

Abstract
Synthetic multiple-target RNA and DNA oligonucleotides were constructed for use as quantification standards for nucleic acid amplification assays for human norovirus genogroup I and II, hepatitis E virus, murine norovirus, human adenovirus, porcine adenovirus and bovine polyomavirus. This approach overcomes the problems related to the difficulty of obtaining practical quantities of viral RNA and DNA from these viruses. The quantification capacity of assays using the standards was excellent in each case (R 2 > 0.998 and PCR efficiency > 0.89). The copy numbers of the standards were equivalent to the genome equivalents of representative viruses (murine norovirus and human adenovirus), ensuring an accurate determination of virus presence. The availability of these standards should facilitate the implementation of nucleic acid amplification-based methods for quantitative virus detection.
Keywords: Foodborne virus, Quantification, Nucleic acid standard, RT real-time PCR
Introduction
Molecular-based methods have become the gold standard for routine detection of viruses in food and environmental samples (Bosch et al. 2011; Croci et al. 2008). A realistic risk assessment strategy to assess the risks created by the contamination of food and the environment by enteric viruses will require a quantitative focus, and therefore accurate virus quantification is necessary. When a nucleic acid amplification-based method is applied for quantitative purposes, known concentrations of nucleic acids are used to construct calibration curves for quantification (Rodríguez-Lázaro et al. 2007). According to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (Bustin et al. 2009), nucleic acids used as standards for quantification can be of several types: purified synthetic RNA or DNA oligonucleotides spanning the complete PCR amplicon, plasmid DNA constructs, cDNA cloned into a plasmid, in vitro transcribed RNA, reference DNA pools and RNA or DNA from biological samples. However, important enteric pathogenic viruses such as human norovirus (hNoV) or hepatitis E virus (HEV) are not culturable in the laboratory (Lees and CEN WG6 TAG4 2010) and therefore, a source of viral nucleic acids necessary to make standard solutions can be very restricted, thus being more convenient to use a synthetic nucleic acid.
The application of synthetic nucleic acid molecules as standards for detection and quantification of organisms whose availability is scarce has been already applied successfully to a wide spectrum of organisms such as genetically modified plants (Hernández et al. 2005; Kuribara et al. 2002; Taverniers et al. 2004) and some pathogenic viruses (Kwiatek et al. 2010; Vester et al. 2010; Workenhe et al. 2008). A similar approach has not been followed for quantitative detection of non-culturable enteric viruses such as hNoV or HEV. However, as the public health implications of their presence in foodstuffs and the environment is becoming increasingly recognised (Anonymous 2008), and methods suitable for their routine monitoring are becoming available (Lees and CEN WG6 TAG4 2010), the availability of specific nucleic acid amplification standards is becoming necessary.
In the present work, we describe the construction of plasmids containing multiple-target sequences, which can be used as standards for quantification of enteric viruses (HEV, hNoV genogroups I and II, bovine polyomavirus—BPyV, human adenovirus—HAdV and porcine adenovirus—PAdV) by (reverse transcription) real-time PCR. The performance in quantitative reactions of these synthetic standards was assessed in comparison to natural nucleic acids extracted from viruses.
Materials and Methods
Viruses and Cell Cultures
Murine norovirus 1 (MNV-1) was propagated in RAW264.7 cells, and titrated by end-point dilution (final stock concentration 4.22 × 106 median tissue culture infective dose (TCID50) ml−1). Human adenovirus type-2 (HAdV-2) was propagated in A549 cells and titrated by the same technique (final stock concentration 2.1 × 107 TCID50 ml−1). Total viral RNA or DNA was extracted from infected cultures using QIAamp viral RNA mini kit (QIAGEN, GMBH, Inc., Hilden, Germany), following manufacturer’s instructions. MNV-1 was supplied by Prof. Herbert W. Virgin IV, Washington University School of Medicine, US according to the MTA signed within the EU project VITAL, and HAdV-2 was supplied by Prof. Rosina Girones, University of Barcelona, Spain.
