

Abstract
The principal effect of TGF-β1 on mesenchymal cells is its stimulation of ECM synthesis. Previous reports indicated the significance of the autocrine TGF-β loop in the pathogenesis of scleroderma. In this study, we focused on Smad7 and Smurfs, principal molecules in the negative regulation of TGF-β signaling, to further understand the autocrine TGF-β loop in scleroderma. Scleroderma fibroblasts exhibited increased Smad7 levels compared with normal fibroblasts in vivo and in vitro. Smad7 constitutively formed a complex with the TGF-β receptors, and the inhibitory effect of Smad7 on the promoter activity of human α2(I) collagen and 3TP-lux was completely impaired in scleroderma fibroblasts. Furthermore, the protein stability of TGF-β receptor type I was significantly increased in scleroderma fibroblasts compared with normal fibroblasts. There was no significant difference in Smurf1 and Smurf2 levels between normal and scleroderma fibroblasts, and the transiently overexpressed Smurf1 and/or Smurf2 did not affect TGF-β receptor type I protein levels in scleroderma fibroblasts. These results indicate that the impaired Smad7-Smurf–mediated inhibitory effect on TGF-β signaling might contribute to maintaining the autocrine TGF-β loop in scleroderma fibroblasts. To our knowledge, this is the first report of a disturbed negative regulation of TGF-β signaling in fibrotic disorders.
Introduction
Systemic sclerosis or scleroderma is an acquired disorder that typically results in fibrosis of the skin and internal organs (1). Although the pathogenesis of this disease is unclear, it includes inflammation, autoimmune attack, and vascular damage, leading to the activation of fibroblasts (2, 3). The reason for the presence of abnormal fibroblasts in scleroderma is not yet known, but it is possible that they develop from a subset of cells that have escaped normal control mechanisms (4, 5).
TGF-β1 is a multifunctional cytokine that regulates the growth, differentiation, and function of various cell types (6). The principal effect of TGF-β1 on mesenchymal cells is its stimulation of ECM deposition. TGF-β1 has been shown to increase the expression of collagen types I, III, VI, VII, and X, fibronectin, and proteoglycans (4, 7–12). Stimulation of ECM production by TGF-β1 is further enhanced by its inhibitory effect on matrix degradation, decreasing the synthesis of proteases and increasing levels of protease inhibitors (13). Although the mechanism of dermal fibroblast activation in scleroderma is presently unknown, many of the characteristics of scleroderma fibroblasts resemble those of healthy fibroblasts stimulated by TGF-β1 (13, 14), suggesting that the dermal fibroblast activation in scleroderma may be a result of stimulation by autocrine TGF-β. This notion is supported by our recent findings that (a) scleroderma fibroblasts express elevated levels of TGF-β receptors, and this correlates with elevated levels of α2(I) collagen mRNA (15); and (b) the blockade of TGF-β signaling with anti–TGF-β Ab’s or anti–TGF-β1 antisense oligonucleotide abolished the increased expression of human α2(I) collagen mRNA in scleroderma fibroblasts (16).
TGF-β1 initiates signaling through the ligand-dependent activation of a complex of heterodimeric transmembrane serine and threonine kinases consisting of TGF-β receptor type I (TβRI) and TGF-β receptor type II (TβRII). Upon binding TGF-β, the receptors rotate within the complex, resulting in the phosphorylation and activation of TβRI by the constitutively active and autophosphorylated TβRII. TGF-β1 signals from the receptor to the nucleus using a set of Smads proteins. The activated TβRI directly phosphorylates Smad2 and Smad3. Once activated, Smad2 and Smad3 associate with Smad4 and translocate to the nucleus, where such a complex regulates transcriptional responses of target genes together with additional DNA-binding cofactors (17). The inhibitory Smad7 associates with ligand-activated TβRI and interferes with the phosphorylation of Smad2 and Smad3 by preventing their interaction with activated TβRI (18). Since the expression of Smad7 is induced by TGF-β1, Smad7 inhibits TGF-β signaling by a negative feedback system (19, 20). Recently, Smad7 has been shown to function as an adaptor protein that recruits ubiquitin ligases, termed Smurfs, to the TGF-β receptor complex to promote its degradation through proteasomal and lysosomal pathways (21, 22). Thus, Smad7 plays a crucial role in a negative feedback loop to control TGF-β activity.
Recently, the marked overexpression of Smad7 was reported in the inflamed intestine of patients with inflammatory bowel disease. Furthermore, blocking Smad7 with a specific antisense oligonucleotide restores TGF-β signaling and, importantly, allows TGF-β to inhibit proinflammatory cytokine production (23). This indicates the importance of Smad7 in the pathogenesis of inflammatory disorders and suggests that Smad7 could be a target for the treatment of disorders with disturbed TGF-β signaling.
This study was undertaken to clarify the role of Smad7 and Smurfs in the pathogenesis of scleroderma. First, we compared the expression levels of Smad7 in normal and scleroderma fibroblasts. We then investigated the subcellular localization of Smad7 and the inhibitory effect of this protein on human α2(I) collagen promoter activity and 3TP-lux promoter activity in scleroderma fibroblasts. Furthermore, we compared the protein stability of TβRI between normal and scleroderma fibroblasts and investigated the effect of transiently overexpressed Smurfs on the levels of TβRI protein in scleroderma fibroblasts. The results suggest that the impaired inhibitory effect of Smad7 and Smurfs on TGF-β signaling might contribute to maintaining the autocrine TGF-β loop in scleroderma fibroblasts. To our knowledge, this is the first report of a disturbed negative regulation of TGF-β signaling in fibrotic disorders.
Methods
Reagents.
Ab’s for Smad2/Smad3, Smad4, Smad7, TβRI, TβRII, Smurf1, and Smurf2 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, California, USA). Ab for phospho-Smad2 was purchased from Upstate Biotechnology Inc. (Lake Placid, New York, USA). Ab’s for β-actin and Flag were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Recombinant human TGF-β1 was purchased from R&D Systems Inc. (Minneapolis, Minnesota, USA).
Cell cultures.
Human dermal fibroblasts were obtained by skin biopsy from the affected areas (dorsal forearm) of eight patients with diffuse cutaneous scleroderma and less than 2 years of skin thickening (16). Control fibroblasts were obtained by skin biopsy from eight healthy donors. Institutional approval and informed consent were obtained from all subjects. Control donors were matched with each scleroderma patient for age, sex, and biopsy site, and control and patient samples were processed in parallel. Primary explant cultures were established in 25-cm2 culture flasks in MEM with 10% FCS, 2 mM L-glutamine, and 50 μg/ml of amphotericin. Fibroblast cultures independently isolated from different individuals were maintained as monolayers at 37°C in 95% air and 5% CO2 and were studied between the third and sixth subpassages. The cell line Mv1Lu was purchased from American Type Culture Collection (Manassas, Virginia, USA) and maintained as described above.
Cell lysis and immunoblotting.
Fibroblasts were grown to subconfluence in MEM with 10% FCS. After 24 hours of serum starvation, cells were solubilized in lysis buffer (1% Triton X-100 in 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 3 mM MgCl2, 1 mM CaCl2 containing 10 μg/ml of leupeptin, pepstatin, and aprotinin, 1 mM PMSF, and 1 mM Na3VO4). Each lysate normalized for protein content was subjected to SDS-PAGE and transferred to nitrocellulose membranes (24). Membranes were probed with the primary Ab’s, washed, and incubated for 60 minutes with HRP-conjugated secondary Ab’s. After washing, visualization was performed using ECL (Amersham Biosciences, Buckinghamshire, United Kingdom). The densities of bands were measured with a densitometer.
