

Abstract
A.SW mice, which are known to be prone to mercury (Hg)-induced immune nephritis, were assessed for their ability to develop autoimmunity to brain antigens after developmental exposure to Hg. Maternal drinking water containing subclinical doses of 1.25μM methyl Hg (MeHg) or 50μM Hg chloride (HgCl2) were used to evaluate developmental (exposure from gestational day 8 to postnatal day 21) induction of immune responses to brain antigens. Only HgCl2 induced autoantibody production; the HgCl2-exposed offspring showed an increased number of CD4+ splenic T cells expressing CD25 and Vβ 8.3 chains, and the brain-reactive immunoglobulin G (IgG) antibodies were predominantly against nuclear proteins (30 and 34 kD). The antibodies were deposited in all brain regions. Although male and female A.SW mice exposed to HgCl2 showed deposition of IgG in multiple brain regions, inflammation responses were observed only in the cerebellum (CB) of female A.SW mice; these responses were associated with increased levels of exploratory behavior. The developmental exposure to MeHg also induced inflammation in the CB and increased exploratory behavior of the female A.SW mice, but the change did not correlate with increased IgG in the brain. Interestingly, the non–Hg-exposed female A.SW mice habituated (adapted to the information and/or stimuli of a new environment) more than the male A.SW mice during exploratory behavior assessment, and the Hg exposure eliminated the habituation (i.e., no changes in behavior with subsequent trials), making the female behaviors more like those of the male A.SW mice. Additionally, gender differences in A.SW brain cytokine expressions prior to Hg exposure were eliminated by the Hg exposure.
Keywords: autoimmunity, brain antigens, mercury
The heavy metal mercury (Hg) is widely used in consumer products, such as lamps, thermometers, barometers, and manometers. In many countries, elemental Hg is still used in amalgams for dental restoration (Guzzi et al., 2008). Elemental Hg from the earth’s crust is released into water; through vaporization, the elemental Hg is transformed into inorganic Hg. Inorganic Hg can be converted by organisms, including marine plants and microorganisms, into organic methyl Hg (MeHg), which is consumed by fish. Humans are exposed to MeHg mainly through eating fish, shellfish, and marine mammals and to inorganic Hg through inhalation of exhausted air from coal-fired power plants and incinerators (Jedrychowski et al., 2007; Nakagawa et al., 1997; Trasande et al., 2005, 2006).
Experimental rats and mice exposed to Hg have been reported to develop T cell–dependent renal autoimmunity that is restricted to major histocompatibility complex (MHC) haplotypes; the mouse haplotype most susceptible to Hg-induced autoimmune disease is H-2s (Abedi-Valugerdi et al., 1997, 2001; al-Balaghi et al., 1996; Hultman and Enestrom, 1988, 1992; Hultman et al., 1993; Pelletier et al., 1987; Vas and Monestier, 2008). Additionally, it has been shown, in a C57BL/6J mouse model, that prenatal exposure to a low dose of MeHg can cause deficits in motor abilities, coordination, overall activity, and reference memory (Montgomery et al., 2008). Developmental exposure to MeHg altered learning behavior and induced depression-like behavior in male C57BL/6 mice (Onishchenko et al., 2007). The effects of HgCl2 on rat behavior were shown to be dependent on when the rats were exposed; rats exposed on postnatal day (PND) 1 were more sensitive than were those exposed later in development (Peixoto et al., 2007). In a cohort study, prenatal MeHg exposure was significantly associated with neurobehavioral changes, whereas postnatal MeHg exposure induced no such effects (Debes et al., 2006). Developmental MeHg exposure in a rat model induced acute hippocampal cell death, reductions in neurogensis, and learning deficits during puberty, via unknown mechanisms (Falluel-Morel et al., 2007). Hg can interact with neurons by direct effects or indirectly through effects on astrocytes, although disruptions of thyroid hormones, metallothioneins, and glutathione also have been suggested to be involved (Aschner et al., 1997, 2006; Eddins et al., 2008; Kaur et al., 2006; Soldin et al., 2008; Yoshida et al., 2006). Astrocytes have also been proposed to have a protective role in MeHg-induced neurotoxicity, based on an in vitro test (Morken et al., 2005). Low-dose long-term exposure to MeHg can influence brain functions, via lipid peroxidation, alterations to Na+/K+-ATPase activities, and increased nitric oxide (NO) levels (Huang et al., 2008). Prenatal MeHg exposures of mice also hampered the glutathione antioxidant system and induced long-lasting oxidative stress in the brain (Stringari et al., 2008). The level of autoimmune antigen-antibody complexes induced by HgCl2 was decreased in mice with a deficiency of activating Fc receptors (FcγRI and FcγRIII), whereas the level of these complexes increased in mice lacking the inhibitory Fc receptor, FcγRIIB (Martinsson et al., 2008).
Antibodies against brain antigens have been demonstrated in patients with various neurological diseases, such as neuropsychiatric lupus, Parkinson’s disease, Alzheimer’s disease, schizophrenia, and autism spectrum disorders (ASD) (Carvey et al., 1991; Lawrence et al., 2007; Schott et al., 1996; Silva et al., 2004; Tanaka et al., 1989; Yang et al., 1994). Central nervous system (CNS) inflammation and presence of maternal antibodies specific to fetal brain proteins have been associated with ASD (Braunschweig et al., 2008; Croen et al., 2008). Epidemiological studies provide evidence that genetic factors are not the only factors that contribute to ASD (Bello, 2007; Taniai et al., 2008).
Microglial cells and mast cells, considered to be the immune cells of the CNS, can influence neuronal functions (Nautiyal et al., 2008; Takeuchi et al., 2008). Thus, CNS immune cells, as well as genetic factors, such as MHC haplotype, could be involved in Hg-induced behavior changes. During development, the brain is well known to be vulnerable to Hg exposure (Monnet-Tschudi et al., 1996). Although antibodies against brain antigens have been demonstrated in patients with neurological diseases, there has been no evaluation, to date, of Hg-induced autoantibodies against brain antigens. To evaluate the mechanistic connection between developmental Hg exposure and behavior changes resulting from immune activities, we hypothesized that upon developmental exposure to Hg (organic and/or inorganic): (1) the fetal brain is damaged; (2) neural proteins or peptides are released into the circulation and subsequently into the mother’s circulation; (3) the maternal immune system is then triggered to produce antibodies to the fetal brain antigens; and (4) the maternal antibodies disrupt normal developmental neuronal connections due to CNS inflammation. In our experiments, A.SW (H-2s) dams were treated with 1.25μM MeHg or 50μM HgCl2 in their drinking water, from gestational day (GD) 8 to PND 21. Sera anti-brain immunoglobulin G (IgG), Vβ expression of splenic T cells, IgG deposition in brain sections, molecular weights of brain antigens, and cytokines were determined in offspring and dams after weaning of pups at PND 21. At PND 70, exploratory behaviors of these offspring were measured. A/WySnJ (H-2a) mice were selected as the control strain; they differ genetically from the A.SW strain mainly at the MHC locus. Hg induced significant immune and behavioral changes in A.SW mice, in comparison to the effects in exposed A/WySnJ mice, and A.SW mice also displayed gender differences.
MATERIALS AND METHODS
Animals, Hg treatments, and organ collection.
A.SW (H-2s) and A/WySnJ (H-2a) mice, 7–9 week of age from Jackson Laboratory (Bar Harbor, ME), were bred for these experiments. Mice were housed in the Association for Assessment and Accreditation of Laboratory Animal Care-approved Wadsworth Center Animal Facility with controlled temperature (23±°C), humidity, and an artificial 12-h light and 12-h dark cycle, and this study was approved by Wadsworth Center’s Institutional Animal Care and Use Committee. Two females and one male were housed together with free access to water and food. At GD 8 (based on the appearance of vaginal plug), females were taken out and placed in individual cages. Preliminary experiments indicated that the maximal Hg doses that do not affect pup survival in A.SW mice were 1.25μM MeHg or 50μM HgCl2 in drinking water (Table 1). The internal doses of Hg in the dams’ whole blood and survival pups’ whole brain were measured by Atomic absorption spectroscopy (Table 1). In evaluation of the developmental toxicity of Hg on neurons through immune dysfunction, the dose selected should be sufficient to cause immune dysfunction, yet not be lethal. Thus, our experiments utilized 1.25μM MeHg or 50μM HgCl2 in dd H2O, which was provided ad libitum from GD 8 to PND 21. At PND 21, males and females were separated by gender and litter, in which male pups from the same litter were put in one cage and the female pups from the same litter were put in another cage. Dams and minimally one male and one female pup per litter were randomly selected and euthanized by CO2 exposure when the offspring were at PND 21. Bloods and organs (brains, kidneys, livers, and spleens) were collected after perfusion with PBS. Organs were stored in −80°C until use except that spleens were used immediately. Bloods were stored at 4°C for 24 h, and sera were collected after centrifugation at 12,000 × g for 10 min and then stored at −80°C until use. MeHg or HgCl2 exposure was stopped at the same time point, PND 21, for the mice used in assessment of exploratory behavior at PND 70.
