targeted cancer therapeutic agents. In this study, we investigated the role of JAK/STAT3 signaling in 6-bromoindirubin-3'-oxime (6BIO) mediated growth inhibition of human melanoma cells and assessed 6BIO as an anticancer drug candidate. We found that 6BIO is a pan-JAK inhibitor that induced apoptosis of human melanoma cells. 6BIO directly inhibited JAK family kinase activity both in vitro and in cancer cells. Apoptosis of human melanoma cells induced by 6BIO was associated with reduced phosphorylation of JAKs and STAT3 in both a dose- and time-dependent manners. Consistent with inhibition of STAT3 signaling, the anti-apoptotic protein Mcl-1 was down-regulated. In contrast to the decreased levels of phosphorylation of JAKs and STAT3, phosphorylation levels of the AKT and MAPK signaling proteins were not inhibited in cells treated with 6BIO. Importantly, 6BIO suppressed tumor growth in vivo with low toxicity in a mouse xenograft model of melanoma. Taken together, these results demonstrate that 6BIO is a novel pan-JAK inhibitor that can selectively inhibit STAT3 signaling and induced tumor cell apoptosis. Our findings support further development of 6BIO as a potential anti-cancer therapeutic agent that targets JAK/STAT3 signaling in tumor cells.
Keywords: bromoindirubin, JAK inhibitor, STAT3 signaling, apoptosis, melanoma
Introduction
Janus kinase (JAK) family includes JAK1, JAK2, JAK3 and TYK2. While JAK1, JAK2 and TYK2 are ubiquitously expressed in mammalian cells, JAK3 is restricted to hematopoietic cells (1-2). JAK family kinases activate Signal Transducer and Activator of Transcription (STAT) proteins in response to different cytokines and growth factors (2-5). In normal cells, JAK/STAT signaling is tightly regulated. However, in cancer cells, it is persistently activated due to the aberrant activation of JAK family kinases or other tyrosine kinases. As one of the more recently recognized oncogenic signaling pathways, JAK/STAT3 signaling has been shown to be important for tumorigenesis. Among the seven STAT family members, STAT3 is most frequently aberrantly activated in human cancer cells. Persistent activation of JAK/STAT3 signaling contributes to the malignancy of tumors by promoting tumor cell proliferation and survival, angiogenesis and immune evasion (6-10). Thus, JAK/STAT3 signaling is a promising molecular target for cancer therapy.
Recently, small-molecule inhibitors of JAK/STAT3 signaling have been identified for development of cancer therapeutics. Discovery of the JAK2 V617F mutation in myeloproliferative disease has prompted development of selective JAK2 inhibitors for treatment of hematologic disorders (11-13). Clinical studies of JAK2 inhibitors are currently ongoing (14-15). The role of constitutive JAK2 kinase activity in myeloproliferative neoplastic growth also provides the rationale for investigating JAK inhibitors in solid tumors. Small-molecule inhibitors of JAK/STAT3 signaling have been reported to suppress cancer cell growth both in vitro and in vivo (16-22).
Indirubin, a bis-indole alkaloid, is the active ingredient of Danggui Longhui Wan, a traditional Chinese medicine for treatment of chronic myelocytic leukemia (CML) (23-24). Indirubins, namely indirubin and some indirubin derivatives (IRDs) are known to be inhibitors of cyclin-dependent kinases (CDKs), glycogen synthase kinase-3 (GSK-3), and glycogen phosphorylase b. In addition, some IRDs induce apoptosis in human cancer cells through inhibition of SRC/STAT3 signaling (25-29), suggesting STAT3 signaling might be a potential target for IRDs.
Bromo-IRDs are novel synthetic IRDs with improved potency (30-33). In this study, we have identified 6BIO as a pan-JAK inhibitor, selectively targeting JAK/STAT3 signaling in human melanoma cells. Among a series of synthetic IRDs, 6BIO shows the best anticancer activity, and induces apoptosis of melanoma cells. 6BIO suppressed tumor growth with low toxicity in an A2058 human melanoma xenograft mouse model. We investigated the effects of 6BIO particularly on human melanoma cells due to a need for more effective therapeutics for this tumor site. Dacarbazine, the only FDA-approved drug for treatment of metastatic melanoma, has approximately a 15% response rate (34-35). Thus, 6BIO as a pan-JAK inhibitor represents a promising lead compound for development of new anticancer therapeutics, especially for melanoma.