RNA preparation and Northern blot analysis.
Fibroblasts were grown to subconfluence as described above and then incubated for 24 hours in serum-free medium before addition of the appropriate reagent. Total RNA was extracted using an acid guanidinium thiocyanate-phenol-chloroform method (25, 26). Poly(A)+ RNA was extracted from total RNA using an oligotex TM-dT30 Super kit (Takara Shuzo Co., Kyoto, Japan) and analyzed by Northern blotting (25, 26). Two micrograms of poly(A)+ RNA was subjected to electrophoresis on 1% agarose/formaldehyde gels and blotted onto a nylon filter. The filter was UV cross-linked, prehybridized, and hybridized with a DNA probe for GAPDH or an RNA probe for Smad7. The membrane was then washed and exposed to x-ray film. The x-ray film was scanned as described above.
Treatment of dermal fibroblasts with anti–TGF-β Ab’s or antisense TGF-β1 oligonucleotide.
We used a panspecific neutralizing TGF-β Ab (R&D Systems Inc.), which has been shown to inhibit TGF-β activity, specifically. We also used a TGF-β1 19-mer antisense oligonucleotide (GAGGGCGGCATGGGGAGG), which overlaps the promoter and transcriptional start site of the TGF-β1 gene. This same sequence, which is specific for the TGF-β1 isoform, has been found to be sufficient to block TGF-β1 transcription in vitro (27) and in vivo (28). A sense oligonucleotide served as a control. Fibroblasts were grown to confluence and the culture medium was removed, washed with MEM to remove excess TGF-β1, and replaced with serum-free MEM. Various amounts of TGF-β Ab’s or antisense or sense TGF-β1 oligonucleotide were added, and RNA isolation was performed 48 hours later.
Immunohistochemical staining.
Immunohistochemical staining was performed using a VectaStain ABC kit (Vector Laboratories, Burlingame, California, USA) (29). Two-micrometer thick paraffin-embedded sections were mounted on slides, deparaffinized with xylene, and rehydrated through a graded series of ethyl alcohol and PBS. The sections were then incubated with anti-Smad7 Ab overnight at 4°C. The immunoreactivity was visualized using diaminobenzidine. The sections were then counterstained with hematoxylin.
Immunofluorescence.
Normal and scleroderma fibroblasts were grown in four-well LAB TEK chambers (Nalge Nunc International, Naperville, Illinois, USA) to subconfluence as described above. After 24 hours of serum starvation, cells were fixed with 3.7% formaldehyde, permeabilized with 0.5% Triton X-100 in PBS (PBST), and blocked with 10% FCS in PBST (30). Cells were stained with primary Ab’s, washed, and incubated with FITC-conjugated donkey anti-goat IgG (Chemicon International, Temecula, California, USA) and rhodamine-conjugated donkey anti-rabbit IgG (Chemicon International). Nuclei were stained with DAPI. For immunofluorescence using Mv1Lu cells, cells were grown in four-well LAB TEK chambers, transfected with the expression vector for Smad7, and incubated an additional 48 hours. Cells were then fixed and stained as described above. In some experiments, Mv1Lu cells were treated with 2 ng/ml of TGF-β1 for the last 3 hours prior to fixation. To visualize the fluorescence, a Zeiss microscope was used. Necessary controls were performed; that is, cells were stained with only FITC-conjugated donkey anti-goat IgG or rhodamine-conjugated donkey anti-rabbit IgG to check for nonspecific binding of the secondary Ab’s. Nonspecific binding was not detected, and the results were omitted from the figures for clarity.
Immunoprecipitation.
Each extract prepared as described above was precleared with 10 μl of protein G-Sepharose. The supernatant was then incubated with 10 μl of protein G-Sepharose beads conjugated to Smad7-specific Ab or TβRI-specific Ab, and the immunoprecipitates were washed four times with lysis buffer (31). After the last wash, the beads were resuspended in 30 μl of sample buffer and boiled for 5 minutes. Each immunoprecipitate was subjected to immunoblotting.
Plasmid construction.
Generation of a –772COL1A2/chloramphenicol acetyltransferase (–772COL1A2/CAT) construct consisting of a human collagen α2(I) gene fragment (+58 to –772 bp relative to the transcription start site) linked to the CAT reporter gene was done as previously described (25). The expression vector for Smad7 was a gift from Peter ten Dijke (The Netherlands Cancer Institute, Amsterdam, The Netherlands) (19). The 3TP-lux, which contains part of the promoter region of the plasminogen activator inhibitor-1 gene and three tandem repeats of AP-1-binding sites of the collagenase 1 gene, was provided by John Massague (Memorial Sloan-Kettering Cancer Center, New York, New York, USA). Expression vectors for Flag-tagged Smurf1 and Flag-tagged Smurf2 were gifts from Kohei Miyazono (University of Tokyo, Tokyo, Japan) (22) and Jeffrey L. Wrana (Mount Sinai Hospital, Toronto, Ontario, Canada) (21), respectively. Plasmids were purified twice on CsCl gradients. At least two different plasmid preparations were used for each experiment.
Transient transfection.
Fibroblasts were grown to 50% confluence in 100-mm dishes as described above. After a 4-hour incubation with serum-free medium, cultures were transfected with 2 μg of the –772COL1A2/CAT or 3TP-lux construct, along with 0.1 μg of the Smad7 expression vector or corresponding empty construct, employing FuGENE 6 (Roche Diagnostics Corp., Indianapolis, Indiana, USA) (24). To control for minor variations in transfection efficiency, 1 μg of pSV-β-galactosidase vector (Promega Corp., Madison, Wisconsin, USA) was included in all transfections. After 24 hours of incubation, the appropriate reagent was added. Cells were harvested 48 hours after transfection. To determine the promoter activity of –772COL1A2/CAT, each extract normalized for protein content was incubated with butyl-CoA and 14C-chloramphenicol for 90 minutes at 37°C. Butylated chloramphenicol was extracted using an organic solvent (a 2:1 mixture of tetramethylpentadecane and xylene) and quantitated by scintillation counting. To determine the promoter activity of 3TP-lux, each extract normalized for protein content was assayed for luciferase activity. Every experiment was performed in duplicate.
Pulse-chase analysis.
Normal and scleroderma dermal fibroblasts were grown until nearly confluent in 100-mm dishes as described above. For pulse labeling, cells were incubated in cysteine- and methionine-free MEM for 30 minutes and then pulsed in the same medium containing [35S]cysteine and [35S]methionine (1 mCi/ml) for 30 minutes. Pulse-labeled cells were chased for 0, 3, or 6 hours in serum-free medium. At each time point, the cells were placed on ice and rinsed with cold PBS, lysed in lysis buffer, and incubated for an additional 30 minutes on ice. For immunoprecipitation assays, aliquots (1 mg) of protein extracts were preadsorbed with protein G–agarose beads (Santa Cruz Biotechnology Inc.) for 2 hours at 4°C on a vertical rotator, incubated with 2 μg of anti-TβRI Ab for 1 hour, and, finally, with protein G–agarose beads for 2 hours. The beads were spun down, washed three times with lysis buffer, resuspended in the sample buffer for electrophoresis, boiled for 3 minutes, and spun briefly. The supernatants were analyzed by SDS-PAGE and autoradiography. The densities of bands were measured with a densitometer.