TABLE 1.
Litter Survival and Internal Doses for Organic and Inorganic Hg Treatment
| Strain (haplotype) | MeHg (μM) | HgCl2 (μM) | ||||||||||||||||
| dd H2O | 0.5 | 1.25 | 2.5 | 50 | 100 | |||||||||||||
| Litter survival | Blood Hg | Brain Hg | Litter survival | Blood Hg | Brain Hg | Litter survival | Blood Hg | Brain Hg | Litter survival | Blood Hg | Brain Hg | Litter survival | Blood Hg | Brain Hg | Litter survival | Blood Hg | Brain Hg | |
| A.SW (H-2s) | 3/3 | 4.2 ± 0.4 | <MDL | 2/2 | 171.3 ± 19.9 | <MDL | 3/4 | 372.1 ± 160 | 0.03 ± 0.01 | 1/7 | 815.4 ± 283 | 0.05 ± 0 | 2/3 | 296.8 ± 116 | 0.27 ± 0.25 | 2/2 | 542.4 ± 315 | 0.27 ± 0.03 |
| A/WySnJ (H-2a) | 2/2 | 10.2 ± 1.7 | <MDL | 2/2 | 133.7 ± 30.4 | <MDL | 2/2 | 425.3 ± 106 | 0.03 ± 0.01 | 2/3 | 1197 ± 173 | 0.12 ± 0.07 | 2/2 | 474.3 ± 352 | 0.30 ± 0.26 | 1/1 | 787.9 | 0.2 |
Note. Pregnant female A.SW and A/WySnJ mice were treated with dd H2O, MeHg (0.5, 1.25, or 2.5μM) or HgCl2 (50 or 100μM) since GD 8. The numerator indicates the number of dams had produced litters and the denominator indicates the number of pregnant dams that were assayed. Blood Hg indicates the Hg concentration in dams’ whole blood (micrograms per liter); brain Hg indicates the Hg concentration in PND 21 pups’ whole brains (micrograms per gram of protein); MDL, minimal detection level.
Enzyme-linked immunosorbent assay.
ELISA was used to measure sera IgG to brain antigens. Each brain was homogenized in the presence of 2 ml of homogenization buffer containing 1% NP-40, 50mM Tris-Cl (pH = 7.6) (Sigma, St Louis, MO), 150mM NaCl, 2mM EDTA, 1mM Na-orthovanadate, 5mM NaF, and 20 μg of proteinase inhibitor cocktail (Sigma). The homogenates were sonicated for 1 min. The supernatant of homogenates was collected and utilized after centrifugation at 12,000 × g for 30 min at 4°C. Protein concentrations in the supernatant were measured by BCA Protein Assay Kit (Pierce, Rockford, IL) according to the manufacturer’s instructions. Homogenates were diluted with 0.1M carbonate buffer (pH = 9.5, coating buffer) to 200 μg of protein per milliliter. Fifty microliters of the diluted homogenate was coated into each well of 96-well ELISA plates, incubated at 4°C overnight, and then washed three times with PBS containing 0.05% Tween 20 (PBST; Sigma). After that, the plates were incubated with 5% fish gelatin (Sigma) (200 μl/well) in PBS at room temperature (RT) for 2 h, and then washed three times. Rat anti-mouse CD16/CD32 (0.5 μg) (Fc block; BD Pharmingen, San Jose, CA) in 45 μl PBST containing 1% fish gelatin (assay buffer) was added per well and incubated for 15 min at RT. Next, 5 μl of mouse serum were added to each well in the presence of Fc block, incubated at RT for 2 h, and the plates were then washed six times. Fifty microliters of peroxidase-conjugated goat anti-mouse IgG γ-chain specific (Roche, Basel, Switzerland) (1:1000 diluted with assay buffer) was added per well, incubated at RT for 1 h in the dark, and then washed six times. Fifty microliters of 3,3′,5,5′-tetramethylbenzidine liquid substrate (ELISA substrate; Sigma) was added per well and incubated at RT for 40 min in the dark. Finally, 25 μl of 1M H2SO4 were added to each well to stop the reaction. The plates were read optical density at 450 nanometer (OD 450) on an ELISA analyzer (Bio-Tek, Winooski, VT).
For IgG concentration in different brain regions, whole brains from PND 21 offspring selected from H2O (control), MeHg, and HgCl2 groups were dissected into substantia nigra (SN), hypothalamus (HT), frontal cortex (FCTX), striatum (STR), cortex (CTX), hippocampus (HC), and cerebellum (CB). Protein extraction and concentration determinations were described earlier. Goat anti-mouse IgG γ-chain–specific antibodies (5 μg; Sigma) in 100 μl of coating buffer were coated into each well in 96-well ELISA plates overnight at 4°C and then washed six times. The plates were incubated with 5% bovine serum albumin (BSA) (Sigma) in PBS at RT for 2 h, and the plates were then washed three times. Hundred microliters of brain section homogenates (1:50 diluted with PBST containing 1% BSA) were added into each well, incubated at RT for 2 h, and the plates were then washed six times. Horseradish peroxidase–conjugated goat anti-mouse IgG whole molecule antibodies (100 μl; Sigma) (1:1000 diluted with PBST containing 1% BSA) were added into each well, incubated for 1 h at RT in the dark, and the plates were then washed six times, and 100 μl of ELISA substrate was added to the well with incubation for 5 min at RT in the dark. Then, 50 μl of 1M H2SO4 was added to each well to stop the reaction. The plates were read at OD 450 on the ELISA analyzer. The concentration of IgG in each brain region homogenate was calculated as nanograms of IgG per milligram of protein.
Flow cytometry.
At PND 21, spleens of offspring were harvested and single-cell suspensions were prepared; 5 ml of red blood cell (RBC) lysis buffer containing 0.017M Tris and 0.75% NH4Cl (pH = 7.4) was used to lyse RBC with incubation on ice for 5 min. Cell numbers were adjusted to 1 × 106 cells in 100 μl of PBS containing 0.1% NaN3 in each 5 ml polystyrene tube and 1 μg of Fc block was added, and the tubes were then incubated on ice for 10 min. After that, 2 μl of PerCP-conjugated rat anti-mouse CD4, 2 μl of Phycoerythrin-conjugated rat anti-mouse CD8, 2 μl of allophycocyanin (APC)–conjugated rat anti-mouse CD25, and 2 μl of Fluorescein isothiocyanate-conjugated mouse anti-mouse Vβ (Mouse Vβ TCR Screening Panel) (all from BD Pharmingen) were added and incubated on ice for 30 min in the dark. Cells were washed with 1 ml of PBS containing 0.1% NaN3 once and then the supernatant was removed. Cell pellets were resuspended with 400 μl of PBS containing 1% paraformaldehyde (PFA) (Fisher, Fair Lawn, NJ) and kept at 4°C with foil covered. The samples were assayed on Flow Cytometry (BD FacsAria IIu, San Diego, CA) within 3 days.
Immunohistochemistry.