Materials and Methods
Reagents
The preparation of IRDs has been described previously (30). The compounds were dissolved in DMSO as 20 mmol/L and stocked in -20 °C before use for in vitro experiments and treatments in cells. For in vivo experiments, 6BIO was freshly prepared in 30% Solutol (Basf) as 10 mg/mL. Anti-Survivin was from Novus. Anti-β-Actin was from Sigma. Horeseradish peroxidase (HRP)-labeled anti-mouse and anti-rabbit secondary antibodies were from GE Healthcare. All other antibodies were from Cell Signaling.
Cell lines and culture
Cell lines of A2058, G361, SK-MEL-5 and SK-MEL-28 human melanoma, DU145 and LNCaP prostate cancer, U266 myeloma and MDA-MB-468 breast cancer were obtained from American Type Culture Collection. MDA-MB-468 cells were maintained in DMEM medium. Other types of cells were maintained in RPMI 1640 medium. The cell culture medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S).
Cell viability assay
Human cancer cells were seeded at 96-well plates with 5000 cells per well. After 24 h incubation, cells were treated with IRDs or DMSO as vehicle control for 48 h. MTS reagent (CellTiter 96 AQueousOne Solution Cell proliferation Assay, Promega) was added to each well according to the supplier's protocol. Absorbance was measured at 490 nm within 4h using a micro-plate reader (BIO-RAD). The values of cell viability were calculated as percentages of absorbance from treated samples to absorbance from the vehicle control.
Flow cytometric analysis of apoptosis
Cells were seeded on 6-well plates with 50,000 cells per well in 3 ml of RPMI 1640 medium supplemented with 10% FBS and 1% P/S. After 24 h incubation, the cells were treated with 6BIO or DMSO for 24 h or 48 h. After treatment, all cells were collected, and apoptotic cells were detected by flow cytometry using Annexin V-FITC Apoptosis Detection Kit (BD Biosciences) according to the supplier's instruction.
Western blot analysis
Cell lysates were prepared in RIPA buffer supplemented with inhibitors of proteases (Roche Diagnostics GmbH, Germany) and sodium orthovanadate, an inhibitor of phosphotases (Aldrich). Protein concentrations were determined using Biomate spectrometer (Thermo) and protein assay (Bio-Rad). 40 μg of each protein sample was resolved in 8% or 8-16% gradient SDS-PAGE gels (Pierce). After gel electrophoresis, the proteins were transferred to Hybond-C membranes (Amersham). The membranes were blocked in 5% non-fat milk in PBS containing 0.1% Tween 20 (PBST) at room temperature for 1-3 hours followed by an overnight incubation at 4 °C with primary antibodies in PBST containing 5% non-fat milk. The membranes were then washed with PBST and incubated with HRP-conjugated secondary antibody in 5% non-fat milk/PBST solution for 1-3 hours at room temperature, or overnight at 4 °C. Specific proteins were detected by exposure to X-ray film using Super Signal west pico chemiluminescent substrate or Super Signal west dura extended duration substrate (Pierce).
Kinase assay
Recombinant JAK kinases, substrates and 33P-labeled ATP were used for the in vitro kinase assays. The recombinant JAK catalytic domains were tagged with GST and purified from insect cells. The substrate was prepared in freshly prepared Base Reaction Buffer (20 mmol/L pH 7.5 Hepes, 10 mmol/LMgCl2, 1 mmol/L EGTA, 0.02% Brij-35, 0.02 mg/ml BSA, 0.1 mmol/L Na3VO4, 2 mmol/L DTT, 1% DMSO). Then required cofactors and kinase were added into the substrate solution. Compounds in DMSO were delivered into the kinase reaction mixture, and then 33P-labeled ATP (specific activity 0.01 μCi/μl final) was added into the reaction mixture to initiate the reaction. Kinase reaction mixture was incubated for 120 min at room temperature. Reactions were spotted onto P81 ion exchange paper (Whatman) for measurement of radioactivity.
