

Abstract
Phthalates and other endocrine-disruptive chemicals are manufactured in large quantities for use as plasticizers and other commercial applications, resulting in ubiquitous human exposure and thus, concern regarding their toxicity. Innate defense against small molecule exposures is controlled in large part by the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR). The human CAR gene undergoes multiple alternative splicing events resulting in the CAR2 and CAR3 variant receptors. Recent studies from our laboratory show that CAR2 is potently and specifically activated by di(2-ethylhexyl) phthalate (DEHP). We hypothesized that alternative splicing is a mechanism for increasing CAR’s functional diversity, broadening the human receptors’ repertoire of response to environmental xenobiotics. In these studies, we examine the interaction of alternatively spliced CARs and PXR with a range of suspected endocrine disruptors, including phthalates, bisphenol A (BPA), and 4-N-nonylphenol (NP). Transactivation and two-hybrid studies in COS-1 cells revealed differential selectivity of endocrine-disrupting chemicals for the variant CAR and PXR. Ex vivo studies showed DEHP and di-isononyl phthalate potently induced CYP2B6 and CYP3A4 expression in human hepatocytes. Mutation analysis of CAR2, in silico modeling, and ligand docking studies suggested that the SPTV amino acid insertion of CAR2 creates a unique ligand-binding pocket. Alternative gene splicing results in variant CAR receptors that selectively recognize phthalates and BPA. The interaction of phthalates with CAR and PXR suggests a xenobiotic response that is complex and biologically redundant.
Keywords: nuclear receptors, constitutive androstane receptor, pregnane X receptor, phthalates, endocrine disrupting compounds, liver
The innate defense against exposure to xenobiotics is controlled in large part by the expression of a milieu of metabolic enzymes and transporters. A critical part of this defense system is the xenobiotic exposure–induced upregulation of these enzymes and transporters, which is accomplished through transcriptional activation (Xu et al., 2005). Nuclear receptors play a vital role in this process. Nuclear receptors are superfamily of receptors that are ligand activated, and upon activation, bind to specific sequences proximal to target genes, resulting in increased gene expression. Two important members of this family are the constitutive androstane receptor (CAR) (Baes et al., 1994) and the pregnane X receptor (PXR) (Kliewer et al., 1998).
Together CAR and PXR orchestrate the expression of phases I, II, and III metabolism enzymes (Wei et al., 2000, 2002) by heterodimerizing with the retinoid X receptor α (RXRα) and binding specific DNA response elements upstrem of target genes. The battery of CAR target genes includes certain cytochrome P450s, UDP-glucuronosyltransferase, sulfotransferase, glutathione S-transferase, aldehyde dehydrogenase, and ATP-binding cassette transporter families (Maglich et al., 2002; Ueda et al., 2002). In addition to xenobiotics, CAR is also involved in regulation of metabolism of endogenous compounds, such as bile acids (Guo et al., 2003) and steroid hormones (Xie et al., 2003). More recently, these receptors have been implicated in the regulation of liver energy metabolism (Kodama et al., 2004; Konno et al., 2008; Masson et al., 2008). CYP2B6 and CYP3A4 are the prototypical target genes for CAR (Sueyoshi et al., 1999) and PXR (Lehmann et al., 1998), respectively, but there is considerable cross talk between the two receptors on the regulatory elements of these genes (Pascussi et al., 2003).
Our laboratory initially identified that alternative splicing events are important contributors to CAR expression (Auerbach et al., 2003), as corroborated by others (Jinno et al., 2004; Lamba et al., 2004; Ross et al., 2010). Two prominent CAR splice variants are CAR2, generated by an alternative splice acceptor site in intron 6, resulting in a four amino acid insertion (SPTV) in the vicinity of the receptor’s ligand-binding pocket (LBP), and CAR3, generated by an alternative splice acceptor site in intron 7, leading to a five amino acid insertion (APYLT) in the receptor’s ligand-binding/heterodimerization domain (Auerbach et al., 2003). The CAR2 and CAR3 transcripts are prominently expressed in human liver and primary hepatocytes, with combined levels ranging up to ∼50% of total CAR (Dekeyser et al., 2009; Jinno et al., 2004; Ross et al., 2010). Although immunologically indistinguishable with currently available antibody reagents, the variant receptor proteins are stable in in vitro expression systems, in bacteria, and in a host of transiently and stably transfected mammalian cells (Auerbach et al., 2003; Chen et al., 2010; Dekeyser et al., 2009). While the reference human CAR (referred to hereafter as CAR1) has high constitutive activity in the absence of exogenous ligand, CAR2 and CAR3 require ligand for activity. The presence of multiple variants of CAR (Arnold et al., 2004; Jinno et al., 2004; Lamba et al., 2004; Savkur et al., 2003) provides potential for increased diversity of the CAR gene. Notably, the CAR2 site is not conserved in marmoset, mouse, or rat; thus, these species are incapable of generating a CAR2-like protein (Dekeyser et al., 2009).