Whole brains from PND 21 untreated A.SW mice were fixed with 4% PFA in PBS overnight at 4°C. Then, the brains were sectioned into four slices from dorsal to ventral and fixed again with 4% PFA in PBS for 5 h at RT. The sections were processed with a VIP 2000 tissue processor Model 4618 (Tissue-Tek, Holly, MI). The brain sections were embedded with paraffin, cut into slices of 7-μm thickness, and floated onto charged microscopic slides. The slides were warmed at 56°C for 35 min and then cooled down prior to use. The slides were incubated with xylenes for 15 min, rehydrated through a graded alcohol series (100, 95, 70, and 50%) for 10 s each, and finally Milli-Q (MQ) water. Citrate buffer (0.01M sodium citrate, pH = 6) was heated till boiling, and the sections were microwaved in the citrate buffer for 3 min at full power followed by 7 min at ¾ power. The slides were put into PBS to cool. The sections were then incubated with 200 μl of PBS containing 3% goat serum and 0.1% BSA for 30 min at RT. Next, they were incubated with 200 μl of serum (from dams exposed or nonexposed to Hg; IgG concentration was diluted to 80 μg/ml with PBS containing 3% goat serum and 0.1% BSA; commercial purified mouse IgG [Sigma] was used as negative control) in the presence of 2 μg of Fc block, for 2 h at RT, and then washed three times with PBS. The sections were incubated with 200 μl of Alexa Fluor 488–conjugated chicken anti-mouse IgG (1:300 dilution by PBS containing 3% goat serum and 0.1% BSA) (Molecular Probes, Eugene, OR) for 1 h at RT in the dark and then washed six times with PBS. Next, they were incubated with 200 μl of 4',6-diamidino-2-phenylindole (DAPI) (Sigma) (1 μg/ml diluted with PBS containing 3% goat serum and 0.1% BSA) for 5 min at RT in the dark and then washed six times with PBS. Finally, the sections were assayed by fluorescence microscopy (Nikon Eclipse 50i; Nikon Instruments, Inc., Lakeland, FL).
Immunoprecipitation protocol.
We used immunoprecipitation of IgG to assess the molecular masses of the brain antigens bound to the IgG. Because these brain antigens were found in nuclei, nuclear proteins were extracted from a normal PND 21 A.SW mouse brain. Extraction was following the paper published by Cox. B and Emili. A (Cox and Emili, 2006), through which nuclear proteins slightly bound to DNA (Nuc-S) and nuclear proteins tightly bound to DNA (Nuc-T) were yielded. Brain-reactive IgG in sera from HgCl2-treated A.SW dams were used to capture the brain antigens (dam sera from water group and commercial purified mouse IgG were selected as controls). Protocol followed the manufacturer’s instructions in Immunoprecipitation Kit-Dynabeads Protein G (Invitrogen, Carlsbad, CA). After elution, the supernatant containing murine IgG and brain antigens was boiled for 5 min in the presence of buffer containing 2% SDS (Bio-Rad Laboratories Inc., Hercules, CA), 10% glycerol, 1.5% dithiothreitol, 0.05% bromophenol blue, and 0.25M Tris-HCl (pH = 6.8) (heated by water at 90°C for 10 min before use). Then, the denatured proteins were loaded into 12% SDS-polyacrylamide gel electrophoresis (PAGE). Resolving gel was composed of 1.7 ml of MQ H2O, 2.0 ml of 30% acrylamide (Bio-Rad Laboratories Inc.) mix containing 0.8% N,N′-methylene-bis-acrylamide (Bis) (Bio-Rad Laboratories Inc.), 1.3 ml of 1.5M Tris (pH = 8.8), 50 μl of 10% SDS, 50 μl of 10% ammonium persulfate (APS) (Sigma), and 2 μl of N,N,N′,N′-tetramethylethylenediamine (TEMED) (Sigma). Stacking gel was made of 0.68 ml of MQ H2O, 0.17 ml of 30% acrylamide mixture, 0.13 ml of 1.0M Tris (pH = 6.8), 10 μl of 10% SDS, 10 μl of 10% APS, and 1 μl of TEMED. Kaleidoscope prestained standards (Bio-Rad Laboratories Inc.) were used as markers. After electrophoresis by 120 V for 120 min, gels were fixed with 100 ml of fixation buffer containing 50% methanol, 7% acetic acid in MQ H2O, for 30 min at RT. The fixation procedure was repeated once. The gels were incubated with 50 ml of Sypro Ruby gel stain buffer (Bio-Rad Laboratories, Inc.) and shaken at RT in the dark overnight. The gels were washed with 100 ml of wash buffer containing 10% methanol, 7% acetic acid in MQ H2O for 30 min at RT, and then rinsed with 100 ml of MQ H2O for 5 min twice. After that, the gels were assayed on LAS3000 imager (Fujifilm, Tokyo).
Luminex.
PND 21 A.SW pup brains were dissected as described earlier. Cytokines granulocyte-macrophage colony–stimulating factor (GM-CSF), interferon-gamma (IFN-γ), interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12(p70), IL-13, monocyte chemotactic protein-1 (MCP-1), and tumor necrosis factor-alpha (TNF-α) in the supernatants of brain sections homogenates were assayed with a Milliplex Map Kit (Millipore Corporation, Billerica, MA). The protocol followed the manufacturer’s instructions. Plates were run on a Luminex 100 IS analyzer.
Exploratory behavior.
At PND 70, mice were tested for exploratory behavior in a single 45-min session, using Versamax animal activity monitors (42-cm long, 42-cm wide, and 30-cm high; AccuScan Inc., Columbus, OH) enclosed in melamine sound–attenuating chambers (65-cm long, 55-cm wide, and 55-cm high; Med Associates, St Albans, VT). Mice were transported to the testing room 1 h before the start of testing. For the test, the mouse was placed in the center of the dark activity monitor. At the end of testing, the mouse was returned to its home cage, and the activity monitor was cleaned with 70% ethanol.
Statistics.
Data (mean ± SD) in the three treatment groups were first compared, using one-way ANOVA. If there was a significant difference (p < 0.05), Dunnett’s t-test was used to compare each treatment group with the corresponding control group. In the cytokine expression and open-field test analyses, male and female comparisons were performed with two-way ANOVA, followed by Student-Newman-Keuls (SNK) test if there were significant differences.
RESULTS
IgG against Brain Antigens Was Induced in HgCl2-Treated A.SW Mice
We first determined whether MeHg or HgCl2 exposure enhanced production of IgG against brain antigens in sera from the dams and/or the offspring. We used brains from non–Hg-treated mice as the source of brain antigens (embryonic day [ED] 14, PND 1, PND 21, or PND 70) and a dam’s brain. The HgCl2-treated A.SW mice produced IgG (sera from dams, PND 21, and PND 70 offspring) to brain antigens regardless of age, whereas no enhanced production of anti-brain antibodies was seen in either the H2O (vehicle control) group or the MeHg group. Neither HgCl2 nor MeHg induced IgG antibodies to brain antigens in mice of the A/WySnJ control strain (Fig. 1). Brain-reactive IgG was also detected in the sera from the HgCl2-treated A.SW dams when the brain antigens used were from Hg-treated PND 21 A.SW offspring or vehicle control PND 21 BALB/c or A/WySnJ offspring (data not shown).
FIG. 1.

IgG to brain antigens in sera from Hg-treated dams and offspring. A.SW and A/WySnJ dams were sacrificed on weaning of the pups (PND 21), and their sera were collected and assayed (A). Sera from offspring were collected at PND 21 and 70 for male (B) and female (C) mice. Serum anti-brain antibodies were quantified by ELISA with brain antigens from brains of untreated dams and their offspring, which were harvested at ED 14, PND 1, PND 21, and PND 70. Each bar represents the mean values of IgG in sera of mice from three to five separate litters; * indicates a significant difference compared with the appropriate H2O group (p < 0.05).
HgCl2-Induced T-Cell Activation (CD4+/CD25+), Preferentially of Vβ 8.3+ T Cells
Hg-induced antigen-antibody complex deposition in kidneys has been reported to be T-cell dependent. Here, we investigated whether a particular T-cell β-chain variable region was preferentially expanded with spleens from PND 21 offspring; CD4+ T cells were considered activated based on CD25 expression. The activated CD4+ T cells were screened for Vβ expression. Because male and female A.SW mice had enhanced IgG to brain antigens after HgCl2 exposure, male and female A.SW and A/WySnJ mice were randomly selected from separate litters for Vβ expression screening. Although there was no significant increase in the number of CD4+ T cells expressing CD25 for the HgCl2-treated A.SW offspring (Fig. 2A), the percentage of the CD25+ CD4+ T cells from these mice expressing Vβ 8.3 chains was preferentially and significantly increased (Fig. 2B). There were no significant changes in the percentages of any Vβ subsets for the MeHg-treated A.SW offspring (Fig. 2B) or for the experimental or control (H2O) groups of the A/WySnJ mice (Fig. 2C).
FIG. 2.