Construction of a Plasmid for Transcription of Synthetic RNA
A synthetic DNA molecule was designed to contain target sequences for reverse transcription real-time PCR (RT-qPCR) assays for HEV (Jothikumar et al. 2006), hNoV GI (Svraka et al. 2007) and hNoV GII (da Silva et al. 2007). The oligonucleotide was synthesised (Eurofins MWG Operon, Ebersberg, Germany) and cloned into a pCR 2.1-TOPO plasmid (Invitrogen, Breda, The Netherlands). Then, the target sequence for a RT-qPCR assay for murine norovirus (MNV-1) (Baert et al. 2008) was added to the 3′ end of the plasmid. This was done by modifying the primers to add tails containing sites for XhoI (primer FwORF1/ORF2) and ApaI (primer RvORF1/ORF2). The final sequence was 424 bp (Fig. 1). The recombinant plasmid was designated as pCR2.1TOPO-rSTD.
Fig. 1.

Graphic representation of pCR2.1TOPO-rSTD containing the sequence of the synthetic rFBV1 RNA. The length of the plasmid pCR2.1TOPO-rSTD is 4295 bp. The viral insert was flanked by NotI and ApaI sites. The sequences of the RT-qPCR assays are shown (hNoV GII—within box, hNoV GI—italics, HEV—bold and MNV-1—underlined. The sequences corresponding to the TOPO vector are in normal type
Production of the Synthetic RNA
Six micrograms of pCR2.1TOPO-rSTD clone were linearised by digestion with HindIII enzyme (New England Biolabs, Ipswich, MA, USA) prior to in vitro transcription. The linearised plasmid was subsequently purified using the QIAquick PCR purification kit (QIAGEN, GMBH, Inc., Hilden, Germany) and in vitro transcribed using T7 RNA polymerase (Riboprobe in vitro transcription system, Promega, Madison, WI, USA) following the manufacturer’s instructions. As viral target DNA was reverse cloned in pCR-2.1 TOPO vector, transcription was performed in the antisense direction to generate a ssRNA(+). Residual DNA was removed by digestion with 35 U of RNase-free DNase contained in the kit. Subsequently, RNA purification was carried out using RNeasy kit (QIAGEN, GMBH, Inc., Hilden, Germany). Parallel qPCR and RT-qPCR assays for each virus verified that residual DNA had been removed (data not shown). To check the integrity of the RNA, an aliquot was electrophoresed in a native 1.5% agarose gel. To verify that the sequence of the insert was correct, direct sequencing using two pairs of flanking primers M13 Forward/M13 Reverse and FwORF1/ORF2/COG2R was performed using the kits Big Dye v3.1 or v1.1 (Applied Biosystems, Foster City, CA, USA) in an ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) following manufacturer’s instructions. Transcription reactions were repeated several times to obtain a higher amount of RNA, and after the integrity and purity was assessed, the RNA solutions were pooled, aliquoted in suitable volumes and stored at −80°C. The synthesised RNA molecule was designated rFBV1.
Construction of the Synthetic DNA
A synthetic DNA molecule was designed to contain target sequences for qPCR assays for BPyV (Hundesa et al. 2010), HAdV (Hernroth et al. 2002) and PAdV (Hundesa et al. 2009). The oligonucleotide was synthesised (Eurofins MWG Operon, Ebersberg, Germany) and cloned into a pCR 2.1-TOPO plasmid (Invitrogen, Breda, The Netherlands). The final sequence was 228 bp (Fig. 2). To verify that the sequence of the insert was correct, direct sequencing using the two flanking primers M13 Forward and M13 Reverse was performed as for synthetic RNA. The plasmid was cloned in E. coli, and a purified solution prepared using QIAGEN plasmid Midi kit (QIAGEN, GMBH, Inc., Hilden, Germany). The DNA solutions were pooled, aliquoted in suitable volumes and stored at −80°C. The synthesised DNA was designated as pFBV2.
Fig. 2.