Although controversial, phthalates, bisphenol A (BPA), and nonylphenols are among the many chemicals that have been linked to endocrine disruption (Diamanti-Kandarakis et al., 2009). Because of their wide use in a variety of commercial and industrial applications, they are ubiquitous in the environment and the potential for human exposure is high (Bonefeld-Jorgensen et al., 2007; Schettler, 2006). For example, in 2006, phthalate consumption reached 1,000,000 tons in Western Europe alone (Wittassek and Angerer, 2008). In a previous report, we demonstrated that di(2-ethylhexyl) phthalate (DEHP) is a highly potent agonist of CAR2 but is not a strong agonist of CAR1 or CAR3 (Dekeyser et al., 2009). This observation provided strong support for the hypothesis that alternative splicing of the CAR gene is a mechanism for increasing the functional diversity of the receptor. The current studies further examine phthalates and other endocrine disruptive compounds as activators of xenobiotic metabolism pathways through human CAR variants and PXR and to examine specific properties of CAR2 that allow for its high-affinity binding to DEHP and di-isononyl phthalate (DiNP).
MATERIALS AND METHODS
Chemicals.
DEHP (CAS #117-81-7), 5α-androstan-3α-ol (ANDRO; CAS #7657-50-3), DiNP (CAS #68515-48-0), dimethyl sulfoxide (DMSO; CAS #67-68-5), phenobarbital (PB; CAS #50-06-6) bisphenol A (CAS #80-05-7), and nonylphenol (CAS #104-40-5) were purchased from Sigma-Aldrich (St Louis, MO). Diethyl phthalate (DEP; CAS #84-66-2), di-n-butyl phthalate (DnBP; CAS #84-74-2), and di-isobutyl phthalate (DiBP, CAS #84-69-5) were purchased from Alfa Aesar (Ward Hill, MA). 6-(4-Chlorophenyl)imidazo[2,1-b] [1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl) oxime (CITCO; CAS #338404-52-7) was obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA). Rifampicin (RFPM) was purchased from VWR Biosciences. TO901317 (TO; CAS #293754-55-9) was obtained from Cayman Chemical Co. (Ann Arbor, MI). All compounds were > 98% pure except DiNP, which is manufactured as mixture of 9-carbon isomers. Structures are shown in Figure 1.
FIG. 1.

Structures of the compounds used in this study. All compounds were supplied at >98% purity and were isomerically pure, except DiNP, which was a mixture of 9-carbon isomers.
Cell culture and transfections.