Analysis of half-lives.
Pulse-chase experiments were analyzed by plotting the log of the densitometer volume for each band (with background from a blank lane on the gel subtracted) versus chase time and by obtaining a slope m for the plot by regression analysis. Turnover was assumed to be a first-order process, with t1/2 = 0.693/k, where k = –2.3 m. All half-life measurements are from at least five independent experiments yielding correlation coefficients greater than 0.9.
Statistical analysis.
Statistical analysis was carried out with the Mann-Whitney test for comparison of means. P values less than 0.05 were considered to be significant.
Results
Comparison of expression levels of TβRI, TβRII, Smad2, Smad3, Smad4, and Smad7 between normal and scleroderma fibroblasts.
First, we compared the endogenous expression levels of TβRI, TβRII, Smad2, Smad3, Smad4, and Smad7 proteins between normal and scleroderma fibroblasts by immunoblotting. As shown in Figure 1, scleroderma fibroblasts expressed increased levels of the TβRI protein (2.5-fold increase; P < 0.01) and of the TβRII protein (2.8-fold increase; P < 0.01). These results were consistent with our previous report (16). Regarding Smads, there was no significant difference in the expression levels of Smad2, Smad3, or Smad4 between normal and scleroderma fibroblasts, whereas the expression levels of Smad7 were markedly elevated in scleroderma fibroblasts compared with normal fibroblasts (3.5-fold increase; P < 0.01). These bands were confirmed to be Smad7 protein by performing a competition assay using Smad7-blocking peptide (data not shown). We also investigated the mRNA levels of Smad7 in normal and scleroderma fibroblasts. As shown in Figure 2, the levels were significantly elevated in scleroderma fibroblasts compared with normal fibroblasts (2.5-fold increase; P < 0.01).
Figure 1.

Expression levels of TβRI, TβRII, Smad2, Smad3, Smad4, and Smad7 in normal and scleroderma fibroblasts. (a) Whole-cell lysates were electrophoresed through a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with the indicated Ab’s. One representative of five independent experiments is shown. (b) The protein levels quantitated by scanning densitometry are shown relative to those in normal fibroblasts (100 arbitrary units [AU]). Data are expressed as the mean ± SD of five independent experiments. NS, normal skin fibroblasts; SSc, scleroderma fibroblasts.
Figure 2.

Comparison of the Smad7 mRNA levels between normal and scleroderma fibroblasts. Northern blot analysis of Smad7 mRNA was performed using 2 μg of Poly(A)+ RNA. Levels of GAPDH mRNA are shown as a loading control. One representative of five independent experiments is shown (a). Smad7 mRNA levels quantitated by scanning densitometry and corrected for the level of GAPDH in the same samples are shown relative to those in normal fibroblasts (100 AU) (b). Data are expressed as the mean ± SD of five independent experiments.
Increased phosphorylation of Smad2 in scleroderma fibroblasts.
We previously demonstrated that the activation of dermal fibroblasts in scleroderma may be a result of stimulation by autocrine TGF-β signaling (16). To confirm this notion, we analyzed the state of Smad2 phosphorylation. As shown in Figure 3, no apparent phosphorylation of Smad2 was observed in untreated normal fibroblasts. In contrast, Smad2 was constitutively phosphorylated in scleroderma fibroblasts, but the levels ranged from weak to moderate. These observations were consistent across all of the normal and scleroderma fibroblasts. These results strongly support the notion described above. We also investigated the TGF-β–induced phosphorylation of Smad2. There was no significant difference in the state of Smad2 phosphorylation between normal and scleroderma fibroblasts after TGF-β1 stimulation.
Figure 3.

The phosphorylation levels of Smad2 in normal and scleroderma fibroblasts either treated or untreated [TGF-β1 (–)] with TGF-β1. Cell extracts were prepared from confluent quiescent fibroblasts either treated or untreated with 2 ng/ml of TGF-β1 for 1 hour, electrophoresed through a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with anti–phospho-Smad2 Ab (pSmad2). After stripping, total levels of Smad2 were determined using anti-Smad2/Smad3 Ab. One representative of five independent experiments is shown.
Upregulated expression of Smad7 depends on the stimulation by autocrine TGF-β in scleroderma fibroblasts.
To examine the possibility that the upregulated expression of Smad7 in scleroderma fibroblasts is due to stimulation by autocrine TGF-β, we first investigated the effect of exogenous TGF-β1 on the expression of Smad7 mRNA. Consistent with previous reports, normal fibroblasts showed a markedly increased expression of Smad7 mRNA on TGF-β1 stimulation. In contrast, Smad7 expression was not significantly affected by TGF-β1 in scleroderma fibroblasts (Figure 4a). The mRNA levels of Smad7 in scleroderma fibroblasts were equivalent to those in normal fibroblasts treated with 2 ng/ml of TGF-β1 for 3 hours. Next, we investigated the effect of anti–TGF-β Ab’s on the expression of Smad7 mRNA. Upon treatment with anti–TGF-β Ab’s, normal fibroblasts showed no reduction in Smad7 mRNA. In contrast, scleroderma fibroblasts treated with anti–TGF-β Ab’s showed a marked reduction in the expression levels of Smad7 mRNA (Figure 4b). Treatment with preimmune rabbit IgG had no effect on the expression of Smad7 mRNA. We further examined the effect of a TGF-β1 antisense oligonucleotide. This antisense oligonucleotide caused little decrease in the expression of Smad7 mRNA in normal fibroblasts. Scleroderma fibroblasts treated with TGF-β1 antisense oligonucleotide, however, showed a marked reduction in the expression of Smad7 mRNA (Figure 4b). The sense oligonucleotide had no such inhibitory effect. These results indicate that the upregulated Smad7 expression in scleroderma fibroblasts depends on the stimulation by autocrine TGF-β.
Figure 4.

Effects of TGF-β1 stimulation, anti–TGF-β Ab, or TGF-β1 antisense oligonucleotide on Smad7 mRNA expression in normal and scleroderma fibroblasts. (a) Smad7 mRNA levels in normal and scleroderma fibroblasts treated or untreated with TGF-β1 (2 ng/ml) for 3 hours were investigated by Northern blot analysis. Representative results are shown (upper panels). Smad7 mRNA levels quantitated by scanning densitometry and corrected for the level of GAPDH in the same samples are shown relative to those in normal fibroblasts without TGF-β1 stimulation (100 AU) (lower panel). Data are expressed as the mean ± SD of five independent experiments. (b) Cells were treated with the indicated reagent for 48 hours, and Smad7 mRNA levels were investigated by Northern blot analysis. Representative results are shown (upper panels). Smad7 mRNA levels quantitated by scanning densitometry and corrected for the level of GAPDH in the same samples are shown relative to those in normal fibroblasts (100 AU) (lower panels). Data are expressed as the mean ± SD of five independent experiments.
Comparison of Smad7 expression levels in dermal tissue between scleroderma and healthy controls.
To compare the expression levels of Smad7 between normal and scleroderma dermal tissues, we performed immunohistochemical staining using paraffin-embedded sections. No expression of Smad7 was detected in normal dermal fibroblasts (Figure 5a), whereas the expression was detected strongly in scleroderma fibroblasts (Figure 5b). Regarding the epidermis, blood vessels, and infiltrated cells, there was no immunoreactivity for Smad7 in either normal or scleroderma dermal sections. These results were consistent across all of the normal and scleroderma dermal sections.