Graphic representation of pFBV2 containing the sequence of the synthetic DNA. The length of the plasmid is 4,159 bp. The viral insert was flanked by ApaI and NotI sites. The sequences of the qPCR assays are shown (BPyV—bold, HAdV-2—italics and PAdV—underlined. The sequences corresponding to the TOPO vector are in normal type
Quantification of RNA and DNA
RNA and DNA concentrations were determined by UV spectrophotometry in a Nanodrop ND-1000 spectrophotometer (ThermoScientific, Wilmington, NC, USA). The measurements were performed in duplicate and concentration in g was converted to molecule number using the following formulae:
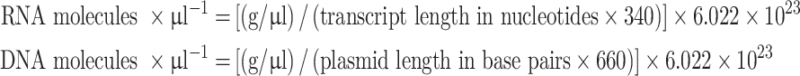 |
RT-qPCRs and qPCRs
All RT-qPCRs and qPCRs were run in an LC 480 II instrument (Roche, Mannheim, Germany). For RT-qPCR assays, 5 μl of the RNA solution were added to 15 μl of master mix consisting of 7.4 μl Light Cycler 480 Master Hydrolysis probes (Roche, Mannheim, Germany), and 1.3 μl activator (50 mM). Primers and probes concentrations were those described before for MNV-1 (Baert et al. 2008) and HEV (Jothikumar et al. 2006). For hNoV GI and GII assays, forward primer was added at a concentration of 500 nM, reverse primer at 900 nM and probe at 250 nM. Cycling conditions consisted of 63°C for 30 min followed by denaturation at 95°C for 5 min, and 45 cycles of denaturation at 95°C for 15 s, and annealing, amplification and detection at 60°C for 1 min. For HEV, cycling conditions were 63°C for 30 min followed by denaturation at 95°C for 5 min 45 cycles of 10 s at 95°C, annealing at 55°C for 20 s and amplification and detection at 72°C for 15 s. For MNV-1, cycling conditions were 63°C for 30 min followed by denaturation at 95°C for 5 min and 40 cycles of denaturation at 95°C for 15 s and annealing, amplification and detection at 60°C for 1 min.
For qPCR assays, 5 μl of the DNA solution were added to 20 μl of master mix consisting of 12.5 μl Light Cycler 480 Probes Master (Roche, Mannheim, Germany). Primers and probes concentrations were those described previously (Hernroth et al. 2002; Hundesa et al. 2010; Hundesa et al. 2009). Cycling conditions for HAdV and PAdV assays consisted of denaturation at 95°C for 10 min, and 45 cycles of denaturation at 95°C for 15 s, and annealing, amplification and detection at 60°C for 1 min. Cycling conditions for BPyV were denaturation at 95°C for 10 min followed by 45 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 1 min.
Results
Evaluation of the Performance of the RT-qPCR Assays Using Synthesised RNA
The results of the performance of the four RT-qPCR assays (hNoVGI, hNoVGII, HEV and MNV-1) using tenfold dilutions (from 106 to 101 RNA molecules) of the rFVB1 are shown in Table 1. The capacity for quantification using rFVB1 for each RT-qPCR assay was also calculated based on the linearity and PCR efficiency (E) (Rodríguez-Lazaro et al. 2005). Both parameters were very close to the optimal in all RT-qPCR assays (R 2 ≥ 0.998 and E ≥ 0.89; Table 1) demonstrating that the use of the synthetic RNA for constructing standard quantification curves is an excellent approach. The limits of quantification (LOQ) were 1 × 101 rFVB1 copies per reaction in all the assays, with the exception of hNoV GII and HEV where the LOQ was 1 × 102 copies per reaction (Table 1). In addition, reliable quantification was possible over a dynamic range at least of five logs.
Table 1.
Quantitative detection of synthetic (hNoVGI, hNoVGII, HEV and MNV-1) and native MNV-1 RNA
| Approx. molecules/reactiona | Synthetic RNA | Virus RNA | |||
|---|---|---|---|---|---|
| hNoVGI b | hNoVGIIc | HEVd | MNV-1e | MNV-1f | |
| C p valueg | C p value | C p value | C p valueg | C p valueg | |