Culture conditions for maintenance of COS-1 cells (ATCC, Manassas, VA) were previously published (Auerbach et al., 2005). COS-1 cells were used since they are devoid of endogenous CAR expression/activity as demonstrated in previous reports (Auerbach et al., 2005, 2007) and by the current study, as no CAR reporter activity is detected in COS-1 cells transfected with empty expression vectors. For transfection and chemical treatments, the same medium was used except dextran/charcoal-treated fetal bovine serum (FBS) (HyClone, Logan, UT) replaced normal FBS. All transfections for luciferase reporter and mammalian two-hybrid assays were performed in a 48-well format. The vectors CMV2-CAR1, CMV2-CAR2, CMV-CAR2A, CMV-CAR2D, 3.1-RXRα, 2B6-XREM-PBREM luciferase reporter, and 3A4-XREM-pER6 luciferase reporter were described previously (Auerbach et al., 2007). The vector CMV2-CAR3 and mammalian two-hybrid vectors were also reported previously (Auerbach et al., 2005). Experimental treatments were performed in triplicate or quadruplicate, and all experiments were repeated at least once. Transfections for transactivation and mammalian two-hybrid assays were preformed as previously described (Auerbach et al., 2005), except in the two-hybrid assays, equal amounts (20 ng) of the pM-CAR (Gal4 DNA-binding domain) and VP16-SRC1 (activation domain) vectors were used. Since RXRα is CAR and PXRs’ heterodimer partner, a 3.1-RXRα ligand-biding domain vector was included in the mammalian two-hybrid assays. All test compounds were diluted in DMSO and levels never exceeded 0.2% (vol/vol). As positive controls, CITCO for CAR variants (Maglich et al., 2003) or RFPM (Bertilsson et al., 1998) or TO (Mitro et al., 2007) for PXR were included. Because CAR1 is constitutively active, ANDRO (10μM), a mouse CAR (Forman et al., 1998) and human CAR1 (Auerbach et al., 2007) inverse agonist, was included to decrease its activity, which can be restored in the presence of an agonist. All chemical treatments were for 24 h and luciferase assays were performed as previously described (Dekeyser et al., 2009).
Human primary hepatocyte culture.
Normal human hepatocytes were obtained through Dr Stephen Strom from the Liver Tissue Cell Distribution System, Pittsburgh, PA, funded by NIH Contract #N01-DK-7-0004/HHSN267200700004C. Cell culture conditions were published previously (Goyak et al., 2008). The medium was replaced every 24 h. Hepatocytes were treated with vehicle control (0.1% DMSO), PB (1mM), CITCO (2μM), DEHP (0.1, 1, or 10μM), or DINP (0.1, 1, or 10μM) 72 h after the first medium change. After a 48-h treatment, cells were harvested in 250 μl of ice-cold cell lysis buffer (20mM Tris-HCl, pH 7.5, containing 100mM NaCl, 0.5% NP40, and 1X protease cocktail inhibitor) and incubated on ice for 30 min. The cell lysates were sonicated and centrifuged to collect supernatants. Total protein concentration was determined with the Pierce 660 Protein Assay using a Nanodrop 2000 spectrophotometer (Thermo Scientific).
Western blot analyses.
Cell lysate supernatants (25 μg) from treated human primary hepatocytes were separated on 10% SDS-polyacrylamide gels and electrophoretically transferred onto Immuno-Blot PVDF membranes (Bio-Rad). The membranes were blocked in 5% nonfat dry milk in 1× Tris-buffered saline containing 0.1% Tween-20 (0.1% TBST). The membranes were then incubated with specific antibodies against human CYP2B6 or CYP3A4 (obtained from Dr Harry C. Gelboin, National Institutes of Health), diluted 1:1000 in 2% nonfat dry milk in 0.1% TBST, respectively. β-Actin (Santa Cruz, CA) was used as internal control. Blots were washed and incubated with horseradish peroxidase goat anti-mouse IgG antibody diluted 1:5000 in 2% nonfat dry milk in 0.1% TBST. Blots were developed using Pierce Super-signal ECL Western blotting detection reagents (Thermo Scientific).
Ligand-binding domain modeling and ligand docking.
The Phyre (Protein homology/analogy recognition engine) modeling program was used to generate CAR2 and CAR3 ligand-binding domain models with the assistance of Dr Lawrence Kelley (Kelley and Sternberg, 2009). The quality of the models was assessed with RAMPAGE (http://mordred.bioc.cam.ac.uk/∼rapper/rampage.php; Lovell et al., 2003). The structures of the LBP of the CAR1 structure (Xu et al., 2004) and the Phyre generated CAR2 and CAR3 models were then characterized using Pocket-Finder (http://bmbpcu36.leeds.ac.uk/pocketfinder/) (Laurie and Jackson, 2005). ICM MolSoft Browser Pro (http://www.molsoft.com/) was used to visualize CAR and CAR3 predicted structures and CAR1 models and LBP.