In vivo therapeutic efficacy testing
BALB/c mice (at 6-8 weeks old) were purchased from National Cancer Institute for toxicity study. Immunodeficient NOD/SCID/IL2Rgamma null (NSG) mice (female at 6-8 weeks old) were purchased from The Jackson Laboratory for xenograft model. Animal experimental protocol was approved by the IACUC of the Beckman Research Institute at City of Hope Medical Center. A2058 human melanoma cells at 5×106 cells in serum free medium were inoculated subcutaneously into the dorsal area of NSG mice to create xenograft model. When tumors became palpable, 6BIO or vehicle control was administered via oral gavage once daily at 50 mg/kg body weight. Tumor growth was monitored every other day. Tumor volumes were measured every 3 to 4 days. Tumor volumes were calculated using the formula: 0.5 × (larger diameter) × (small diameter) 2.
Statistics
A two-sided t test was used to evaluate statistical significance of differences between treated and control groups. P < 0.05 was considered statistically significant.
Results
Bromoindirubin derivatives inhibit cell viability in human cancer cells
We screened the synthetic IRDs using MTS cell viability assays in A2058 cells, which are derived from metastatic lymph node melanoma. Two bromo-IRDs, MLS-2407 (6BIO) and MLS-2408, showed the most potent anti-cancer activity (Table 1). 6BIO is an IRD with double modifications of oxime- and bromo-groups, whereas MLS-2412 is an IRD with double modifications of oxime- and fluoro-groups at 3′- and 6-positions respectively. MLS-2446 and -2406 are IRDs with single modification of oxime-group or bromo-group at 3′-or 6-position. Among 6BIO, MLS-2446, -2406 and -2412, 6BIO decreased the viability of melanoma cells to 31% at 10 μmol/L, whereas MLS-2446, -2406, and -2412 showed marginal anti-cancer activities (Table 1), indicating that modifications with both 3′-oxime- and 6-bromo-groups are important for anti-cancer potency. 6BIO also inhibits cell viability in other human cancer cells (Supplementary Fig. S1). Thus, 6BIO was selected for further study.
Table 1.
Primary screening of synthetic IRDs in human melanoma cells
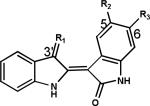
|
Compound | R1 | R2 | R3 | Cell Viability (%) |
| MLS-2402 | O | H | H | 109 ± 8 | |
| MLS-2410 | O | H |
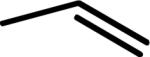
|
106 ± 3 | |
| MLS-2403 |
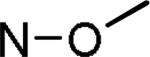
|
H | H | 102 ± 3 | |
| MLS-2446 | N-OH | H | H | 80 ± 5 | |
| MLS-2406 | O | H | Br | 93 ± 1 | |
| MLS-2414 | O | CH3 | Br | 92 ± 5 | |
| MLS-2417 | O | NO2 | Br | 116 ± 6 | |
| MLS-2407 | N-OH | H | Br | 31 ± 6 | |
| MLS-2418 | N-OH | NO2 | Br | 60 ± 4 | |
| MLS-2408 |
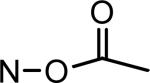
|
H | Br | 26 ±5 | |
| MLS-2419 |
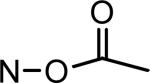
|
NO2 | Br | 59 ± 1 | |
| MLS-2409 | O | H | I | 115 ± 2 | |
| MLS-2411 | O | H | F | 108 ± 2 | |
| MLS-2412 | N-OH | H | F | 92 ± 2 | |
| MLS-2415 | O | Cl | Cl | 81 ± 3 | |
| MLS-2416 |
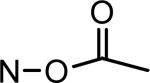
|
Cl | Cl | 89 ± 2 |
*MTS cell viability assays were performed in A2058 human melanoma cells with treatment of IRDs at 10 μmol/L for 48 h. Cell viability was determined as percentage of the vehicle control. MLS-2407 = 6BIO; MLS-2402 = indirubin.