Figure 5.
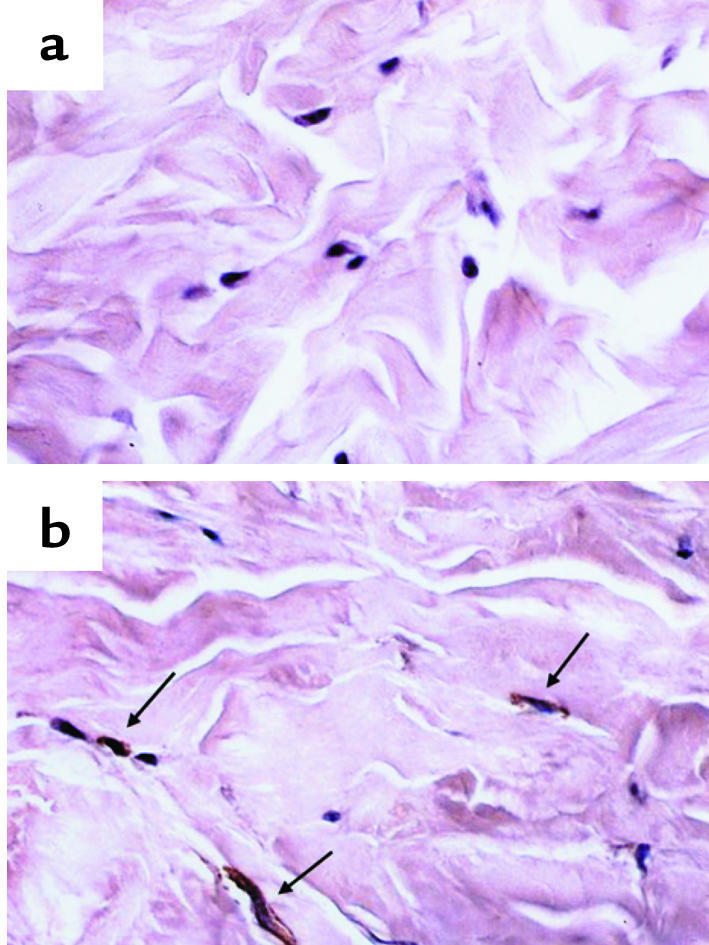
Expression levels of Smad7 in normal and scleroderma dermal tissue. Paraffin sections of normal dermal tissue (a) and scleroderma dermal tissue (b) were subjected to immunohistochemical analysis with anti-Smad7 Ab, as described in Methods. The immunoreactivity for Smad7 is indicated by arrows.
Subcellular localization of Smad7 and TGF-β receptors in normal and scleroderma fibroblasts.
Next, we investigated the subcellular localization of Smad7 in normal and scleroderma fibroblasts based on immunofluorescence. In normal fibroblasts, no immunoreactivity for Smad7, TβRI, or TβRII was detected (Figure 6, a, b, and c, respectively). In contrast, in scleroderma fibroblasts, strong immunoreactivity for Smad7 was detected as spots in the cytoplasm without any stimulation (Figure 6, d and g). The absence of Smad7 in the nucleus was confirmed by staining with 4′,6-diamidino-2-phenylindole. To further understand the subcellular localization of Smad7, we also stained scleroderma fibroblasts using anti-TβRI or anti-TβRII Ab. These two receptors also showed a spotted pattern in the cytoplasm (Figure 6, e and h, respectively), and the immunoreactivity for TGF-β receptors completely colocalized with that for Smad7 (Figure 6, f and i, respectively) in scleroderma fibroblasts. In normal fibroblasts, however, we could not detect immunoreactivity for Smad7, TβRI, or TβRII even after stimulation with TGF-β1 for 1, 3, 6, 12, and 24 hours (data not shown). These observations were consistent across all of the normal and scleroderma fibroblasts. Finally, we analyzed the subcellular localization of exogenously overexpressed Smad7 in Mv1Lu cells to confirm the specificity of the anti-Smad7 Ab used in this study. Smad7 was localized mainly in the nucleus of transfected cells without any stimulation and was exported from the nucleus to cytoplasm in response to TGF-β1 (Figure 6, j and k, respectively). This finding is consistent with previous reports (18, 19), suggesting that the anti-Smad7 Ab used in this study recognized Smad7 proteins specifically. Taken together, the results of these immunofluorescence experiments imply that the upregulated Smad7 constitutively forms a complex with TGF-β receptors in scleroderma fibroblasts.
Figure 6.

Subcellular localization of Smad7 in normal and scleroderma fibroblasts. (a–i) The subcellular distribution of Smad7, TβRI, and TβRII was visualized by immunofluorescence. Smad7 was visualized with polyclonal goat anti-Smad7 Ab and FITC-conjugated donkey anti-goat IgG (green). TβRI and TβRII were detected with polyclonal rabbit anti-TβRI and TβRII Ab’s and TRITC-conjugated donkey anti-rabbit IgG (red), respectively. Colocalization of Smad7 and TGF-β receptors (overlay) appears as yellow. (j and k) Mv1Lu cells were transfected with the expression vector for Smad7. After a 48-hour incubation, Smad7 was visualized as described above (j). In some experiments, cells were treated with TGF-β1 (2 ng/ml) for the last 3 hours (k).
Smad7 constitutively makes a complex with TGF-β receptors in scleroderma fibroblasts.
To confirm the finding described above, we performed immunoprecipitation. As an initial experiment, we performed immunoprecipitation using normal fibroblasts either treated or untreated with TGF-β1 (left panels in Figure 7a). Anti-TβRI Ab precipitated Smad7 at detectable levels in normal fibroblasts treated with TGF-β1. In contrast, it could not precipitate Smad7 in untreated normal fibroblasts. These results are consistent with the notion that Smad7 associates with TβRI after TGF-β1 stimulation. Since total levels of Smad7 also elevated after TGF-β1 stimulation, however, these results do not rule out the possibility that the association of Smad7 with TβRI depends on the level of Smad7. To further investigate this point, we performed immunoprecipitation using fibroblasts transfected with the Smad7 expression vector (right panels in Figure 7a). Anti-TβRI Ab precipitated Smad7 at detectable levels in transfectants treated with TGF-β1, while the precipitated Smad7 levels were low in transfectants without TGF-β1 stimulation. These results indicate that Smad7 forms a complex with TβRI in response to TGF-β1 in normal fibroblasts. Next, we compared the association of Smad7 with TβRI between normal and scleroderma fibroblasts. As shown in the upper panels in Figure 7b, anti-TβRI Ab precipitated Smad7 at detectable levels in scleroderma fibroblasts. Reverse immunoprecipitation using anti-Smad7 Ab was also performed, and similar results were obtained (lower panels in Figure 7b). These results were consistent across all of the normal and scleroderma fibroblasts. Taken together with the results of the immunofluorescence analysis, these findings indicate that the upregulated Smad7 constitutively forms a complex with TGF-β receptors in scleroderma fibroblasts.
Figure 7.