For the docking simulations of DEHP into the Phyre CAR2 structural model, AutoDock4 (Morris et al., 2009) and AutoDock Vina (Trott and Olson, 2010) were used with AutoDockTools graphical user interface. Prior to docking, all polar hydrogens were added to the molecules. The ligands were allowed to rotate freely around all active torsions during docking, whereas the protein structures remained rigid. A 22 × 22 × 22 Å grid box was used to center the docking algorithm on the ligand-binding site. All other settings were defaults in AutoDock Vina. The resulting ligand-docking configurations were analyzed for lowest binding energy (kilocalories per mole) and visualized using and Molsoft Browser Pro.
Statistical analysis.
Statistics and EC50 values were obtained using GraphPad Prism 5 (GraphPad Software, San Diego, CA). For determining differences in activation of CAR or PXR by various treatments, two-way ANOVAs were performed, followed by a Bonferroni test for comparison to controls.
RESULTS
BPA Activates CAR1 and CAR3
Based on our previous finding that DEHP is a potent activator of CAR2, we examined the ability of BPA and NP, both plasticizers with reported estrogenic activity, to activate CAR variants and PXR. At 0.1 and 10μM, BPA reactivated reporter activity through CAR1 to levels about equal to CITCO, whereas 10μM BPA activated CAR3 to levels about 60% of CITCO-induced reactivation (Fig. 2). BPA, even at the higher dose, showed no activation of the promoter through CAR2 and only a small effect on PXR. NP (0.1 and 10μM) activated the 2B6-XREM-PBREM reporter through CAR1 to levels about 45% of CITCO-induced activation with no effect on CAR2, CAR3, or PXR.
FIG. 2.

Activation of the 2B6-XREM-PBREM reporter by CAR1, CAR2, CAR3, or PXR after treatment with bisphenol A or nonylphenol. Results shown here represent a single transfection experiment, with all treatments in quadruplicate. COS-1 cells were transfected with the 3.1-RXRα expression vector, the 2B6-XREM-PBREM reporter, the pRL-CMV vector for normalization of transfection efficiency and either CMV2-CAR1, CMV2-CAR2, CMV2-CAR3, or CMVs-PXR. All treatments were for 24 h, and the data are represented as normalized luciferase values and each data point represents the mean (±SEM). *p < 0.05 compared with androstanol control; **p < 0.05 compared with DMSO control.
CAR and PXR Recognize a Broad Range of Phthalates
We next examined the abilities of DEP, DnBP, DiBP, and DiNP to activate the 2B6-XREM-PBREM reporter through CAR1, CAR2, CAR3, and PXR (Figs. 3A–D). No activity was observed in cells transfected with the empty CMV2 vector (data not shown). For CAR1, 10μM ANDRO was included in all treatments except DMSO. DEP showed little or no activation of the 2B6-XREM-PBREM reporter through any of the CAR variants or PXR, whereas DnNP and DiBP were weak activators of CAR1, CAR2, CAR3, and PXR. In contrast, DiNP displayed maximal activation of the 2B6-XREM-PBREM reporter (compared with CITCO) through CAR2 (Fig. 3B), with no activation of CAR1 or CAR3. DiNP was a weak activator of PXR through the 2B6-XREM-PBREM reporter. We also tested phthalate activation of the 3A4-XREM-pER6 reporter through PXR, and the results were essentially identical to those with the 2B6-XREM-PBREM (Supplementary fig. S1).
FIG. 3.

Activation of the 2B6-XREM-PBREM reporter by CAR1, CAR2, CAR3, and PXR after treatment with phthalates. Results shown here represent a single transfection experiment, with all treatments in quadruplicate. COS-1 cells were transfected with 3.1-RXRα expression vector, the 2B6-XREM-PBREM reporter, the pRL-CMV vector for normalization of transfection efficiency and either CMV2-CAR1 (A and E), CMV2-CAR2 (B and F), CMV2-CAR3 (C and G), or CMV2-PXR (D and H). Panels (A), (B), (C), and (D) show activity of various phthalates with each of the receptors. Panels (E), (F), (G), and (H) show a more complete dose-response curve comparing DEHP, DiNP, and the positive control CITCO for CAR1, CAR2, and CAR3 or TO901317 for PXR. All treatments were for 24 h, and the data are represented as normalized luciferase values and each data point represents the mean (±SEM). *p < 0.05 compared with androstanol control for CAR1 or DMSO control for CAR2, CAR3, and PXR.