6BIO inhibits viability of human melanoma cells, phosphorylation of STAT3 and its upstream tyrosine kinases
In primary screening assays, A2058 human melanoma cells were sensitive to 6BIO. We further compared the effects of 6BIO on A2058, G361, SK-MEL-5 and SK-MEL-28 human melanoma cell lines (Fig. 1). Cell viability was inhibited in all four melanoma cell lines. IC50 values are approximately 5 μmol/L for A2058 and G361 cells, and 12 μmol/L for SK-MEL-5 and SK-MEL-28 cells (Fig. 1A). Previous studies showed that human melanoma cells harbor persistently activated STAT3, which contributes to tumorigenesis and metastasis (6). Phosphorylation of JAK2, SRC and STAT3 was inhibited at 10 μmol/L of 6BIO 4 h after treatment in melanoma cells (Fig. 1B). Phosphorylation of SRC was inhibited in G361, SK-MEL-5 and SK-MEL-28 melanoma cell lines, whereas phosphorylation of JAK2 was inhibited in A2058 and G361 cell lines and only partially inhibited in SK-MEL-5 and SK-MEL-28 that are more resistant cell lines. Phosphorylation of SRC was not inhibited in A2058 human melanoma cells even though phosphorylation of JAK2 was substantially inhibited (Fig. 1B). Consistent inhibition of phosphorylation of JAK2 in all melanoma cell lines suggests that JAK family kinases may be relevant targets in melanoma cells.
Figure 1.

Effects of 6BIO on human melanoma cell lines. A, MTS cell viability assays were performed in A2058, G361, SK-MEL-5, and SK-MEL-28 human melanoma cells. Cells were treated with 6BIO at various concentrations for 48 h. B, human melanoma cells were treated with 10 μmol/L of 6BIO for 4 h. Cells were lysed for Western blot analysis using antibodies specific to p-Jak2, Jak2, p-Src, Src, p-Stat3, Stat3, and β-Actin.
6BIO inhibits phosphorylation of JAK family kinases in cells and JAK enzymatic activity in vitro
Human melanoma cells express JAK1, JAK2 and TYK2. We further investigated effects of 6BIO on JAK1, JAK2 and TYK2 in A2058, SK-MEL-5, SK-MEL-28, and G361 cells. In A2058 cells, 6BIO inhibited phosphorylation of JAK1, JAK2 and TYK2 in a dose-dependent manner at both 4 h (Fig. 2A) and 24 h (Fig. 2B) after treatment, and in a time-dependent manner (Fig. 2C). Inhibition of phosphorylation of JAK1, JAK2 and TYK2 was observed in A2058 cells as early as started at 5 min after treatment with 6BIO of 10 μmol/L (Supplementary Fig. S2A). Reduction of levels of phosphorylated JAK1, JAK2 and TYK2 was also detected in SK-MEL-5, SK-MEL-28, and G361 cells at 4 h after treatment with 6BIO of 10 μmol/L (Supplementary Fig. S2B). To determine whether 6BIO is a pan-JAK inhibitor, we further investigated the effect of 6BIO on JAK enzymatic activity in an in vitro kinase assay. Activities of JAK family kinases were inhibited in a dose-dependent manner. The IC50 values are 0.03 μmol/L for TYK2, 1.5 μmol/L for JAK1, 8.0 μmol/L for JAK2 (Fig. 2D), and 0.5 μmol/L for JAK3 (Supplementary Fig. S3). These findings suggest that 6BIO is a novel pan-JAK small-molecule inhibitor.
Figure 2.

6BIO inhibits phosphorylation of JAK family kinases in cells and JAK enzymatic activity in vitro. A, A2058 human melanoma cells were treated with 6BIO at various concentrations for 4 h. Cells were lysed for Western blot analysis using antibodies specific to p-Jak1, Jak1, p-Jak2, Jak2, p-Tyk2, Tyk2, and β-Actin . B, A2058 human melanoma cells were treated with 6BIO at various concentrations for 24 h. Cells were lysed for Western blot analysis using antibodies specific to p-Jak1, Jak1, p-Jak2, Jak2, p-Tyk2, Tyk2, and β-Actin. C, A2058 human melanoma cells were treated with 10 μmol/L of 6BIO for various lengths of time. Cells were lysed for Western blot analysis using antibodies specific to p-Jak1, Jak1, p-Jak2, Jak2, p-Tyk2, Tyk2, and β-Actin. D, in vitro kinase assays of JAK1, JAK2 and TYK2 were conducted using recombinant JAK proteins, substrates and 33P-labeled ATP in the presence of 6BIO at various concentrations. Radioactivity was measured for determination of kinase activity.