Constitutive complex formation of Smad7 with TβRI in scleroderma fibroblasts. (a) In the left panels, confluent quiescent normal fibroblasts were incubated in the absence or presence of TGF-β1 (2 ng/ml) for 24 hours. In the right panels, normal fibroblasts were transfected with the Smad7 expression vector (2 μg) and incubated for 48 hours prior to cell extraction. Some transfectants were stimulated with TGF-β1 (2 ng/ml) for the last 3 hours. Cell lysates (1 mg of protein/sample) were subjected to immunoprecipitation (IP) using anti-TβRI Ab. Immunoprecipitates were subjected to immunoblotting (Blot) using anti-Smad7 Ab. After stripping, total TβRI levels were determined by immunoblotting. Total Smad7 levels were determined by immunoblotting using whole-cell lysates (20 μg of protein/sample). (b) Cell lysates prepared from confluent quiescent fibroblasts were subjected to immunoprecipitation and immunoblotting as described above (upper panels). Reverse immunoprecipitation using anti-Smad7 Ab was also performed (lower panels). Each figure shows one representative of five independent experiments.
Impaired inhibitory effect of Smad7 on human α2(I) collagen promoter activity and 3TP-lux transcriptional activity in scleroderma fibroblasts.
Next, we compared the inhibitory effect of Smad7 on the transcriptional activity of the –772COL1A2 promoter between normal and scleroderma fibroblasts either untreated or treated with 2 ng/ml of TGF-β1 (Figure 8a). The basal –772COL1A2 promoter activity of scleroderma fibroblasts was about three times higher than that of normal fibroblasts. TGF-β1 increased –772COL1A2 expression about threefold in normal fibroblasts, while scleroderma fibroblasts did not show a significant increase in –772COL1A2 expression. With the transient overexpression of Smad7, the basal –772COL1A2 promoter activity was slightly decreased and the stimulation of –772COL1A2 promoter activity by TGF-β1 was completely abrogated in normal fibroblasts. In contrast, the transient overexpression of Smad7 had no significant inhibitory effect on the –772COL1A2 promoter activity in scleroderma fibroblasts with or without TGF-β1 stimulation. These results show that the inhibitory effect of Smad7 on the human α2(I) collagen promoter activity is completely impaired in scleroderma fibroblasts. To confirm this finding, we compared the effects of exogenous overexpressed Smad7 on the promoter activity of 3TP-lux, which contains TGF-β response elements for plasminogen activator inhibitor–1, between normal and scleroderma fibroblasts either untreated or treated with TGF-β1. The basal 3TP-lux promoter activity of scleroderma fibroblasts was about 18 times higher than that of normal fibroblasts. TGF-β1 stimulation significantly increased luciferase activity in normal and scleroderma fibroblasts (11.1-fold increase, P < 0.01, and 2.3-fold increase, P < 0.01, respectively). With the transient overexpression of Smad7, the basal luciferase activity was slightly decreased and the stimulation of luciferase activity by TGF-β1 was significantly abrogated in normal fibroblasts. In contrast, the transient overexpression of Smad7 had no significant inhibitory effect on the luciferase activity in scleroderma fibroblasts with or without TGF-β1 stimulation (Figure 8b). Taken together with the results of CAT experiments, these findings show that the inhibitory effect of Smad7 on TGF-β signaling is completely impaired in scleroderma fibroblasts.
Figure 8.

The inhibitory effect of Smad7 in normal and scleroderma fibroblasts. Cells were transfected with 2 μg of the –772COL1A2/CAT (a) or 3TP-lux construct (b), along with 0.1 μg of Smad7 or the corresponding empty construct. After a 24-hour incubation, the indicated reagent was added. Cells were harvested 48 hours after transfection. Values represent the promoter activity relative to that of untreated normal fibroblasts transfected with empty vector, which was set at 100 (a) or 10 AU (b). The mean and SEM results from five separate experiments are shown.
Comparison of the stability of TβRI and the expression levels of Smurf1/Smurf2 between normal and scleroderma fibroblasts.
As described above, we demonstrated that upregulated Smad7 constitutively associates with TGF-β receptors in scleroderma fibroblasts and that it is unable to block TGF-β signaling. A defect of Smurf-dependent degradation of TβRI is one obvious explanation for this finding. To address this point, the protein turnover of TβRI was monitored in normal and scleroderma fibroblasts for 6 hours in metabolic pulse-chase experiments. As shown in Figure 9, the half-life of TβRI protein was significantly longer in scleroderma fibroblasts than normal fibroblasts (4.34 ± 0.35 hours versus 1.88 ± 0.13 hours, P < 0.05), which strongly supports the hypothesis described above. To determine whether the disturbed TβRI turnover in scleroderma fibroblasts is due to decreased expression levels of Smurf1/Smurf2, we also evaluated Smurf expression levels. As shown in Figure 10, there was no significant difference in the expression levels of Smurf1 or Smurf2 between normal and scleroderma fibroblasts. This implies that the increased stability of TβRI protein in scleroderma fibroblasts can be attributed to the impaired functions of Smurfs rather than to the decreased expression of Smurfs.
Figure 9.

Comparison of the stability of TβRI protein between normal and scleroderma fibroblasts. Cells were incubated in cysteine- and methionine-free MEM for 30 minutes and then pulsed in the same medium containing [35S]cysteine and [35S]methionine (1 mCi/ml) for 30 minutes. Pulse-labeled cells were chased for 0, 3, or 6 hours in serum-free medium. Cell extracts (1 μg) were subjected to immunoprecipitation using anti-TβRI Ab, and immunoprecipitates (IP) were analyzed by SDS-PAGE and autoradiography. One representative of five independent experiments is shown (a). The densities of bands were measured with a densitometer, and the protein half-life was calculated as described in Methods. The mean and SEM from five separate experiments are shown (b).
Figure 10.

Comparison of the expression levels of Smurfs between normal and scleroderma fibroblasts. Cell lysates prepared from confluent quiescent fibroblasts were subjected to immunoblotting using the indicated Ab’s. One representative of five independent experiments is shown (a). The protein levels quantitated by scanning densitometry are shown relative to those in normal fibroblasts (100 AU) (b). Data are expressed as the mean ± SD of five independent experiments.
The overexpression of Smurf1 and/or Smurf2 does not affect the levels of TβRI protein in scleroderma fibroblasts.