The strong activation of CAR2 by DEHP and DiNP prompted us to directly compare these two phthalates in transactivation assays over a range of doses and to confirm our results with CAR1, CAR3, and PXR (Figs. 3E–H). For CAR1 and CAR3, DEHP and DiNP showed little activation of the 2B6-XREM-PBREM reporter (Figs. 3E and G). In contrast, DEHP and DiNP activated the reporter through CAR2 with a much higher affinity than CITCO. Furthermore, DEHP exhibited approximately three times higher affinity than DiNP (Fig. 3F). We used TO as a positive control for PXR, as it is much more potent than RFPM (Mitro et al., 2007); TO does not activate the CAR variants (Supplementary fig. S2). DEHP and DiNP activated the 2B6- (Fig. 3H) and 3A4-XREM-pER6 (Supplementary fig. S3) through PXR with equal affinity; however, they were much less potent than TO. The EC50 values for activation of 2B6-XREM-PBREM each of the compounds tested with CAR2, CAR3, and PXR are shown in Table 1. For CAR1, EC50 values were not determined due to the inherent problem of its constitutive activity.
TABLE 1.
Estimated EC50 Values for Activation of 2B6-XREM-PBREM through CAR2, CAR3, and PXRa
| Ligand | CAR2 | CAR3 | PXR |
| DEP | NA | 267.6b | 495.6 |
| DnBP | 24.1 | 8.0 | 12.5 |
| DiBP | 17.1 | 11.4 | 9.9 |
| DiNP | 0.34 | NA | 3.6 |
| DEHP | 0.1 | NA | 3.8 |
| CITCO | 0.9 | 0.1 | ND |
| Rifampicin | ND | ND | 2.1 |
| TO901317 | NA | NA | 0.03 |
Note. NA, no or minimal activation; ND, not determined.
Not determined for CAR1 due to constitutive activation.
All values are micromolar.
We also tested DEHP and DiNP activation of CAR and PXR using a mammalian two-hybrid protein interaction assay. Again, DEHP and DiNP showed very little or no activation of CAR1 or CAR3 (data not shown). DEHP and DiNP were both potent activators of CAR2 relative to CITCO, showing the same rank order potency as in the transactivation assay (Fig. 4A). For PXR, DEHP and DiNP exhibited only moderate activity (Fig. 4B). The doses of DEHP and DiNP required for maximal activation of the reporter through PXR were about 10–100× higher than were required for CAR2. These studies suggest that DEHP and DiNP are potent activators of CAR2, with selectivity at lower concentrations for CAR2 over PXR.
FIG. 4.
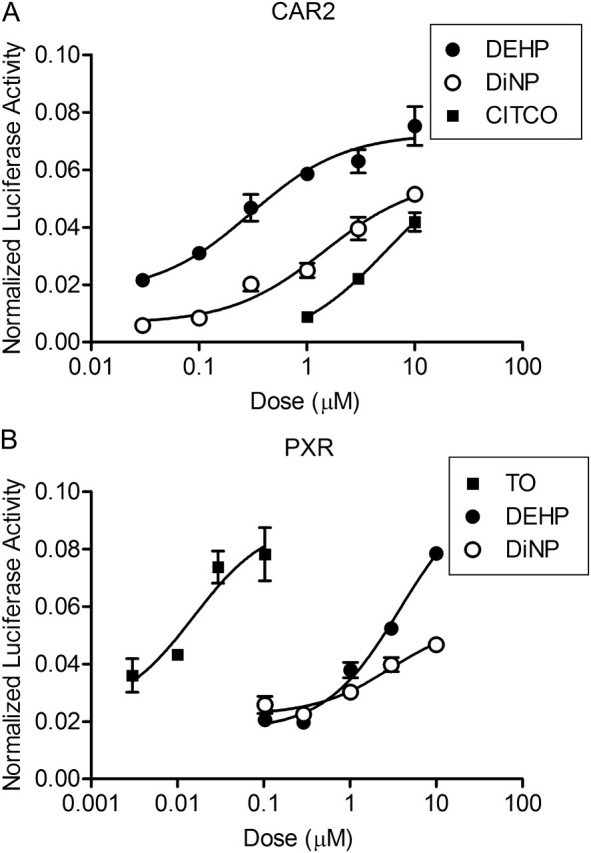
DEHP and DiNP demonstrate selectivity for CAR2 in mammalian two-hybrid assay. COS-1 cells were transfected with CAR or PXR in the pm (GAL4) vector, SRC1 in the VP16 vector, 3.1 RXRα-LBD, pFR-luciferase reporter, and pRL-CMV vector for normalization. Chemical treatments were for 24 h. Data are represented as normalized luciferase values, and each data point represents the mean (±SEM) of quadruplicate treatment wells from a representative transfection experiment.