6BIO selectively inhibits phosphorylation of STAT3 in human melanoma cells
To investigate the selectivity of inhibition on JAK/STAT3 signaling, SRC and other two important signaling proteins, AKT and MAPK (Erk1/2) were examined. Western blots were performed to detect the levels of total and phosphorylated SRC, STAT3, AKT and MAPK (Erk1/2) (Fig. 3). In contrast to inhibition of phosphorylation of JAK family kinases (Fig. 2A), phosphorylation of SRC was not changed (Fig. 3A). Phosphorylation of STAT3 was decreased whereas phosphorylation of AKT and MAPK (Erk1/2) was not inhibited in all of the four human melanoma cell lines (Fig. 3). These observations suggest, among signaling pathways of STAT3, AKT and MAPK, 6BIO selectively inhibits JAK/STAT3 signaling in human melanoma cells.
Figure 3.

6BIO selectively inhibits phosphorylation of STAT3 in human melanoma cells. A, A2058 human melanoma cells were treated with 6BIO at various concentrations for 4 h. Cells were lysed for Western blot analysis using antibodies specific to p-Src, Src, p-Stat3, Stat3, p-Akt, Akt, p-Erk1/2, Erk1/2 and β-Actin. B, SK-MEL-5, SK-MEL-28 and G361 human melanoma cells were treated with 10 μmol/L of 6BIO for 4 h. Cells were lysed for Western blot analysis using antibodies specific to p-Akt, Akt, p-Erk1/2, Erk1/2 and β-Actin.
6BIO induces apoptosis of human melanoma cells associated with down-regulation of anti-apoptotic protein Mcl-1
STAT3 activation is involved in cell survival in human cancer cells (6). We examined STAT3 downstream anti-apoptotic proteins including Mcl-1, Bcl-2, Bcl-xL and Survivin. Expression of Mcl-1 was decreased in a dose-dependent manner in A2058 cells (Fig. 4A) and other human melanoma cell lines (Fig. 4B). Cleaved forms of PARP and caspase-3 were detected at 5 μmol/L 24 h after treatment. The levels of full length PARP and caspase-3 were decreased corresponding to the increased levels of cleaved forms (Fig. 4C). 6BIO induced apoptosis of human melanoma cells in both dose- and time-dependent manners (Fig. 4D). These results demonstrate that 6BIO induces melanoma cell apoptosis at least in part by inhibition of Mcl-1.
Figure 4.

6BIO induces apoptosis associated with down-regulation of anti-apoptotic protein Mcl-1. A, A2058 human melanoma cells were treated with 6BIO at various concentrations for 24 h. Cells were lysed for Western blot analysis using antibodies specific to Mcl-1, Bcl-2, Bcl-xL, Survivin and β-Actin. B, human melanoma SK-MEL-5, SK-MEL-28 and G361 cells were treated with 10 μmol/L of 6BIO for 24 h. Cells were lysed for Western blot analysis using antibodies specific to Mcl-1 and β-Actin. C, human melanoma A2058 cells were treated with 6BIO at various concentrations for 24 h. Cells were lysed for Western blot analysis using antibodies specific to PARP, Caspase-3 and β-Actin. D, A2058 cells were treated with 6BIO at various concentrations for 24 h or 48 h. Apoptosis was analyzed by flow cytometry using Annexin V-FITC staining.
6BIO suppresses melanoma tumor growth in a mouse xenograft model
Anti-cancer activity of 6BIO in vivo was studied with an A2058 human melanoma xenograft mouse model. We first tested the toxicity of 6BIO in BALB/c normal mice. 6BIO at the dose of 100 mg/kg was found safe to the mice (Fig. 5A). Then we used a dose of 50 mg/kg for 6BIO therapy study in NSG mouse xenograft model by oral administration once daily for 2 weeks. As shown in Figure 5B, the tumor growth was significantly suppressed. No side effects were observed in the 6BIO-treated mice. These findings demonstrate the anti-tumor activity of 6BIO in vivo against human melanoma cells.
Figure 5.

6BIO suppresses melanoma tumor growth in a mouse xenograft model. A, Toxicity study with BALB/c normal mice. 6BIO or vehicle control was administered through oral gavage once daily to each group, at doses of 50, 75 and 100 mg/kg body weight. The maximum tolerated dose (MTD) was determined as 100 mg/kg. B, 6BIO suppressed tumor growth of A2058 human melanoma xenografts in NSG mice. A2058 cells (5×106) were inoculated subcutaneously into the dorsal area of NSG mice. When tumors became palpable, 6BIO or vehicle control was administrated via oral gavage once daily. Points, mean (n = 8); bars, SE. P < 0.001.