| 1 × 106 | 14.75 ± 0.24 | 12.45 ± 0.07 | 16.61 ± 0.18 | 15.57 ± 0.04 | 15.59 ± 0.02 |
| 1 × 105 | 17.91 ± 0.15 | 15.89 ± 0.07 | 19.80 ± 0.15 | 19.56 ± 0.04 | 19.32 ± 0.04 |
| 1 × 104 | 21.68 ± 0.30 | 19.35 ± 0.13 | 23.35 ± 0.16 | 23.30 ± 0.04 | 22.95 ± 0.03 |
| 1 × 103 | 25.01 ± 0.28 | 23.11 ± 0.15 | 26.71 ± 0.21 | 27.07 ± 0.05 | 26.65 ± 0.02 |
| 1 × 102 | 28.47 ± 0.37 | 26.92 ± 0.20 | 29.54 ± 0.30 | 30.30 ± 0.03 | 29.85 ± 0.01 |
| 1 × 101 | 32.23 ± 0.45 | 35.56 ± 0.25 | ndh | 33.74 ± 0.04 | 33.09 ± 0.03 |
aEstimated number of synthetic rFBV1 or virus RNA molecules in each RT-qPCR run
bhNoVGI RT-qPCR results from tenfold serial dilutions of synthetic RNA rFBV1. The standard curve was: y = −3.497x + 35.58; and the R 2 and PCR efficiency values were 0.999 and 0.93, respectively
chNoVGII RT-qPCR results from tenfold serial dilutions of synthetic RNA rFBV1. The standard curve was: y = −3.616x + 34.00; and the R 2 and PCR efficiency values were 0.999 and 0.89, respectively
dHEV RT-qPCR results from tenfold serial dilutions of synthetic RNA rFBV1. The standard curve was: y = −3.277x + 36.31; and the R 2 and PCR efficiency values were 0.998 and 1.02, respectively
eMNV-1 RT-qPCR results from tenfold serial dilutions of synthetic RNA rFBV1. The standard curve was: y = −3.624x + 37.60; and the R 2 and PCR efficiency values were 0.998 and 0.89, respectively
fMNV-1 RT-qPCR results from tenfold serial dilutions of RNA purified from MNV-1. The standard curve was: y = −3.508x + 36.85; and the R 2 and PCR efficiency values were 0.998 and 0.93, respectively
gCycle number at which fluorescence intensity equals a fixed threshold. Mean value ± standard error of the mean. The experimental results were statistically significant (P < 0.05) taking into account unavoidable error associated with serial dilutions
hNot detected
The performance of the RT-qPCR assays using tenfold dilutions of rFVB1 were also compared to those assays using tenfold dilutions of native RNA from MNV-1 extracted from infected cells (from 106 to 101 RNA molecules). The performances were very similar, as the linearity and PCR efficiency values were very similar (R 2 of 0.998 and E of 0.93 and 0.89 for native and synthetic MNV-1 RNA, respectively) (Table 1; Fig. 3a). When the C p (cycle to positivity) values obtained using each type of RNA were plotted in a graphic (synthetic RNA C p values vs. MNV-1 RNA C p values) an excellent correlation (slope of 0.967 and R 2 of 0.999) was found (Fig. 3b).
Fig. 3.

Comparison of the performance of the RT-qPCR assays using native and synthetic RNA. a Standard curve generated by tenfold dilutions (from 106 to 101 RNA molecules) of rFVB1 (triangle) and native RNA from MNV-1 extracted from infected cells (square). b Representation of the equivalence of the C p values of tenfold dilutions (from 106 to 101 RNA molecules) of rFVB1 (y-axis) and MNV-1 (x-axis)
Evaluation of the Performance of the qPCR Assays Using Synthetic DNA
The results of the performance of the three qPCR assays (BPyV, PAdV and HAdV) using tenfold dilutions (from 105 to 101 DNA molecules) of the pFBV2 are shown in Table 2. Similarly to the RT-qPCR assays, the capacity for quantification was also calculated based on the linearity and PCR efficiency. Both parameters were very close to the optimal in all qPCR assays (R 2 ≥ 0.96 and E ≥ 0.997; Table 2) demonstrating that the use of the synthetic DNA for constructing standard quantification curves is an excellent approach. The limits of quantification (LOQ) were 1 × 101 pFBV2 copies per reaction in all the assays (Table 2). In addition, reliable quantification was possible over a dynamic range at least of five logs.
Table 2.