DEHP and DiNP Induce CYP2B6 and CYP3A4 Protein in Human Hepatocytes
To support our in vitro findings, DEHP- and DiNP-treated human hepatocytes from four separate donors were assessed for CYP2B6 and CYP3A4 protein levels. A representative immunoblot from one donor is shown in Figure 5. CITCO (2μM) and PB (1mM) were positive controls. DEHP and DiNP resulted in markedly increased CYP2B6 and CYP3A4 protein expression relative to DMSO controls. A dose-response relationship was evident for CYP2B6 and CYP3A4 in the DiNP-treated samples but not in the DEHP-treated samples. This result was similar for all of the donors except one, which exhibited increased CYP2B6 and CYP3A4 relative to DMSO controls; however, the increase was modest and did not exhibit a dose-response relationship for either protein.
FIG. 5.

Representative immunoblot showing expression of CYP2B6 and CYP3A4 in human hepatocytes treated with DEHP or DiNP. Human hepatocytes were treated with varying concentrations of DEHP and DiNP with DMSO, PB, and CITCO as controls for 48 h. Protein (S9 fractions) were isolated from lysed cells and subjected to immunoblot analysis with CYP2B6- or CYP3A4-specific antibodies. Actin is shown for normalization of protein loading.
Mutation of CAR2 Greatly Alters the Response to DEHP and DiNP
To further evaluate the contribution of the CAR2 SPTV insertion DEHP and DiNP activity, two mutant constructs were tested in transactivation assays. The serine 233, a polar uncharged residue, was replaced with either an alanine, a small hydrophobic uncharged residue (S233A and CAR2A), or aspartate, a larger, negatively charged residue (S233D and CAR2D) in order to determine how these differences affect DEHP and DiNP interaction with the CAR2 receptor. CITCO was also included as a reference. The CAR2 mutations had the least impact on CITCO EC50 (Fig. 6A). The CAR2 and CAR2A mutant exhibited approximately equal EC50 for CITCO, whereas the CAR2D mutation resulted in approximately two times increase in EC50. Although the CAR2A mutation appeared to reduce CITCO’s Emax, the CAR2A and CAR2D mutations had a marked impact on the EC50 of DEHP and DiNP. The CAR2A mutant displayed a greatly reduced EC50 (∼10× more potent) for DEHP and DiNP in comparison to the endogenous CAR2 receptor (Figs. 6B and C). The CAR2D substitution markedly decreased DEHP and DiNP activation; the DEHP EC50 value with CAR2D was ∼10× higher than CAR2. An EC50 value for DiNP with CAR2D was not determined because the dose-response curve was not complete; however, higher doses of DiNP are likely not physiologically relevant.
FIG. 6.