Discussion
Indirubin has been extensively derivatized by chemical modifications in order to improve its selectivity profile, kinase inhibitory effect and solubility (26, 30). In particular, modifications have been carried out on positions 5 or 6 on one indole ring and 3′ on the other indole ring. The IRDs shown in Table 1 were synthesized with modifications at 3′-, 5- and 6-positions. 6BIO and MLS-2408 showed the most potent anti-cancer activity. These active bromo-IRDs contain a bromo-group at the 6-position and a hydrophilic group at the 3′-position. Our data show that either 3′- or 6-substitution alone as shown in MLS-2403 and -2446, or -2406, -2409 and -2411, is not sufficient to improve anti-cancer activity. Furthermore, 6BIO inhibited cell viability strongly whereas 6-fluoroindirubin-3′-oxime (6FIO, MLS-2412) is a very weak inhibitor of cell viability (Table 1), indicating bromo-substitution is more important than fluoro-substitution at the 6-position. Substitution at the 5-position with a nitro-group as in MLS-2418 and -2419 reduced anti-cancer activity, compared with 6BIO and MLS-2408. This finding suggests that modification at the 5-position with a nitro-group is unnecessary for anti-cancer activity of 6-bromoindirubins. Comparing the structures and activities of 6BIO, MLS-2446 and -2406, we found that both substitution of an oxime-group at the 3′-position and substitution of a bromo-group at the 6-positon are important to enhance anti-cancer activity.
It is interesting that 6BIO displays differential inhibition of JAK family kinase activity in an in vitro system using recombinant JAK kinases (Fig. 2D). 6BIO inhibits TYK2 most potently (IC50 value: 0.03 μmol/L), whereas the IC50 value is 8 μmol/L for JAK2 kinase activity. The IC50 value of JAK1 kinase activity is similar in vitro as in intact A2058 cells. The differential inhibition of phosphorylated JAKs in human melanoma cells is not as dramatic as the differential inhibition of JAK kinase activity in an in vitro system (Fig. 2A and D). It is striking that 6BIO inhibits phosphorylation of JAK2 strongly in A2058 cells, but it is a weak inhibitor of JAK2 kinase activity in vitro. We have found that 6BIO inhibits phosphorylation of JAK1, JAK2 and TYK2 in cells very effectively even though it is not a strong inhibitor for JAK2 in vitro. This finding is consistent with JAK trans-phosphorylation and reciprocal regulation of JAK kinases in cells. Previous studies showed that interaction between two members among JAK1, JAK2 and TYK2 regulates JAK kinase activity. In particular, it has been found that TYK2 is required for kinase activity of JAK2 and /or JAK1 (4, 36-40). The long term effect of 6BIO on A2058 human melanoma cells showed substantial differences between phosphorylated levels of JAK1, JAK2 and TYK2. Phosphorylation of JAK1 and TYK2 was most potently inhibited, whereas phosphorylation of JAK2 was least inhibited in a dose-dependent manner (Fig. 2B). This inhibition pattern is very similar to the inhibition of in vitro JAK kinase activity (Fig. 2D). Our findings suggest that TYK2 may regulate kinase activity of JAK2 and/or JAK1 in human melanoma cells. In the in vitro system, 6BIO is a strong inhibitor of TYK2, a moderate inhibitor of JAK1, but a weak inhibitor of JAK2. However, it is a pan-JAK inhibitor in intact A2058, SK-MEL-5, SK-MEL-28, and G361 human melanoma cells (Fig. 2, Supplementary Fig. S2B), suggesting reciprocal regulation among JAK1, JAK2 and TYK2 in cells.