We next investigated whether the transient overexpression of Smurf1 and/or Smurf2 affects the levels of TβRI protein in scleroderma fibroblasts. We first examined the effect of Smurf1 and Smurf2 overexpression on the levels of TβRI protein in normal fibroblasts either untreated or treated with TGF-β1. As shown in lanes 1–4 of Figure 11a, the transient overexpression of Smurf1 and Smurf2 did not affect the expression levels of TβRI protein in untreated normal fibroblasts. Coexpression of Smad7 along with Smurf1 and Smurf2 also did not affect the levels of TβRI protein in untreated normal fibroblasts (data not shown). These results are consistent with the notion that the interaction with Smad7, which is located in the nucleus in the absence of TGF-β stimulation, is required for the Smurf-mediated degradation of TβRI protein. The treatment with exogenous TGF-β1, however, significantly reduced the levels of TβRI protein in normal fibroblasts transfected with empty vector (59% decrease, P < 0.05). Moreover, the transient overexpression of Smurf1 and Smurf2 further significantly reduced the levels of TβRI protein in normal fibroblasts treated with TGF-β1 (74% decrease, P < 0.05), indicating that the transiently overexpressed Smurf1 and Smurf2 accelerates the TβRI protein degradation in normal fibroblasts treated with TGF-β1. We next investigated the effect of Smurf1 and Smurf2 overexpression on the levels of TβRI protein in scleroderma fibroblasts. As shown in lanes 5–8 of Figure 11a, the transient overexpression of Smurf1 and Smurf2 or the treatment with exogenous TGF-β1 induced a slight decrease in the TβRI protein levels in scleroderma fibroblasts, but the effect was not significant. Furthermore, a combination of the transient overexpression of Smurf1 and Smurf2 and the exogenous TGF-β1 stimulation also had no significant effect on the levels of TβRI protein in scleroderma fibroblasts. We also examined the TβRI protein levels at 1, 3, 6, and 12 hours after TGF-β1 stimulation and confirmed that the combination of the transient overexpression of Smurf1 and Smurf2 and the treatment with exogenous TGF-β1 had no significant effect on the levels of TβRI protein in scleroderma fibroblasts (data not shown). In these experiments, equal amounts of Smurf1 and Smurf2 were confirmed to be expressed by immunoblotting using anti-Flag Ab. We also investigated the effect of either Smurf1 or Smurf2 on the levels of TβRI protein in normal and scleroderma fibroblasts under the experimental conditions described above and obtained similar results in all experiments (data not shown), suggesting that Smurf1 and Smurf2 have no additive effect on the degradation of TβRI protein in the present assay system. These results were consistent with the hypothesis that the increased stability of TβRI protein in scleroderma fibroblasts can be attributed to the impaired functions of Smurfs.
Figure 11.

The effect of the transient overexpression of Smurf1/Smurf2 on the levels of TβRI protein in normal and scleroderma fibroblasts. Cells were transfected with 1 μg each of the Smurf1 and Smurf2 expression vectors or the corresponding empty constructs. After a 48-hour incubation, the levels of TβRI or exogenous overexpressed Smurf1/Smurf2 were determined by Western blot analysis using anti-TβRI Ab or anti-Flag Ab, respectively. In some experiments, cells were treated with TGF-β1 (2 μg/ml) for the last 24 hours. One representative of five independent experiments is shown (a). The protein levels quantitated by scanning densitometry are shown relative to those in untreated normal fibroblasts transfected with empty vector (100 AU) (b). Data are expressed as the mean ± SD of five independent experiments.
Smurfs can interact with Smad7 in scleroderma fibroblasts.
Finally, we confirmed the interaction of Smad7 with Smurfs in scleroderma fibroblasts by immunoprecipitation. As shown in the upper panels in Figure 12a, anti-Smad7 Ab precipitated Smurf1 and Smurf2 at detectable levels in normal fibroblasts treated with TGF-β1, but not in untreated normal fibroblasts. We also performed immunoprecipitation using normal fibroblasts transfected with the Smad7 expression vector and obtained similar results, suggesting that Smad7 forms a complex with Smurfs in response to TGF-β1 in normal fibroblasts. In contrast, anti-Smad7 Ab precipitated Smurf1 and Smurf2 at detectable levels in scleroderma fibroblasts (lower panels in Figure 12a). This finding was confirmed by reverse immunoprecipitation using anti-Smurf1 Ab or anti-Smurf2 Ab (Figure 12b). These observations were consistent across all of the normal and scleroderma fibroblasts. These results rule out the possibility that the impaired Smad7-Smurf–mediated negative regulation of TGF-β signaling in scleroderma fibroblasts is caused by the disturbed interaction of Smad7 with Smurfs.
Figure 12.

Smad7 can interact with Smurfs in scleroderma fibroblasts. (a) Whole-cell lysates (1 mg of protein/sample) prepared from confluent quiescent normal fibroblasts or normal fibroblasts transfected with the Smad7 expression vector were subjected to immunoprecipitation using anti-Smad7 Ab. In some experiments, cells were stimulated with TGF-β1 (2 ng/ml) for 3 hours. Immunoprecipitates were subjected to immunoblotting using anti-Smurf1 Ab (left panels) or anti-Smurf2 Ab (right panels). After stripping, total Smad7 levels were determined by immunoblotting. Total Smurf1 or Smurf2 levels were determined by immunoblotting using whole-cell lysates (20 μg protein/sample). The same experiments were performed using whole-cell lysates prepared from normal and scleroderma fibroblasts (lower panels). (b) Reverse immunoprecipitation using anti-Smurf1 Ab (upper panels) or anti-Smurf2 Ab (lower panels) was performed.
Discussion
TGF-β signaling has been implicated in the primary pathogenesis of fibrotic disorders. Regarding scleroderma, it was reported that scleroderma fibroblasts of the involved area did not secrete increased levels of TGF-β1 (16, 32). The mechanism of tissue fibrosis in such diseases remains to be clarified. We have reported the overexpression of TβRI and TβRII in scleroderma fibroblasts, indicating one possible mechanism of autocrine TGF-β activity by the overexpression of TβRI or TβRII (15, 16). In support of this, the transient overexpression of TβRI and/or TβRII induced a three- to fourfold increase of α2(I) collagen promoter activity, and this was sensitive to anti–TGF-β Ab (15). Recently, we demonstrated that the blockade of endogenous TGF-β signaling using anti–TGF-β Ab or TGF-β1 antisense oligonucleotide diminished the increased collagen production in scleroderma fibroblasts (16). These previous findings established the notion that autocrine TGF-β signaling might play a central role in the pathogenesis of scleroderma. To further test this notion, we focused on Smad7 and Smurfs in scleroderma fibroblasts because impaired negative regulation can lead to the constitutive activation of signal transduction.
In this study, we first demonstrated that there was a marked overexpression of Smad7 in scleroderma fibroblasts due to stimulation by autocrine TGF-β. Next, we investigated the subcellular localization of Smad7 in scleroderma fibroblasts since Smad7 must form a complex with TGF-β receptors to function as a dominant intracellular regulator of the TGF-β pathway (18). As shown by immunofluorescence, the immunoreactivity for Smad7 in scleroderma fibroblasts showed a spotted pattern in the cytoplasm, and this staining pattern completely colocalized with the immunoreactivity for TGF-β receptors. Furthermore, we confirmed the interaction of Smad7 with TβRI by immunoprecipitation in scleroderma fibroblasts. Previous reports indicated that TGF-β induced endocytosis and the clustering of the TGF-β receptor complex into intracellular punctate domains such as early endosomes (33, 34). These results indicate that the upregulated Smad7 constitutively forms a complex with the activated TGF-β receptors in scleroderma fibroblasts. The interaction of Smad7 with TβRI, however, leads to the reduction of TGF-β signaling activity (18), a finding that seems to be contradictory to the notion that an autocrine TGF-β loop is crucial for the pathogenesis of scleroderma. To explain this discrepancy, we propose two hypotheses: (a) the expression level of Smad7 is not sufficient to block the activated TGF-β signaling in scleroderma fibroblasts, and (b) the inhibitory effect of Smad7 on TGF-β signaling is impaired in scleroderma fibroblasts. To confirm this point, we compared the inhibitory effect of exogenous overexpressed Smad7 on collagen gene expression and 3TP-lux between normal and scleroderma fibroblasts. The results indicated that the exogenous overexpression of Smad7 had no inhibitory effects on collagen gene expression and 3TP-lux promoter activity in scleroderma fibroblasts. These findings suggest that the inhibitory effect of Smad7 on TGF-β signaling is completely impaired in scleroderma fibroblasts.