Vβ expression by activated CD4+/CD25+ splenic T cells from PND 21 A.SW and A/WySnJ offspring. Single-cell suspensions were tested CD4+ T cells activation (A) and screened for Vβ usage after exposure to H2O, MeHg, or HgCl2 from ED 8 to PND 21 in A.SW (B) and A/WySnJ (C) offspring. Each bar represents mean of mice from three separate litters; * indicates a significant difference compared with counterpart H2O group (p < 0.05).
IgG Deposition in Various Brain Regions
The presence of IgG deposited in individual regions of perfused brains from PND 21 A.SW and A/WySnJ mice was assayed by ELISA. Elevated levels of IgG were present in all assayed brain regions of the HgCl2-treated A.SW mice only (Fig. 3). There were no differences between left brain and right brain hemispheres (data not shown).
FIG. 3.

IgG deposition in different brain regions. Brains of perfused PND 21 A.SW and A/WySnJ offspring were dissected into SN, HT, FCTX, STR, CTX, HC, and CB, and each region was assayed for presence of IgG by sandwich ELISA with goat anti-mouse IgG (γ-chain specific) as capture antibody and horseradish peroxidase-goat anti-mouse IgG whole molecule as detection antibody. IgG deposition in brain sections was assayed for PND 21 A.SW male mice (A), PND 21 A.SW female mice (B), PND 21 A/WySnJ male mice (C), and PND 21 A/WySnJ female mice (D). Each bar represents mean of three separate litters; * indicates a significant difference compared with counterpart H2O group.
Localizations of Brain Antigens
To investigate the location of these brain antigens in various brain regions, we performed immunohistochemistry. Brain sections from normal PND 21 A.SW mice were adhered to charged slides, and sera from A.SW dams were used as the primary antibodies. The brain antigens appeared to be located in the cell nuclei, and they could be found in each brain region assayed (Fig. 4). No difference was observed between male brains and female brains.
FIG. 4.
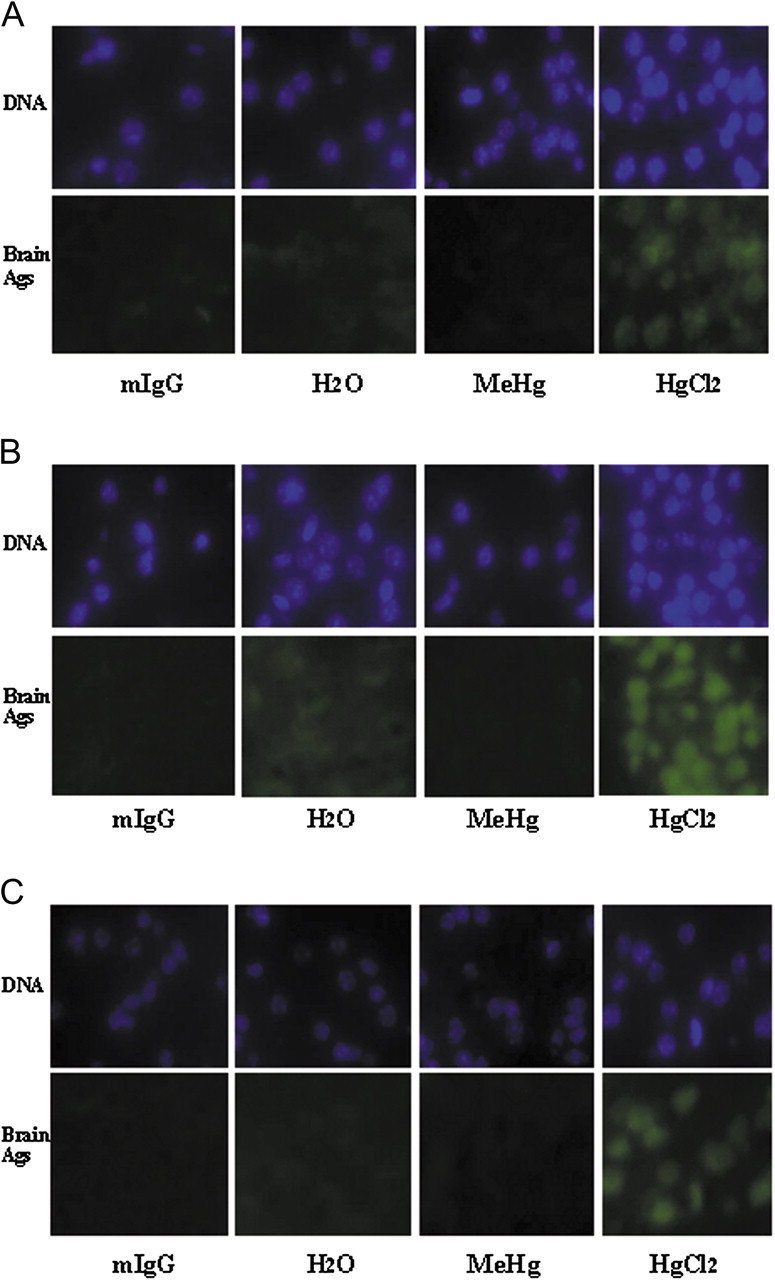
Brain antigens are located in cell nuclei in sections from various brain regions. The sections from PND 21 A.SW brains were applied to charged slides and sera from A.SW dams were added as primary antibodies. Alexa Fluor 488–conjugated anti-mouse IgG was used as detecting antibodies. DAPI was used to stain the DNA. The signal in the upper panel of each pair indicates the DNA and that in the lower panel indicates presence of bound IgG from the dams’ sera. Pictures were taken from different brain regions, (A) FCTX, (B) HC, and (C) CB. Each panel is representative of two separate experiments. mIgG, commercial purified mouse IgG
Molecular Mass Values of the Brain Antigens Detected by the HgCl2-Induced Antibodies
Because autoantibodies to brain antigens were independent of age, only the HgCl2-treated A.SW mice produced IgG to these antigens, and the brain antigens were located in nuclei, nuclear proteins from a normal PND 21 A.SW brain were used as the source of antigens. Sera from HgCl2-treated A.SW dams were used for antibodies; sera from A.SW dams from the H2O group and commercial murine IgG were used as controls. The immunoprecipitated immune complexes from the mixture of IgG and brain antigens were assayed by SDS-PAGE to determine the size of the brain antigens immunoprecipitated with the IgG. Two specific bands from the Nuc-S were observed for the HgCl2-treatment group (30 and 34 kD) (Fig. 5).
FIG. 5.

Molecular mass values of brain antigens bound by sera IgG from HgCl2-treated A.SW dams. Nuclear proteins extracted from a normal PND 21 A.SW offspring brain were immunoprecipitated by A.SW dam sera IgG conjugated by magnetic beads. After dissociation by boiling in SDS buffer for 5 min, the supernatant was loaded onto 12% SDS gel (reducing conditions), and electrophoresed; the gel was stained with SYPRO Ruby. Stained gels were assayed by the Fuji LAS3000 imaging system. mIgG: commercial purified mouse IgG.
Inflammation in Various Brain Regions
To assess whether the IgG deposition induced by HgCl2 in A.SW brains caused inflammation, we assayed the brains for cytokine expression at PND 21. Female A.SW mice showed significantly increased GM-CSF, IL-1β, and MCP-1 in the CB after MeHg treatment, whereas MCP-1 was significantly increased in the CB of the HgCl2-treated group (Fig. 6). Male A.SW mice had only significantly increased IL-1β in the FCTX after HgCl2 treatment. These data indicate that for female A.SW mice, inflammation occurred preferentially in the CB, whereas for male A.SW mice, inflammation occurred preferentially in the FCTX. IL-12(p70) was decreased in the HT of male A.SW mice treated with MeHg and in STR of female A.SW mice treated with HgCl2. IL-10, IL-13, IL-2, and IL-7 were not changed among the three groups. IFN-γ, IL-4, IL-5, IL-6, and TNF-α were not detectable (data not shown). Interestingly, vehicle control (H2O) A.SW males and females showed significant differential expression of some cytokines in the brain, and MeHg or HgCl2 exposure eliminated some of these significant differences (Table 2).
FIG. 6.

CNS cytokine expression in PND 21 A.SW brain regions. Brain regions (SN, HT, FCTX, STR, CTX, HC, and CB) from PND 21 A.SW male (A, B, and C) and female (D, E, and F) offspring were assayed. Each bar represents the mean values of mice from three separate litters; * indicates a significant difference with respect to the counterpart H2O group.
TABLE 2.