As a pan-JAK inhibitor, 6BIO also inhibits phosphorylation of SRC in human melanoma cells. As shown in Figure 1B, phosphorylation of both JAK2 and SRC was inhibited in G361, SK-MEL-5, and SK-MEL-28 cells by 6BIO at 10 μmol/L 4 h after treatment, suggesting cooperation between JAK2 and SRC in G361, SK-MEL-5 and SK-MEL-28 cells to inhibit STAT3 signaling (41). In addition, in A2058 cells, phosphorylation of SRC was inhibited in a dose-dependent manner 24 h after treatment (Supplementary Fig. S2C), but it was not inhibited at 4 h after treatment (Fig. 3A). This observation may indicate a secondary inhibition of SRC by 6BIO in A2058 cells. As shown in Figure 3, comparing with phosphorylation of SRC, AKT and MAPK, phosphorylation of STAT3 was inhibited in a dose-dependent manner by 6BIO 4 h after treatment. This finding suggests the selective inhibition of JAK/STAT3 signaling mediated by 6BIO in human melanoma cells. However, we cannot conclude that 6BIO is an absolutely selective inhibitor of JAK/STAT3 signaling. Indirubin, an ATP-mimic, was originally identified as an inhibitor of CDKs. Later, indirubin and its derivatives have been found to inhibit other kinases, including GSK3-β and SRC. Similarly, we cannot rule out other possible targets of 6BIO such as CDKs, GSK3-β, FLT3, PDGFR, SRC and VEGF-R (31). In summary, we demonstrate that 6BIO is a pan-JAK inhibitor and inhibits the JAK/STAT3 signaling pathway, but we do not exclude inhibition of other pathways.
We have discovered that 6BIO induces apoptosis of human melanoma cells associated with inhibition of JAK/STAT3 signaling as a pan-JAK inhibitor. Two other important signaling proteins, AKT and MAPK, are not involved in the induction of apoptosis. SRC may be an indirect target of 6BIO. Our findings suggest TYK2 may regulate the kinase activity of other JAK family members such as JAK2 and / or JAK1. In addition, 6BIO is a strong inhibitor of JAK3 (Supplementary Fig. S3). Since JAK3 is exclusively expressed in hematopoietic cells, there are no further data regarding JAK3 in melanoma cells, although 6BIO may be a promising drug candidate for treatment of blood malignancies. Importantly, 6BIO suppresses tumor growth in an A2058 human melanoma xenograft mouse model. In summary, 6BIO is promising for development as a novel therapeutic agent targeting JAK/STAT3 signaling in melanoma.
Supplementary Material
Grant support
Supported in part by R01-CA115674 from NIH (R. Jove)
References
- 1.Yamaoka K, Saharinen P, Pesu M, Holt VE, 3rd, Silvennoinen O, O'Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5:253. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 3.O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–31. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 5.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 6.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 7.Gao SP, Bromberg JF. Touched and moved by STAT3. Sci STKE. 2006;2006:pe30. doi: 10.1126/stke.3432006pe30. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–27. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 11.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 12.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–42. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–20. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Verstovsek S. Therapeutic potential of JAK2 inhibitors. Hematology Am Soc Hematol Educ Program. 2009:636–42. doi: 10.1182/asheducation-2009.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayyah J, Sayeski PP. Jak2 inhibitors: rationale and role as therapeutic agents in hematologic malignancies. Curr Oncol Rep. 2009;11:117–24. doi: 10.1007/s11912-009-0018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–9. [PubMed] [Google Scholar]
- 17.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SM. Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity. Oncogene. 2005;24:3236–45. doi: 10.1038/sj.onc.1208470. [DOI] [PubMed] [Google Scholar]
- 19.Liby K, Voong N, Williams CR, Risingsong R, Royce DB, Honda T, et al. The synthetic triterpenoid CDDO-Imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin Cancer Res. 2006;12:4288–93. doi: 10.1158/1078-0432.CCR-06-0215. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)-->signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008;68:2920–6. doi: 10.1158/0008-5472.CAN-07-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Ma X, Yan S, Shen S, Zhu H, Gu Y, et al. 17-hydroxy-jolkinolide B inhibits signal transducers and activators of transcription 3 signaling by covalently cross-linking Janus kinases and induces apoptosis of human cancer cells. Cancer Res. 2009;69:7302–10. doi: 10.1158/0008-5472.CAN-09-0462. [DOI] [PubMed] [Google Scholar]