Quantitative detection of synthetic (PAdV, BPyV and HAdV) and native HAdV DNA
| Approx. molecules/reactiona | Synthetic DNA | Virus DNA | ||
|---|---|---|---|---|
| PAdVb | BPyVc | HAdVd | HAdVe | |
| C p valuef | C p valuef | C p valuef | C p valuef | |
| 1 × 105 | 22.46 ± 0.10 | 23.96 ± 0.07 | 22.08 ± 0.04 | 22.15 ± 0.02 |
| 1 × 104 | 25.83 ± 0.13 | 27.61 ± 0.12 | 25.49 ± 0.04 | 25.49 ± 0.04 |
| 1 × 103 | 29.29 ± 0.11 | 30.98 ± 0.12 | 28.84 ± 0.05 | 28.92 ± 0.03 |
| 1 × 102 | 32.32 ± 0.07 | 34.23 ± 0.04 | 32.11 ± 0.05 | 32.06 ± 0.02 |
| 1 × 101 | 35.95 ± 0.09 | 36.81 ± 0.23 | 35.92 ± 0.10 | 35.44 ± 0.06 |
aEstimated number synthetic pFBV2 or virus DNA molecules in each qPCR run
bPAdV qPCR results from tenfold serial dilutions of synthetic DNA pFBV2. The standard curve was: y = −3.347x + 42.55; and the R 2 and PCR efficiency values were 0.999 and 0.99, respectively
cBPyV qPCR results from tenfold serial dilutions of DNA pFBV2. The standard curve was: y = −3.232x + 43.64; and the R 2 and PCR efficiency values were 0.999 and 1.04, respectively
dHAdV qPCR results from tenfold serial dilutions of DNA pFBV2. The standard curve was: y = −3.430x + 42.60; and the R 2 and PCR efficiency values were 0.999 and 1.02, respectively
eHAdV qPCR results from tenfold serial dilutions of DNA purified from HAdV-2. The standard curve was: y = −3.315x + 42.07; and the R 2 and PCR efficiency values were 0.999 and 1.00, respectively
fCycle number at which fluorescence intensity equals a fixed threshold (Rodríguez-Lázaro et al. 2003). Mean value ± standard error of the mean. The experimental results were statistically significant (P < 0.05) taking into account unavoidable error associated with serial dilutions
The performance of the qPCR assays using tenfold dilutions of pFBV2 were also compared to those assays using tenfold dilutions of native DNA from HAdV-2 extracted from infected cells (from 105 to 101 DNA molecules). The performances were very similar, as the linearity and PCR efficiency values were very similar (R 2 of 0.999 and E of 1.00 and 0.96 for native and synthetic HAdV-2 DNA, respectively) (Table 2; Fig. 4a). When the C p values obtained using each type of DNA were plotted in a graphic (synthetic DNA C p values vs. HAdV-2 DNA C p values) an excellent correlation (slope of the curve of 0.965 and R 2 value of 0.999) was found (Fig. 4b).
Fig. 4.

Comparison of the performance of the qPCR assays using native and synthetic DNA. a Standard curve generated by tenfold dilutions (from 106 to 101 DNA molecules) of pFVB2 (triangle) and native DNA from HAdV-2 extracted from infected cells (square). b Representation of the equivalence of the C p values of tenfold dilutions (from 106 to 101 DNA molecules) of pFVB2 (y-axis) and HAdV-2 (x-axis)
Discussion
Accurate quantification of viruses is important to determine not only the level of contamination of food, surfaces, waters, etc., but also to determine any reduction of virus contamination after disinfection treatments. It can also be used to determine a possible linkage of virus levels to risk of infection or outbreaks (Lees and CEN WG6 TAG4 2010). Probably because of the difficulty of obtaining suitable RNA, several previously published methods have used DNA containing virus-complementary sequences as quantification standards. However, this approach is far from optimal, as the reverse transcription step is thus not considered (Boeuf et al. 2005; Terlizzi et al. 2010; Vester et al. 2010; Workenhe et al. 2008).
Absolute quantification will be reliable only if the standard and the unknown samples are retrotranscribed (only for RNA molecules) and amplified with the same efficiency (Boeuf et al. 2005). So standard curves obtained after amplification of tenfold serial dilutions of purified viral RNA or DNA and external RNA or DNA standards were compared (Tables 1, 2; Figs. 1, 2). RNA transcribed from linearised plasmid pCR2.1TOPO-rSTD was found to give more reliable viral RNA copy number estimation than RNA transcribed from circular pCR2.1TOPO-rSTD (data not shown). It has been suggested that an acceptable RT-qPCR standard curve should have a correlation coefficient (R 2) ≥ 0.98 and a slope value (s) between 3.6 and 3.1, corresponding to reaction efficiencies (E) between 0.9 and 1.1 (La Rosa et al. 2010). For rFBV1 and pFBV2 standards, all R 2 and E values conformed to these acceptable limits. Any observed differences in the capacity for quantification of the individual assays using the standards were slight, and can be attributed to the effect of variations in the individual nucleic acid sequences (Boeuf et al. 2005).