Hg Treatment Eliminated the Preexisting Significant Gender Differences in IL-13, IL-12(p70), and IL-10 Expression between PND 21 A.SW Males and Females
| H2O |
MeHg |
HgCl2 |
|||||
| Cytokine | Regiona | Male | Female | Male | Female | Male | Female |
| IL-13 | SN | 19.4 ± 4.1b | 2.3 ± 2.1 | 12.9 ± 14.1 | 1.1 ± 1.9 | 5.0 ± 8.7 | 1.4 ± 2.5 |
| HT | 4.4 ± 1.8b | 0.3 ± 0.2 | 0.7 ± 0.7 | 0.1 ± 0.1 | 0.7 ± 1.2 | 0 | |
| IL-12(p70) | HT | 3.4 ± 0.4b | 0.1 ± 0.2 | 0.4 ± 0.4 | 0 | 1.7 ± 1.1 | 0 |
| IL-10 | CB | 0b | 0.8 ± 0 | 0 | 0.4 ± 0.4 | 0 | 0.1 ± 0.1 |
Only brain regions with a preexisting difference in the expression of a cytokine between males and females are shown.
The values represent the mean ± SD of picograms of cytokine per milligram of brain protein for male and female pups from three separate litters (n = 3). Significant differences between vehicle control (H2O) males and females existed only for IL-13, IL-12, and IL-10, and Hg exposure eliminated these differences.
Behavior Changes Were Induced in PND 70 A.SW Females by MeHg or HgCl2
To assess whether the IgG and cytokine expression changes in the brains caused any long-lasting behavior modifications, we measured exploratory behavior at PND 70.
A.SW and A/WySnJ mice from the two experimental Hg groups and the H2O control group were tested for 45 min, over three cycles of 15 min. A.SW female mice of both the MeHg and HgCl2 groups showed significantly increased activity compared with the activity of the corresponding H2O group, with regard to horizontal activities (HACTV), number of movements (MOVNO), movement time (MOVTIME), and total distance (TOTDIST). These behavior changes were not observed for either A/WySnJ mice or male A.SW mice (Fig. 7). The significant changes in behaviors of A.SW females were due to the elimination, by Hg exposure, of habituation (Bolivar et al., 2000) in the third 15-min period.
FIG. 7.

Exploratory behavior of PND 70 mice was assayed after different Hg exposures. Each bar represents behavioral assessment of five to eight mice from separate litters. The behavior was assayed in 15-min cycles for a total testing period of 45 min. Multiple behavioral parameters can be obtained with this assay but only those activities (HACTV, horizontal activities; MOVNO, number of movements; MOVTIME, time of movements [seconds]; and TOTDIST, total distance [centimeters]) that displayed a significant change are shown. Behavior changes of PND 70 A.SW and A/WySnJ offspring were tested; * indicates a significant difference of each cycle or the total 45 min compared with the counterpart H2O group; # indicate a significant difference between males and females within the H2O group.
DISCUSSION
The present study was designed to investigate immunologic responses in the brains of Hg-exposed mice. Developmental exposure to HgCl2 (50μM) induced T-cell activation, preferentially of Vβ chain 8.3, and brain-reactive IgG to nuclear proteins (30 and 34 kD); the IgG was deposited in all brain regions examined. Although male and female A.SW mice exposed to HgCl2 had IgG deposited in multiple brain regions, expression of several cytokines was enhanced only in the CB of female A.SW mice, whereas male A.SW mice only had increased IL-1β in the FCTX. The Hg exposures led to modified exploratory behavior only in the female A.SW mice, and the effect appears to correlate with MCP-1 expression in the CB.
Hg-induced autoimmunity is known to be a gene-controlled disease (Abedi-Valugerdi et al., 2001; Hultman et al., 1993). We detected IgG to brain antigens in A.SW, but not A/WySnJ mice, that had been HgCl2 exposed. Both MeHg and HgCl2 induce autoantibodies to renal antigens. It has been suggested that MeHg-induced autoimmunity is due to formation of Hg2+ through demethylation of MeHg in vivo, whereas MeHg has a direct inhibitory effect on the immune system (Havarinasab et al., 2007; Hirokawa and Hayashi, 1980; Hultman and Hansson-Georgiadis, 1999; Ilback et al., 1991; Ohi et al., 1976). The IgG to brain antigens induced in A.SW mice by the HgCl2 exposure reacted to antigens derived from the brains of unexposed control mice of various ages and antigens from brains of PND 21 MeHg- or HgCl2-treated A.SW mice and untreated PND 21 brains from A/WySnJ or BALB/c mice; this pattern of reactivity indicates that the brain antigens are normal proteins not age specific and are distributed among multiple strains of mice. Because Hg-induced autoantibody production to the kidney was suggested to be driven by Hg2+-modified fibrillarin and because the autoantibodies react to both normal and Hg-adducted fibrillarin (Hultman et al., 1989; Pollard et al., 1997), we assume that the brain antigens in normal A.SW mice were first modified by Hg2+; the structurally altered proteins then trigger the production of autoantibodies capable of reacting with both the normal and the modified brain antigens. In the MeHg-treated A.SW mice, we assume that the amount of Hg2+ derived from MeHg was insufficient to induce the IgG to brain antigens. The MeHg exposure was substantially lower than the HgCl2 exposure dose because doses of MeHg > 1.25μM were toxic. Because the brain-reactive IgG still could be detected in the sera of PND 70 A.SW mice treated with HgCl2, these autoantibodies must have been produced by the offspring themselves. IgG in dam serum can pass through the placenta (Kim et al., 2009), and the intact blood-brain barrier (BBB) is not fully developed until 2 weeks after birth (Ribatti et al., 2006). Therefore, it is reasonable to propose that the brain-reactive IgG in the sera from A.SW dams treated with HgCl2 could bind to the fetal brain antigens during the gestation period and could initiate autoimmunity in the offspring.
Hg-induced autoimmunity has been suggested to be due to T cell–dependent polyclonal B-cell production of autoantibodies, in which T-cell helper activity is critical (Pelletier et al., 1987; Stuber and Strober, 1996). We investigated activation of CD4+ T cells and their T-cell receptor (TCR) usage involved in the response to Hg be assessing Vβ expression of CD4+CD25+ splenic cells. The absence of a significant increase in the total number of CD25+ CD4+ T cells in the HgCl2-treated A.SW mice may be due to inclusion of activated T cells and Treg cells in this general subpopulation. We can only suggest that the increased number of Vβ 8.3+ CD25+ CD4+ T cells represent activated CD4+ T cells responding to autoantigen or Hg-modified self-antigens. However, it is interesting that HgCl2, but not MeHg, treatment of the A.SW mice caused an increase in Vβ 8.3 expression. Fillion et al. (1997) reported that there was no change in Vβ expression in Brown-Norway (BN) rats treated with HgCl2. Although that finding appears to conflict with our results, the two sets of experiments differ. Those researchers measured the Vβ repertoire on rat CD4+ cells from lymph node and that population included all of the CD4+ T cells. In contrast, we screened the Vβ expression on activated (CD25+) murine splenic CD4+ T cells. Jiang and Moller (1996) reported that in vitro treatment of splenic CD4+ T cells from BALB/c mice with HgCl2 increased TCR Vβ 6, 8.2, 8.3, 10, and 14 expressions, and depletion of Vβ 8 profoundly inhibited the HgCl2-induced response. Antigens from blood could be captured, processed, and presented in spleen. Thus, for Hg-induced autoimmunity responses to brain antigens, we propose that after the brain antigens were modified by Hg2+, they were released into the circulation, allowing transport into the spleen. There, they were processed and presented by APCs to T cells with Vβ 8.3 chains, for Vβ 8.3+ T-cell expansion.
In the HgCl2-treated PND 21 A.SW mice, IgG deposition was observed in all of the assayed brain sections. Meanwhile the serum IgG level was also enhanced in HgCl2-treated A.SW mice. Here, we cannot conclude that the IgG deposition in brain sections was caused by brain-reactive IgG, enhanced serum IgG, or both. However, HgCl2-treated F1 offspring (H-2b), which had enhanced serum IgG but not brain-reactive IgG, also had IgG deposition in their brains but no behavior changes (our unpublished data), suggesting enhanced serum IgG could increase brain IgG deposition, and the behavior changes probably were more linked to the brain-reactive IgG.