Previous studies have reported that the mutation of molecules involved in TGF-β signaling, including TβRI, TβRII, Smad2, and Smad4, may play a central role in the pathogenesis of various diseases, especially pancreatic and colorecta cancers (35). The present finding that the inhibitory effect of Smad7 on TGF-β signaling is completely impaired in scleroderma fibroblasts suggests that the inactivating mutation in Smad7 may be a pathogenic event in scleroderma fibroblasts. As shown in transient transfection assay, however, overexpression of WT Smad7 could not inhibit the increased promoter activity of –772COL1A2 and 3TP-lux in scleroderma fibroblasts. These results imply that endogenous Smad7 is not mutated in scleroderma fibroblasts.
Smad7 has been shown to function as an adaptor molecule that links TβRI to the ubiquitin ligases, including Smurf1 and Smurf2 (21, 22). Smurfs promote the degradation and rapid turnover of the TβRI protein as well as enhance the inhibitory effect of Smad7 on TGF-β signaling (21, 22). Taken together with these previous reports, the present findings that the upregulated Smad7 constitutively forms a complex with TGF-β receptors suggests that the degradation of TGF-β receptors by Smurfs may also be disturbed in scleroderma fibroblasts. The results of a pulse-chase experiment in which the stability of TβRI was increased in scleroderma fibroblasts strongly support this hypothesis. We also demonstrated that there was no significant difference in the expression levels of Smurfs between normal and scleroderma fibroblasts, which implies that the increased stability of TβRI protein in scleroderma fibroblasts can be attributed to the impaired functions, rather than decreased expression, of Smurfs. Furthermore, the findings that Smurfs can interact with Smad7 and that the transient overexpression of WT Smurfs does not reduce the levels of TβRI protein suggest that endogenous Smurfs are not mutated in scleroderma fibroblasts. It is important to clarify in the future the disturbed function of Smurfs in scleroderma fibroblasts.
During the preparation of this manuscript, Dong et al. reported that the expression levels of Smad7 were markedly decreased, and exogenous transient overexpression of Smad7 inhibited TGF-β signaling in scleroderma fibroblasts (36). These results seem to be contradictory to the present results. The scleroderma fibroblasts were obtained from unaffected areas (an area above the elbow considered to have normal skin thickness as determined by clinical palpation) in this previous report, whereas they were obtained from an affected area (dorsal forearm) in the our study. In the present study, we demonstrated that the expression levels of TβRI and TβRII were elevated while those of Smad2, Smad3, and Smad4 were not elevated in scleroderma fibroblasts. In contrast, Dong et al. reported that levels of TβRI, TβRII, Smad2, and Smad4 were similar between normal and scleroderma fibroblasts, but the expression level of Smad3 was elevated in scleroderma fibroblasts. We also demonstrated that there was no significant difference in the TGF-β–induced phosphorylation levels of Smad2 between normal and scleroderma fibroblasts, whereas Dong et al. reported that scleroderma fibroblasts showed hyperphosphorylation of Smad2 following TGF-β stimulation compared with normal dermal fibroblasts. These results suggest that the discrepancy between the previous report and ours is due to the difference in the population of scleroderma fibroblasts studied.
Recently, Di Guglielmo et al. proposed a novel model for the regulation of TGF-β signaling. The clathrin-dependent internalization of TGF-β receptors into the early endosome antigen-1–positive endosome, where the anchor protein for Smad2 is abundant, promotes TGF-β signaling, while the lipid raft-caveolar internalization pathway contains the Smad7-Smurf2–bound receptor and is required for rapid receptor turnover. These two pathways are mutually exclusive, and the activation and inhibition of TGF-β signaling is regulated by a balance between them (37). The present finding of the constitutive phosphorylation of Smad2 in scleroderma fibroblasts suggests that the clathrin-dependent internalization is accelerated, while the finding of the constitutive formation of a complex between Smad7 and TGF-β receptors in scleroderma fibroblasts suggests that the lipid raft-caveolar internalization is accelerated. These results seem to contradict the notion that a balance between two distinct internalization pathways regulates the activation and inhibition of TGF-β signaling. One plausible explanation for this apparent discrepancy is that the increased stability of the Smad7-TβRI complex caused by the disturbed Smurf function induces an accumulation of this complex in caveolin-positive vesicles and subsequently disturbs the efficient lipid raft-caveolar internalization. Such an abnormality leads to the relative acceleration of the clathrin-dependent internalization. This hypothesis is supported by present and previous findings: (a) the colocalization of Smad7 and TGF-β receptors observed in scleroderma fibroblasts is not detected in normal fibroblasts treated with TGF-β1, suggesting that the complex of Smad7 and TGF-β receptors is stabilized and accumulated within caveolin-positive vesicles in scleroderma fibroblasts, whereas this complex is rapidly degraded in normal fibroblasts; (b) the inhibition of the lipid raft-caveolar internalization by nystatin results in increased phosphorylation levels of Smad2 in Mv1Lu cells (37); and (c) both the overexpression of catalytically inactive Smurf2 and the inhibition of proteasome activity by N-acetyl-leucyl-leucine norleucinal result in increased stability of the Smad7-TβRI complex and the subsequent accumulation of this complex in raft compartments in HEK 293T and Mv1Lu cells (37). To assess this hypothesis, further studies are needed regarding the dynamics of TGF-β receptors in scleroderma fibroblasts.
In conclusion, we demonstrated that the Smad7-Smurf–mediated negative regulation of TGF-β signaling was completely impaired in scleroderma fibroblasts. These results strengthen the concept that autocrine TGF-β signaling is crucial to the pathogenesis of scleroderma since an impaired negative feedback system could maintain the constitutive activation of signal transduction.
Acknowledgments
We would like to thank Peter ten Dijke, Kohei Miyazono, Jeffrey L. Wrana, and John Massague for providing us with expression vectors for Smad7, Smurf1, Smurf2, and 3TP-lux, respectively. This study was supported in part by a grant for scientific research from the Japanese Ministry of Education (10770391) and by project research for progressive systemic sclerosis from the Japanese Ministry of Health and Welfare.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations: TGF-β receptor type I (TβRI); TGF-β receptor type II (TβRII); Triton X-100 in PBS (PBST); chloramphenicol acetyltransferase (CAT).
References
- 1.LeRoy, E.C. 1992. Systemic sclerosis (scleroderma). In Cecil text book of medicine. 19th edition. J.B. Wyngaarden, L.H. Smith, and J.C. Bennett, editors. WB Saunders. Philadelphia, Pennsylvania, USA. 1530–1535.