The quantification of the DNA viruses was more efficient than the quantification of the RNA viruses (Tables 1, 2). This is probably due to the nature of RNA, and also to the additional RT step, as its efficiency depends on many factors (Levesque-Sergerie et al. 2007). Finally, and most importantly, the copy numbers of rFBV1 are equivalent to the genome equivalents of MNV-1, and the copy numbers of pFBV2 are equivalent to the genome equivalents of HAdV-2. This relationship is confidently expected to pertain also to the other virus species presented in these standards. Thus, when using these standards in monitoring a food or environmental matrix for the viruses, the analyst can be confident that the determination of virus presence he/she obtains is accurate. Thus, the availability of these standards should facilitate the implementation of nucleic acid amplification-based methods for quantitative virus detection.
Acknowledgments
The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. KBBE-213178 (VITAL project). M.D.-V. received a Ph.D. studentship from the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA). The authors thank Dr. Nigel Cook for the critical review of the ms.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Mónica Martínez-Martínez and Marta Diez-Valcarce have contributed equally this work.
Contributor Information
Marta Hernández, Phone: +34-983-415287, FAX: +34-983-410462, Email: ita-herperma@itacyl.es.
David Rodríguez-Lázaro, Phone: +34-983-317383, FAX: +34-983-414780, Email: ita-rodlazda@itacyl.es.
References
- Anonymous. (2008). Viruses in food: Scientific advice to support risk management activities. WHO-FAO Microbiological Risk Assessment Series No. 13. Rome. 79 pp.
- Baert L, Wobus CE, Van Coillie E, Thackray LB, Debevere J, Uyttendaele M. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Applied and Environmental Microbiology. 2008;74:543–546. doi: 10.1128/AEM.01039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeuf P, Vigan-Womas I, Jublot D, Loizon S, Barale JC, Akanmori BD, et al. CyProQuant-PCR: A real time RT-PCR technique for profiling human cytokines, based on external RNA standards, readily automatable for clinical use. BMC Immunology. 2005;6:5. doi: 10.1186/1471-2172-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A, Sanchez G, Abbaszadegan M, Carducci A, Guix S, Le Guyader FS, Netshikweta R, Pintó RM, van der Poel W, Rutjes S, Sano D, Rodríguez-Lázaro D, Kovac K, Taylor MB, van Zyl W, Sellwood J. Analytical methods for virus detection in water and food. Food Analytical Methods. 2011;4:4–13. doi: 10.1007/s12161-010-9161-5. [DOI] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Croci L, Dubois E, Cook N, de Medici D, Schultz AC, China B, Rutjes SA, Hoorfar J, Van der Poel WHM. Current methods for extraction and concentration of enteric viruses from fresh fruit and vegetables: Towards international standards. Food Analytical Methods. 2008;1:73–84. doi: 10.1007/s12161-008-9025-4. [DOI] [Google Scholar]
- da Silva AK, Le Saux JC, Parnaudeau S, Pommepuy M, Elimelech M, Le Guyader FS. Evaluation of removal of noroviruses during wastewater treatment, using real-time reverse transcription-PCR: Different behaviors of genogroups I and II. Applied and Environmental Microbiology. 2007;73:7891–7897. doi: 10.1128/AEM.01428-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández M, Rodríguez-Lázaro D, Ferrando A. Current methodology for detection, identification and quantification of genetically modified organisms. Current Analytical Chemistry. 2005;1:203–221. doi: 10.2174/1573411054021574. [DOI] [Google Scholar]
- Hernroth BE, Conden-Hansson AC, Rehnstam-Holm AS, Girones R, Allard AK. Environmental factors influencing human viral pathogens and their potential indicator organisms in the blue mussel, Mytilus edulis: The first Scandinavian report. Applied and Environmental Microbiology. 2002;68:4523–4533. doi: 10.1128/AEM.68.9.4523-4533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundesa A, Bofill-Mas S, de Maluquer Motes C, Rodriguez-Manzano J, Bach A, Casas M, et al. Development of a quantitative PCR assay for the quantitation of bovine polyomavirus as a microbial source-tracking tool. Journal of Virological Methods. 2010;163:385–389. doi: 10.1016/j.jviromet.2009.10.029. [DOI] [PubMed] [Google Scholar]