Fibrillarin, a small nucleolar protein with a molecular mass of about 34 kD, is the major renal antigen that is reactive with Hg2+-induced autoantibodies (Hultman and Enestrom, 1992; Pollard et al., 1997). In our experiment, two antigen bands with molecular masses of about 30 and 34 kD were found by immunoprecipitation of PND 21 brain nuclear proteins that were slightly bound to DNA, with sera from HgCl2-treated dams. Immunohistochemical results indicate that these brain antigens were nuclear proteins and that the autoantibodies also reacted with kidney and liver homogenates. Autoantibodies to nucleolar proteins of 60–70 kD and 10–15 kD also have been reported in HgCl2-treated mice (Hultman et al., 1992). The 34-kD brain antigen that we observed in HgCl2-treated A.SW mice is probably fibrillarin; further experiments need to be performed to identify the other band (30 kD). Although we still do not know which organ was the original source of these antigens that induced the autoantibodies or whether multiple organs were involved, we suggest that based on Hg detection in the brain and that neurons are sensitive to Hg (Bjorkman et al., 2007; Monnet-Tschudi et al., 1996; Stern et al., 2001), these nuclear brain antigens partially contributed to the production of the brain-reactive IgG.
Although the MeHg-treated A.SW mice had no detectable IgG to brain antigens and did not show elevated IgG levels in their brains, several previous studies have reported that MeHg can directly activate microglial cells from sensitive strains of mice and rats (Eskes et al., 2002; Garg and Chang, 2006). We evaluated whether brain inflammation was induced by MeHg or HgCl2 in A.SW mice. Hg exposure enhanced the expression only of GM-CSF, MCP-1, and IL-1β. GM-CSF, a hematopoietic growth factor produced by a variety of cells, is involved in inflammatory cell recruitment and modulation (Shi et al., 2006). MCP-1, a chemoattractant for monocytes derived from many cell types, including monocytes and epithelial cells, can activate endothelial cells directly and can increase the activity of cytotoxic T cells and natural killer cells (Taub et al., 1996; Werle et al., 2002). Only IL-1β expression was enhanced in A.SW males, whereas GM-CSF, MCP-1, and IL-1β increased in females. Females generally are more prone to the development of autoimmune diseases than males. For the MeHg-treated A.SW females, we suggest that the inflammation in the CB was the result of direct interaction between MeHg and microglia, neurons, and/or astrocytes. The BBB is known to differ between males and females (Oztas, 1998), but we do not believe that BBB differences are involved here because males and females had identical IgG deposition in the brain. The causes of the different inflammation patterns induced between A.SW males and females, treated with MeHg or HgCl2, are unclear. Hormones have been demonstrated to play an important role in immune modulation, but the mechanisms responsible for greater incidence of autoimmune diseases in females than in males remain unclear. Recently, it was reported that estrogen can directly activate activation-induced deaminase (AID) transcription and function in B cells and in breast and ovarian tissue (Pauklin et al., 2009); that finding sheds a new light on the function(s) of estrogen in autoimmune diseases. However, in our experiments, because brain-reactive IgG was induced in both male and female HgCl2-treated A.SW mice, we do not think that the AID activation mechanism is responsible for the gender differences.
IgG deposition occurred throughout the brain after HgCl2 exposure, but inflammation was restricted to the FCTX or CB. We currently have no explanation for why cytokine expression was enhanced only in the FCTX for males and in the CB for females when IgG was deposited in all brain regions. Perhaps, if the brain antigens are mainly nuclear proteins, the brain-reactive IgG can only bind to the brain antigens of Hg-damaged brain cells. In the CB, Purkinje cells, stellate cells, basket cells, and Golgi cells are inhibitory neurons, whereas the granule cells are excitatory neurons, which modify the activation of those inhibitory neurons (Voogd and Glickstein, 1998). Hua et al. (1995) reported that Hg vapor exposure of BN rats induced loss of Purkinje cells in the CB. Leyshon and Morgan (1991) reported that MeHg exposure on rats selectively damaged the CB, with particularly granule cells. Accordingly, we suggest that, in our MeHg-treated PND 21 A.SW females, MeHg activated microglia and damaged granule cells directly. The necrotic granule cells then further activated the microglia, inducing inflammation in the CB and that in turn exacerbated damage to yet more granule cells and inhibitory neurons. In HgCl2-treated PND 21 A.SW female mice, Hg2+ taken up by Purkinje cells might modify their nuclear brain antigens resulting in Purkinje cell necrosis, which triggers brain-reactive IgG and subsequent CB inflammation and damage to other neurons.
Our behavioral analysis of treatment of mice with both MeHg and HgCl2 modified habituation, but only in female A.SW mice, which was consistent with the increased expression of inflammatory cytokines in their CB. The significant difference of habituation behavior between males and females in the H2O group was eliminated by the Hg exposure, which was similar to the changes of several cytokines (IL-13 in the SN and HT, IL-12(p70) in the HT, and IL-10 in the CB). However, the trend for IL-13 and IL-12(p70), which was decreased by Hg exposure, was not in concordance with the activation enhancement of behaviors in Hg-exposed female A.SW mice, suggesting the difference of these cytokines might help explain the sex difference of habituation in normal mice, whereas after Hg exposure the inflammation cytokines especially MCP-1 in the CB may dominate. The mechanism remains to be elucidated.
In conclusion, we have shown that developmental exposure to HgCl2 induces inflammation in the CB and increases exploratory behaviors in female A.SW mice, as a function of production of brain-reactive IgG. In contrast, exposure to MeHg produced effects that were independent of autoantibody production. For Hg-treated male A.SW mice, IgG deposition was seen in all of the assayed brain regions and inflammation was found in the HT in HgCl2 group, yet no change in exploratory behaviors was detected. Moreover, Hg caused the exploratory behaviors of A.SW females to become more like those of males.
FUNDING
National Institutes of Environmental Health Sciences grants R01 ES011135 and R21 ES013857.
Acknowledgments
The authors thank Joan Pedersen-Lane, Renjie Song, and Steve Rich in the Immunology Core Facility for flow cytometry assistance, Dr Parsons lab for Hg concentration measurement, Nancy Andersen for her aid with Luminex analysis, Richard Cole and Robert Stack in the Advanced Light Microscopy and Image Analysis Core for fluorescent microscope assistance, Tapan Mondal for nuclear proteins extraction, and Veronica Miller and Ian Guest for help with the immunohistochemistry. We also thank Adrianna Verschoor for her editorial assistance.