- 22.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoessel R, Leclerc S, Endicott JA, Nobel ME, Lawrie A, Tunnah P, et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol. 1999;1:60–7. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 24.Xiao Z, Hao Y, Liu B, Qian L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in China. Leuk Lymphoma. 2002;43:1763–8. doi: 10.1080/1042819021000006295. [DOI] [PubMed] [Google Scholar]
- 25.Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer's disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem. 2001;276:251–60. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 26.Eisenbrand G, Hippe F, Jakobs S, Muehlbeyer S. Molecular mechanisms of indirubin and its derivatives: novel anticancer molecules with their origin in traditional Chinese phytomedicine. J Cancer Res Clin Oncol. 2004;130:627–35. doi: 10.1007/s00432-004-0579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobs S, Merz KH, Vatter S, Eisenbrand G. Molecular targets of indirubins. Int J Clin Pharmacol Ther. 2005;43:592–4. doi: 10.5414/cpp43592. [DOI] [PubMed] [Google Scholar]
- 28.Nam S, Buettner R, Turkson J, Kim D, Cheng JQ, Muehlbeyer S, et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc Natl Acad Sci U S A. 2005;102:5998–6003. doi: 10.1073/pnas.0409467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meijer L, Guyard N, Skaltsounis AL, Eisenbrand G, editors. Life in Progress ed. Roscoff; France: 2006. Indirubin, the Red Shade of Indigo. [Google Scholar]
- 30.Polychronopoulos P, Magiatis P, Skaltsounis AL, Myrianthopoulos V, Mikros E, Tarricone A, et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J Med Chem. 2004;47:935–46. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 31.Ribas J, Bettayeb K, Ferandin Y, Knockaert M, Garrofe-Ochoa X, Totzke F, et al. 7-Bromoindirubin-3'-oxime induces caspase-independent cell death. Oncogene. 2006;25:6304–18. doi: 10.1038/sj.onc.1209648. [DOI] [PubMed] [Google Scholar]
- 32.Ferandin Y, Bettayeb K, Kritsanida M, Lozach O, Polychronopoulos P, Magiatis P, et al. 3'-Substituted 7-halogenoindirubins, a new class of cell death inducing agents. J Med Chem. 2006;49:4638–49. doi: 10.1021/jm060314i. [DOI] [PubMed] [Google Scholar]
- 33.Ribas J, Yuste VJ, Garrofe-Ochoa X, Meijer L, Esquerda JE, Boix J. 7-Bromoindirubin-3'-oxime uncovers a serine protease-mediated paradigm of necrotic cell death. Biochem Pharmacol. 2008;76:39–52. doi: 10.1016/j.bcp.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 34.Smalley KS, Herlyn M. Targeting intracellular signaling pathways as a novel strategy in melanoma therapeutics. Ann N Y Acad Sci. 2005;1059:16–25. doi: 10.1196/annals.1339.005. [DOI] [PubMed] [Google Scholar]
- 35.Garber K. Cancer research. Melanoma drug vindicates targeted approach. Science. 2009;326:1619. doi: 10.1126/science.326.5960.1619. [DOI] [PubMed] [Google Scholar]
- 36.Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kohlhuber F, Rogers NC, Watling D, Feng J, Guschin D, Briscoe J, et al. A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol Cell Biol. 1997;17:695–706. doi: 10.1128/mcb.17.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gauzzi MC, Velazquez L, McKendry R, Mogensen KE, Fellous M, Pellegrini S. Interferon-alpha-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J Biol Chem. 1996;271:20494–500. doi: 10.1074/jbc.271.34.20494. [DOI] [PubMed] [Google Scholar]
- 39.Rani MR, Leaman DW, Han Y, Leung S, Croze E, Fish EN, et al. Catalytically active TYK2 is essential for interferon-beta-mediated phosphorylation of STAT3 and interferon-alpha receptor-1 (IFNAR-1) but not for activation of phosphoinositol 3-kinase. J Biol Chem. 1999;274:32507–11. doi: 10.1074/jbc.274.45.32507. [DOI] [PubMed] [Google Scholar]
- 40.Potla R, Koeck T, Wegrzyn J, Cherukuri S, Shimoda K, Baker DP, et al. Tyk2 tyrosine kinase expression is required for the maintenance of mitochondrial respiration in primary pro-B lymphocytes. Mol Cell Biol. 2006;26:8562–71. doi: 10.1128/MCB.00497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Turkson J, Carter-Su C, Smithgall T, Levitzki A, Kraker A, et al. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J Biol Chem. 2000;275:24935–44. doi: 10.1074/jbc.M002383200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.