- Hundesa A, de Maluquer Motes C, Albinana-Gimenez N, Rodriguez-Manzano J, Bofill-Mas S, Sunen E, et al. Development of a qPCR assay for the quantification of porcine adenoviruses as an MST tool for swine fecal contamination in the environment. Journal of Virological Methods. 2009;158:130–135. doi: 10.1016/j.jviromet.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Jothikumar N, Cromeans TL, Robertson BH, Meng XJ, Hill VR. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. Journal of Virological Methods. 2006;131:65–71. doi: 10.1016/j.jviromet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Kuribara H, Shindo Y, Matsuoka T, Takubo K, Futo S, Aoki N, et al. Novel reference molecules for quantitation of genetically modified maize and soybean. Journal of AOAC International. 2002;85:1077–1089. [PubMed] [Google Scholar]
- Kwiatek O, Keita D, Gil P, Fernandez-Pinero J, Jimenez Clavero MA, Albina E, et al. Quantitative one-step real-time RT-PCR for the fast detection of the four genotypes of PPRV. Journal of Virological Methods. 2010;165:168–177. doi: 10.1016/j.jviromet.2010.01.014. [DOI] [PubMed] [Google Scholar]
- La Rosa G, Pourshaban M, Iaconelli M, Muscillo M. Quantitative real-time PCR of enteric viruses in influent and effluent samples from wastewater treatment plants in Italy. Annali dell’Istituto Superiore di Sanita. 2010;46:266–273. doi: 10.4415/ANN_10_03_07. [DOI] [PubMed] [Google Scholar]
- Lees D, CEN WG6 TAG4 International standardisation of a Method for detection of human pathogenic viruses in molluscan shellfish. Food and Environmental Virology. 2010;2:146–155. doi: 10.1007/s12560-010-9042-5. [DOI] [Google Scholar]
- Levesque-Sergerie JP, Duquette M, Thibault C, Delbecchi L, Bissonnette N. Detection limits of several commercial reverse transcriptase enzymes: impact on the low- and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Molecular Biology. 2007;8:93. doi: 10.1186/1471-2199-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Lázaro D, Hernández M, Esteve T, Hoorfar J, Pla M. A rapid and direct real time PCR-based method for identification of Salmonella spp. Journal of Microbiological Methods. 2003;54:381–390. doi: 10.1016/S0167-7012(03)00071-X. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lázaro D, Lombard B, Smith H, Rzezutka A, D’ Agostino M, Helmuth R, Schroeter A, Malorny B, Miko A, Guerra B, Davison J, Kobilinsky A, Hernández M, Bertheau Y, Cook N. Trends in analytical methodology in food safety and quality: Monitoring microorganisms and genetically modified organisms. Trends in Food Sciences and Technology. 2007;18:306–319. doi: 10.1016/j.tifs.2007.01.009. [DOI] [Google Scholar]
- Rodríguez-Lazaro D, Pla M, Scortti M, Monzo HJ, Vazquez-Boland JA. A novel real-time PCR for Listeria monocytogenes that monitors analytical performance via an internal amplification control. Applied Environmental Microbiology. 2005;71:9008–9012. doi: 10.1128/AEM.71.12.9008-9012.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svraka S, Duizer E, Vennema H, de Bruin E, van der Veer B, Dorresteijn B, et al. Etiological role of viruses in outbreaks of acute gastroenteritis in The Netherlands from 1994 through 2005. Journal of Clinical Microbiology. 2007;45:1389–1394. doi: 10.1128/JCM.02305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverniers I, Van Bockstaele E, De Loose M. Cloned plasmid DNA fragments as calibrators for controlling GMOs: different real-time duplex quantitative PCR methods. Analytical and Bioanalytical Chemistry. 2004;378:1198–1207. doi: 10.1007/s00216-003-2372-5. [DOI] [PubMed] [Google Scholar]
- Terlizzi, M. E., Bergallo, M., Astegiano, S., Sidoti, F., Gambarino, S., Solidoro, P., Costa, C., et al. (2010). Improvement of HRV quantification using cRNA-based standards for real time RT-PCR. Molecular Biotechnology. doi:10.1007/s12033-010-9343-9. [DOI] [PubMed]
- Vester D, Lagoda A, Hoffmann D, Seitz C, Heldt S, Bettenbrock K, et al. Real-time RT-qPCR assay for the analysis of human influenza A virus transcription and replication dynamics. Journal of Virological Methods. 2010;168:63–71. doi: 10.1016/j.jviromet.2010.04.017. [DOI] [PubMed] [Google Scholar]
- Workenhe ST, Kibenge MJ, Iwamoto T, Kibenge FS. Absolute quantitation of infectious salmon anaemia virus using different real-time reverse transcription PCR chemistries. Journal of Virological Methods. 2008;154:128–134. doi: 10.1016/j.jviromet.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]