References
- Abedi-Valugerdi M, Hansson M, Moller G. Genetic control of resistance to mercury-induced immune/autoimmune activation. Scand. J. Immunol. 2001;54:190–197. doi: 10.1046/j.1365-3083.2001.00932.x. [DOI] [PubMed] [Google Scholar]
- Abedi-Valugerdi M, Hu H, Moller G. Mercury-induced renal immune complex deposits in young (NZB x NZW) F1 mice: characterization of antibodies/autoantibodies. Clin. Exp. Immunol. 1997;110:86–91. doi: 10.1046/j.1365-2249.1997.4901392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Balaghi S, Moller E, Moller G, Abedi-Valugerdi M. Mercury induces polyclonal B cell activation, autoantibody production and renal immune complex deposits in young (NZB x NZW) F1 hybrids. Eur. J. Immunol. 1996;26:1519–1526. doi: 10.1002/eji.1830260717. [DOI] [PubMed] [Google Scholar]
- Aschner M, Lorscheider FL, Cowan KS, Conklin DR, Vimy MJ, Lash LH. Metallothionein induction in fetal rat brain and neonatal primary astrocyte cultures by in utero exposure to elemental mercury vapor (Hg0) Brain Res. 1997;778:222–232. doi: 10.1016/s0006-8993(97)01095-0. [DOI] [PubMed] [Google Scholar]
- Aschner M, Syversen T, Souza DO, Rocha JB. Metallothioneins: mercury species-specific induction and their potential role in attenuating neurotoxicity. Exp. Biol. Med. (Maywood) 2006;231:1468–1473. doi: 10.1177/153537020623100904. [DOI] [PubMed] [Google Scholar]
- Bello SC. Autism and environmental influences: review and commentary. Rev. Environ. Health. 2007;22:139–156. doi: 10.1515/reveh.2007.22.2.139. [DOI] [PubMed] [Google Scholar]
- Bjorkman L, Lundekvam BF, Laegreid T, Bertelsen BI, Morild I, Lilleng P, Lind B, Palm B, Vahter M. Mercury in human brain, blood, muscle and toenails in relation to exposure: an autopsy study. Environ. Health. 2007;6:30. doi: 10.1186/1476-069X-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolivar VJ, Caldarone BJ, Reilly AA, Flaherty L. Habituation of activity in an open field: a survey of inbred strains and F1 hybrids. Behav. Genet. 2000;30:285–293. doi: 10.1023/a:1026545316455. [DOI] [PubMed] [Google Scholar]
- Braunschweig D, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Croen LA, Pessah IN, Van de Water J. Autism: maternally derived antibodies specific for fetal brain proteins. Neurotoxicology. 2008;29:226–231. doi: 10.1016/j.neuro.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvey PM, McRae A, Lint TF, Ptak LR, Lo ES, Goetz CG, Klawans HL. The potential use of a dopamine neuron antibody and a striatal-derived neurotrophic factor as diagnostic markers in Parkinson’s disease. Neurology. 1991;41:53–58. doi: 10.1212/wnl.41.5_suppl_2.53. discussion 59–60. [DOI] [PubMed] [Google Scholar]
- Cox B, Emili A. Tissue subcellular fractionantion and protein extraction for use in mass-spectrometry-based proteomics. Nat. Protoc. 2006;1:1872–1878. doi: 10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- Croen LA, Braunschweig D, Haapanen L, Yoshida CK, Fireman B, Grether JK, Kharrazi M, Hansen RL, Ashwood P, Van de Water J. Maternal mid-pregnancy autoantibodies to fetal brain protein: the early markers for autism study. Biol. Psychiatry. 2008;64:583–588. doi: 10.1016/j.biopsych.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debes F, Budtz-Jorgensen E, Weihe P, White RF, Grandjean P. Impact of prenatal methylmercury exposure on neurobehavioral function at age 14 years. Neurotoxicol. Teratol. 2006;28:363–375. doi: 10.1016/j.ntt.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins D, Petro A, Pollard N, Freedman JH, Levin ED. Mercury-induced cognitive impairment in metallothionein-1/2 null mice. Neurotoxicol. Teratol. 2008;30:88–95. doi: 10.1016/j.ntt.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes C, Honegger P, Juillerat-Jeanneret L, Monnet-Tschudi F. Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotection via interactions with astrocytes and IL-6 release. Glia. 2002;37:43–52. doi: 10.1002/glia.10019. [DOI] [PubMed] [Google Scholar]
- Falluel-Morel A, Sokolowski K, Sisti HM, Zhou X, Shors TJ, Dicicco-Bloom E. Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J. Neurochem. 2007;103:1968–1981. doi: 10.1111/j.1471-4159.2007.04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillion J, Baccala R, Pannetier C, Kuhn J, Druet P, Bellon B. Evidence for heterogeneous TCR V beta repertoire expression in mercury-induced immune disorders in rats. Int. Immunol. 1997;9:263–271. doi: 10.1093/intimm/9.2.263. [DOI] [PubMed] [Google Scholar]
- Garg TK, Chang JY. Methylmercury causes oxidative stress and cytotoxicity in microglia: attenuation by 15-deoxy-delta 12, 14-prostaglandin J2. J. Neuroimmunol. 2006;171:17–28. doi: 10.1016/j.jneuroim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Guzzi G, Fogazzi GB, Cantu M, Minoia C, Ronchi A, Pigatto PD, Severi G. Dental amalgam, mercury toxicity, and renal autoimmunity. J. Environ. Pathol. Toxicol. Oncol. 2008;27:147–155. doi: 10.1615/jenvironpatholtoxicoloncol.v27.i2.70. [DOI] [PubMed] [Google Scholar]
- Havarinasab S, Bjorn E, Nielsen JB, Hultman P. Mercury species in lymphoid and non-lymphoid tissues after exposure to methyl mercury: correlation with autoimmune parameters during and after treatment in susceptible mice. Toxicol. Appl. Pharmacol. 2007;221:21–28. doi: 10.1016/j.taap.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Hirokawa K, Hayashi Y. Acute methyl mercury intoxication in mice—effect on the immune system. Acta Pathol. Jpn. 1980;30:23–32. doi: 10.1111/j.1440-1827.1980.tb01302.x. [DOI] [PubMed] [Google Scholar]
- Hua J, Brun A, Berlin M. Pathological changes in the Brown Norway rat cerebellum after mercury vapour exposure. Toxicology. 1995;104:83–90. doi: 10.1016/0300-483x(95)03143-4. [DOI] [PubMed] [Google Scholar]
- Huang CF, Hsu CJ, Liu SH, Lin-Shiau SY. Neurotoxicological mechanism of methylmercury induced by low-dose and long-term exposure in mice: oxidative stress and down-regulated Na+/K(+)-ATPase involved. Toxicol. Lett. 2008;176:188–197. doi: 10.1016/j.toxlet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility to mercury. I. Autoantibody profiles and systemic immune deposits in inbred, congenic, and intra-H-2 recombinant strains. Clin. Immunol. Immunopathol. 1992;65:98–109. doi: 10.1016/0090-1229(92)90212-7. [DOI] [PubMed] [Google Scholar]
- Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility to mercury. II. Autoantibody profiles and renal immune deposits in hybrid, backcross, and H-2d congenic mice. Clin. Immunol. Immunopathol. 1993;68:9–20. doi: 10.1006/clin.1993.1088. [DOI] [PubMed] [Google Scholar]
- Hultman P, Enestrom S. Mercury induced antinuclear antibodies in mice: characterization and correlation with renal immune complex deposits. Clin. Exp. Immunol. 1988;71:269–274. [PMC free article] [PubMed] [Google Scholar]
- Hultman P, Enestrom S. Dose-response studies in murine mercury-induced autoimmunity and immune-complex disease. Toxicol. Appl. Pharmacol. 1992;113:199–208. doi: 10.1016/0041-008x(92)90115-9. [DOI] [PubMed] [Google Scholar]
- Hultman P, Enestrom S, Pollard KM, Tan EM. Anti-fibrillarin autoantibodies in mercury-treated mice. Clin. Exp. Immunol. 1989;78:470–477. [PMC free article] [PubMed] [Google Scholar]
- Hultman P, Hansson-Georgiadis H. Methyl mercury-induced autoimmunity in mice. Toxicol. Appl. Pharmacol. 1999;154:203–211. doi: 10.1006/taap.1998.8576. [DOI] [PubMed] [Google Scholar]
- Ilback NG, Sundberg J, Oskarsson A. Methyl mercury exposure via placenta and milk impairs natural killer (NK) cell function in newborn rats. Toxicol. Lett. 1991;58:149–158. doi: 10.1016/0378-4274(91)90169-7. [DOI] [PubMed] [Google Scholar]
- Jedrychowski W, Perera F, Rauh V, Flak E, Mroz E, Pac A, Skolicki Z, Kaim I. Fish intake during pregnancy and mercury level in cord and maternal blood at delivery: an environmental study in Poland. Int. J. Occup. Med. Environ. Health. 2007;20:31–37. doi: 10.2478/v10001-007-0002-8. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Moller G. In vitro effects of HgCl2 on murine lymphocytes. II. Selective activation of T cells expressing certain V beta TCR. Int. Immunol. 1996;8:1729–1736. doi: 10.1093/intimm/8.11.1729. [DOI] [PubMed] [Google Scholar]