- 2.Korn JH. Immunologic aspects of scleroderma. Curr. Opin. Rheumatol. 1989;1:479–484. doi: 10.1097/00002281-198901040-00011. [DOI] [PubMed] [Google Scholar]
- 3.Mauch C, Krieg T. Fibroblast-matrix interactions and their role in the pathogenesis of fibrosis. Rheum. Dis. Clin. North. Am. 1990;16:93–107. [PubMed] [Google Scholar]
- 4.Jelaska A, Arakawa M, Broketa G, Korn JH. Heterogeneity of collagen synthesis in normal and systemic sclerosis skin fibroblasts: increased proportion of high collagen-producing cells in systemic sclerosis fibroblasts. Arthritis Rheum. 1996;39:1338–1346. doi: 10.1002/art.1780390811. [DOI] [PubMed] [Google Scholar]
- 5.Mauch C, Kozlowska E, Eckes B, Krieg T. Altered regulation of collagen metabolism in scleroderma fibroblasts grown within three-dimensional collagen gels. Exp. Dermatol. 1992;1:185–190. doi: 10.1111/j.1600-0625.1992.tb00187.x. [DOI] [PubMed] [Google Scholar]
- 6.Wahl SM. Transforming growth factor β: the good, the bad, and the ugly. J. Exp. Med. 1994;180:1587–1590. doi: 10.1084/jem.180.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LeRoy EC. Increased collagen synthesis by SSc skin fibroblasts in vitro: a possible defect in the regulation or activation of the scleroderma fibroblast. J. Clin. Invest. 1974;54:880–889. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckingham RB, Prince RK, Rodnan GP, Taylor F. Increased collagen accumulation in dermal fibroblast cultures from patients with progressive systemic sclerosis (scleroderma) J. Lab. Clin. Med. 1978;92:5–21. [PubMed] [Google Scholar]
- 9.Peltonen J, Kahari L, Uitto J, Jimenez SA. Increased expression of type VI collagen genes in systemic sclerosis. Arthritis Rheum. 1990;33:1829–1835. doi: 10.1002/art.1780331211. [DOI] [PubMed] [Google Scholar]
- 10.Xu WD, LeRoy EC, Smith EA. Fibronectin release by systemic sclerosis and normal dermal fibroblasts in response to TGF-β. J. Rheumatol. 1991;18:241–246. [PubMed] [Google Scholar]
- 11.Rudnicka L, et al. Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. Regulation by transforming growth factor-β. J. Clin. Invest. 1994;93:1709–1715. doi: 10.1172/JCI117154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falanga V, Tiegs SL, Alstadt SP, Roberts AB, Sporn MB. Transforming growth factor-β: selective increase in glycosaminoglycan synthesis by cultures of fibroblasts from patients with progressive systemic sclerosis. J. Invest. Dermatol. 1987;89:100–104. doi: 10.1111/1523-1747.ep12580445. [DOI] [PubMed] [Google Scholar]
- 13.Massague J. Transforming growth factor-β family. Annu. Rev. Cell. Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 14.LeRoy EC, Smith EA, Kahaleh MB, Trojanowska M, Silver RM. A strategy for determining the pathogenesis of systemic sclerosis. Is transforming growth factor β the answer? Arthritis Rheum. 1989;32:817–825. [PubMed] [Google Scholar]
- 15.Kawakami T, et al. Increased expression of TGF-β receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-β signaling to scleroderma phenotype. J. Invest. Dermatol. 1998;110:47–51. doi: 10.1046/j.1523-1747.1998.00073.x. [DOI] [PubMed] [Google Scholar]
- 16.Ihn H, Yamane K, Kubo M, Tamaki K. Blockade of endogenous transforming growth factor β signaling prevents up-regulated collagen synthesis in scleroderma fibroblasts: association with increased expression of transforming growth factor β receptors. Arthritis Rheum. 2001;44:474–480. doi: 10.1002/1529-0131(200102)44:2<474::AID-ANR67>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Derynck R, Zhang Y, Feng XH. Smads: Transcriptional activators of TGF-β responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi H, et al. The MAD-related protein Smad7 associates with the TGF receptor and functions as an antagonist of TGF-β signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 19.Nakao A, et al. Identification of Smad7, a TGF β-inducible antagonist of TGF-β signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 20.Afrakhte M, et al. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-β family members. Biochem. Biophys. Res. Commun. 1998;249:505–511. doi: 10.1006/bbrc.1998.9170. [DOI] [PubMed] [Google Scholar]
- 21.Kavsak P, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF-β receptor for degradation. Mol. Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 22.Ebisawa T, et al. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 23.Monteleone G, et al. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Invest. 2001;108:601–609. doi:10.1172/JCI200112821. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ihn H, Yamane K, Asano Y, Kubo M, Tamaki K. IL-4 up-regulates the expression of tissue inhibitor of metalloproteinase-2 in dermal fibroblasts via the p38 mitogen-activated protein kinase-dependent pathway. J. Immunol. 2002;168:1895–1902. doi: 10.4049/jimmunol.168.4.1895. [DOI] [PubMed] [Google Scholar]
- 25.Ihn H, Ohnishi K, Tamaki T, LeRoy EC, Trojanowska M. Transcriptional regulation of the human α2(I) collagen gene. Combined action of upstream stimulatory and inhibitory cis-acting elements. J. Biol. Chem. 1996;271:26717–26723. doi: 10.1074/jbc.271.43.26717. [DOI] [PubMed] [Google Scholar]
- 26.Ihn H, LeRoy EC, Trojanowska M. Oncostatin M stimulates transcription of the human α2(I) collagen gene via the Sp1/Sp3-binding site. J. Biol. Chem. 1997;272:24666–24672. doi: 10.1074/jbc.272.39.24666. [DOI] [PubMed] [Google Scholar]
- 27.Falanga V, et al. Stimulation of collagen synthesis by anabolic steroid stanozol. J. Invest. Dermatol. 1998;111:1193–1197. doi: 10.1046/j.1523-1747.1998.00431.x. [DOI] [PubMed] [Google Scholar]
- 28.Brunet CL, Sharpe PM, Ferguson MWJ. Inhibition of TGF-β3 (but not TGF-β1 or TGF-β2) activity prevents normal and mouse embryonic palate fusion. Int. J. Dev. Biol. 1995;39:345–355. [PubMed] [Google Scholar]
- 29.Kubo M, Ihn H, Yamane K, Tamaki K. Up-regulated expression of transforming growth factor β receptors in dermal fibroblasts in skin sections from patients with localized scleroderma. Arthritis Rheum. 2001;44:731–734. doi: 10.1002/1529-0131(200103)44:3<731::AID-ANR124>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 30.Ihn H, Tamaki K. Oncostatin M stimulates the growth of dermal fibroblasts via a mitogen-activated protein kinase-dependent pathway. J. Immunol. 2000;165:2149–2155. doi: 10.4049/jimmunol.165.4.2149. [DOI] [PubMed] [Google Scholar]
- 31.Ihn H, Tamaki K. Increased phosphorylation of transcription factor Sp1 in scleroderma fibroblasts: association with increased expression of the type I collagen gene. Arthritis Rheum. 2000;43:2240–2247. doi: 10.1002/1529-0131(200010)43:10<2240::AID-ANR11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 32.Needleman BW, Choi J, Burrows-Mezu A, Fontana JA. Secretion and binding of transforming growth factor β by scleroderma and normal dermal fibroblasts. Arthritis Rheum. 1990;33:650–656. doi: 10.1002/art.1780330507. [DOI] [PubMed] [Google Scholar]
- 33.Gilboa L, Wells RG, Lodish HF, Henis YI. Oligomeric structure of type I and type II transforming growth factor receptors: homodimers form in the ER and persist at the plasma membrane. J. Cell Biol. 1998;140:767–777. doi: 10.1083/jcb.140.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGF-β receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 35.de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-β signaling in cancer. J. Natl. Cancer Inst. 2000;92:1388–1402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- 36.Dong C, et al. Deficient Smad7 expression: A putative molecular defect in scleroderma. Proc. Natl. Acad. Sci. U. S. A. 2002;99:3908–3913. doi: 10.1073/pnas.062010399. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]