- Kaur P, Aschner M, Syversen T. Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology. 2006;27:492–500. doi: 10.1016/j.neuro.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Kim J, Mohanty S, Ganesan LP, Hua K, Jarjoura D, Hayton WL, Robinson JM, Anderson CL. FcRn in the yolk sac endoderm of mouse is required for IgG transport to fetus. J. Immunol. 2009;182:2583–2589. doi: 10.4049/jimmunol.0803247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence DA, Bolivar VJ, Hudson CA, Mondal TK, Pabello NG. Antibody induction of lupus-like neuropsychiatric manifestations. J. Neuroimmunol. 2007;182:185–194. doi: 10.1016/j.jneuroim.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyshon K, Morgan AJ. An integrated study of the morphological and gross-elemental consequences of methyl mercury intoxication in rats, with particular attention on the cerebellum. Scanning Microsc. 1991;5:895–904. [PubMed] [Google Scholar]
- Martinsson K, Carlsson L, Kleinau S, Hultman P. The effect of activating and inhibiting Fc-receptors on murine mercury-induced autoimmunity. J. Autoimmun. 2008;31:22–29. doi: 10.1016/j.jaut.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Monnet-Tschudi F, Zurich MG, Honegger P. Comparison of the developmental effects of two mercury compounds on glial cells and neurons in aggregate cultures of rat telencephalon. Brain Res. 1996;741:52–59. doi: 10.1016/s0006-8993(96)00895-5. [DOI] [PubMed] [Google Scholar]
- Montgomery KS, Mackey J, Thuett K, Ginestra S, Bizon JL, Abbott LC. Chronic, low-dose prenatal exposure to methylmercury impairs motor and mnemonic function in adult C57/B6 mice. Behav. Brain Res. 2008;191:55–61. doi: 10.1016/j.bbr.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Morken TS, Sonnewald U, Aschner M, Syversen T. Effects of methylmercury on primary brain cells in mono- and co-culture. Toxicol. Sci. 2005;87:169–175. doi: 10.1093/toxsci/kfi227. [DOI] [PubMed] [Google Scholar]
- Nakagawa R, Yumita Y, Hiromoto M. Total mercury intake from fish and shellfish by Japanese people. Chemosphere. 1997;35:2909–2913. doi: 10.1016/s0045-6535(97)00351-2. [DOI] [PubMed] [Google Scholar]
- Nautiyal KM, Ribeiro AC, Pfaff DW, Silver R. Brain mast cells link the immune system to anxiety-like behavior. Proc. Natl. Acad. Sci. U.S.A. 2008;105:18053–18057. doi: 10.1073/pnas.0809479105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi G, Fukuda M, Seto H, Yagyu H. Effect of methylmercury on humoral immune responses in mice under conditions simulated to practical situations. Bull. Environ. Contam. Toxicol. 1976;15:175–180. doi: 10.1007/BF01685157. [DOI] [PubMed] [Google Scholar]
- Onishchenko N, Tamm C, Vahter M, Hokfelt T, Johnson JA, Johnson DA, Ceccatelli S. Developmental exposure to methylmercury alters learning and induces depression-like behavior in male mice. Toxicol. Sci. 2007;97:428–437. doi: 10.1093/toxsci/kfl199. [DOI] [PubMed] [Google Scholar]
- Oztas B. Sex and blood-brain barrier. Pharmacol. Res. 1998;37:165–167. doi: 10.1006/phrs.1997.0243. [DOI] [PubMed] [Google Scholar]
- Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J. Exp. Med. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto NC, Roza T, Morsch VM, Pereira ME. Behavioral alterations induced by HgCl2 depend on the postnatal period of exposure. Int. J. Dev. Neurosci. 2007;25:39–46. doi: 10.1016/j.ijdevneu.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Pelletier L, Pasquier R, Vial MC, Mandet C, Moutier R, Salomon JC, Druet P. Mercury-induced autoimmune glomerulonephritis: requirement for T-cells. Nephrol. Dial. Transplant. 1987;1:211–218. [PubMed] [Google Scholar]
- Pollard KM, Lee DK, Casiano CA, Bluthner M, Johnston MM, Tan EM. The autoimmunity-inducing xenobiotic mercury interacts with the autoantigen fibrillarin and modifies its molecular and antigenic properties. J. Immunol. 1997;158:3521–3528. [PubMed] [Google Scholar]
- Ribatti D, Nico B, Crivellato E, Artico M. Development of the blood-brain barrier: a historical point of view. Anat. Rec. B New. Anat. 2006;289:3–8. doi: 10.1002/ar.b.20087. [DOI] [PubMed] [Google Scholar]
- Schott K, Wormstall H, Dietrich M, Klein R, Batra A. Autoantibody reactivity in serum of patients with Alzheimer’s disease and other age-related dementias. Psychiatry Res. 1996;59:251–254. doi: 10.1016/0165-1781(95)02703-3. [DOI] [PubMed] [Google Scholar]
- Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, Zhang Y, Zhang L, Yuan ZR, Tan HS, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. 2006;16:126–133. doi: 10.1038/sj.cr.7310017. [DOI] [PubMed] [Google Scholar]
- Silva SC, Correia C, Fesel C, Barreto M, Coutinho AM, Marques C, Miguel TS, Ataide A, Bento C, Borges L, et al. Autoantibody repertoires to brain tissue in autism nuclear families. J. Neuroimmunol. 2004;152:176–182. doi: 10.1016/j.jneuroim.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Soldin OP, O'Mara DM, Aschner M. Thyroid hormones and methylmercury toxicity. Biol. Trace Elem. Res. 2008;126:1–12. doi: 10.1007/s12011-008-8199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Cox C, Cernichiari E, Balys M, Weiss B. Perinatal and lifetime exposure to methylmercury in the mouse: blood and brain concentrations of mercury to 26 months of age. Neurotoxicology. 2001;22:467–477. doi: 10.1016/s0161-813x(01)00047-x. [DOI] [PubMed] [Google Scholar]
- Stringari J, Nunes AK, Franco JL, Bohrer D, Garcia SC, Dafre AL, Milatovic D, Souza DO, Rocha JB, Aschner M, et al. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol. Appl. Pharmacol. 2008;227:147–154. doi: 10.1016/j.taap.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber E, Strober W. The T cell-B cell interaction via OX40–OX40L is necessary for the T cell-dependent humoral immune response. J. Exp. Med. 1996;183:979–989. doi: 10.1084/jem.183.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Suzuki H, Doi Y, Liang J, Kawanokuchi J, Mizuno T, Sawada M, Suzumura A. Blockade of microglial glutamate release protects against ischemic brain injury. Exp. Neurol. 2008;214:144–146. doi: 10.1016/j.expneurol.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Nakamura K, Takeda M, Tada K, Suzuki H, Morita H, Okado T, Hariguchi S, Nishimura T. Enzyme-linked immunosorbent assay for human autoantibody to glial fibrillary acidic protein: higher titer of the antibody is detected in serum of patients with Alzheimer's disease. Acta. Neurol. Scand. 1989;80:554–560. doi: 10.1111/j.1600-0404.1989.tb03926.x. [DOI] [PubMed] [Google Scholar]
- Taniai H, Nishiyama T, Miyachi T, Imaeda M, Sumi S. Genetic influences on the broad spectrum of autism: study of proband-ascertained twins. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008;147B:844–849. doi: 10.1002/ajmg.b.30740. [DOI] [PubMed] [Google Scholar]
- Taub DD, Ortaldo JR, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. J. Leukoc. Biol. 1996;59:81–89. doi: 10.1002/jlb.59.1.81. [DOI] [PubMed] [Google Scholar]
- Trasande L, Landrigan PJ, Schechter C. Public health and economic consequences of methyl mercury toxicity to the developing brain. Environ. Health Perspect. 2005;113:590–596. doi: 10.1289/ehp.7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasande L, Schechter CB, Haynes KA, Landrigan PJ. Mental retardation and prenatal methylmercury toxicity. Am. J. Ind. Med. 2006;49:153–158. doi: 10.1002/ajim.20268. [DOI] [PubMed] [Google Scholar]
- Vas J, Monestier M. Immunology of mercury. Ann. N. Y. Acad. Sci. 2008;1143:240–267. doi: 10.1196/annals.1443.022. [DOI] [PubMed] [Google Scholar]
- Voogd J, Glickstein M. The anatomy of the cerebellum. Trends Neurosci. 1998;21:370–375. doi: 10.1016/s0166-2236(98)01318-6. [DOI] [PubMed] [Google Scholar]
- Werle M, Schmal U, Hanna K, Kreuzer J. MCP-1 induces activation of MAP-kinases ERK, JNK and p38 MAPK in human endothelial cells. Cardiovasc. Res. 2002;56:284–292. doi: 10.1016/s0008-6363(02)00600-4. [DOI] [PubMed] [Google Scholar]
- Yang ZW, Chengappa KN, Shurin G, Brar JS, Rabin BS, Gubbi AV, Ganguli R. An association between anti-hippocampal antibody concentration and lymphocyte production of IL-2 in patients with schizophrenia. Psychol. Med. 1994;24:449–455. doi: 10.1017/s0033291700027410. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Watanabe C, Kishimoto M, Yasutake A, Satoh M, Sawada M, Akama Y. Behavioral changes in metallothionein-null mice after the cessation of long-term, low-level exposure to mercury vapor. Toxicol. Lett. 2006;161:210–218. doi: 10.1016/j.toxlet.2005.09.007. [DOI] [PubMed] [Google Scholar]