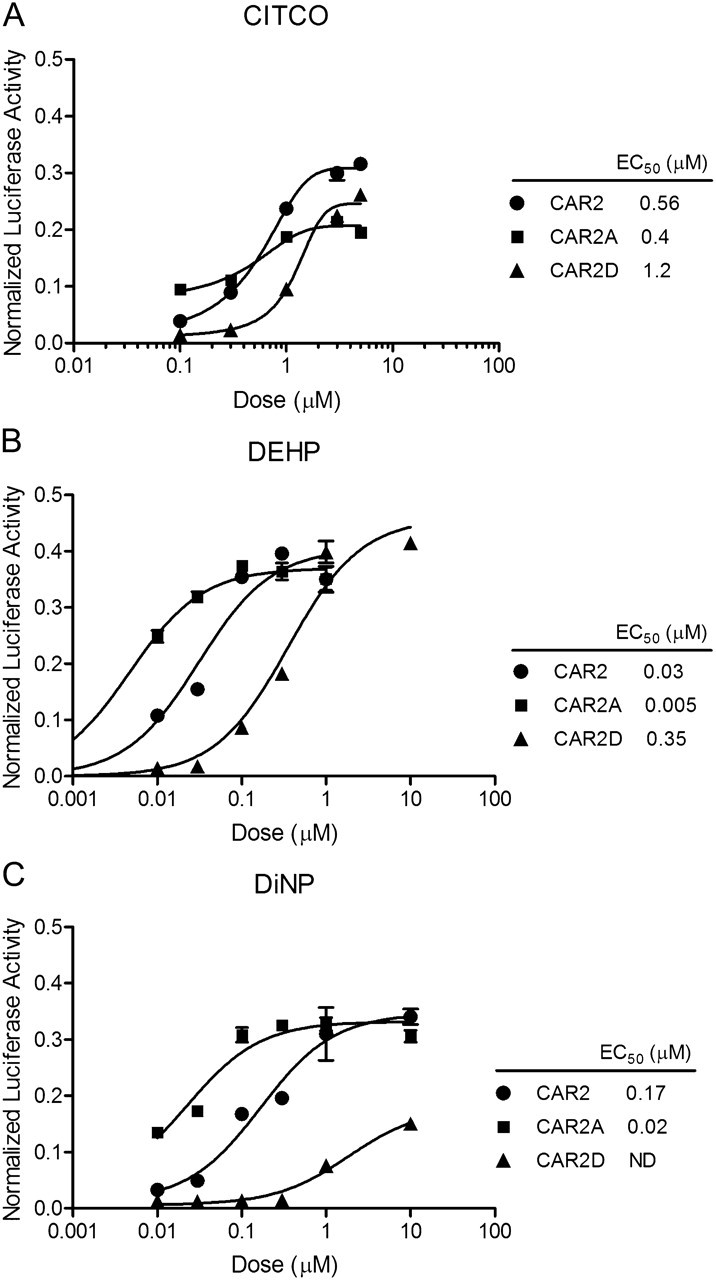
Activation of the 2B6-XREM-PBREM reporter by CAR2, CAR2A, and CAR2D in response to CITCO, DEHP, and DiNP. All transfections included the 3.1-RXRα expression vector, the 2B6-XREM-PBREM reporter, the pRL-CMV vector for normalization of transfection efficiency and either CMV2-CAR2 or CMV2-CAR2A. Cells were treated for 24 h. Data are represented as normalized luciferase values, and each data point represents the mean (±SEM) of quadruplicate treatment wells from a representative transfection experiment.
CAR2 Model Suggests Altered LBP, Relative to CAR1 and CAR3
The Phyre protein modeling program predicted CAR2 structure was superimposed on the CAR1 structure (1XVP) for comparison (Figs. 7A and B). Generally, the main chain coordinates of the CAR structures are very similar, with the exception of the amino acids flanking the SPTV CAR2 insertion. While the SPTV insertion (shown in yellow) aligns with the alpha helix of CAR1 (red), a region on the N-terminal side of the insertion forms an unstructured loop (green). Ramachandran analysis of CAR2 revealed that 94% of the residues were in favored regions, 4% were in allowed regions, and 2% were in outlier regions. Notably, one outlier was the serine of the SPTV insertion; probably because as a pre-proline residue, the phi-psi bond angles were not favored.
FIG. 7.

CAR2 predicted protein structure and CAR1 and CAR2 LBP models. Panels (A) and (B) show two views the Phyre homology model of the human CAR2 receptor ligand-binding domain (aqua) superimposed on the CAR1 ligand-binding domain structure (red). CITCO (blue) is shown in the LBP of CAR1. The CAR2 SPTV insertion is shown in yellow and a region adjacent to the insertion where the structure deviates from CAR1 is shown in green. Panel (C) shows an overlay of PocketFinder computer generated models of the CAR1 (red) and CAR2 (aqua) LBP with their respective ligands, CITCO (red) and DEHP (blue). Panels (D) and (E) show the individual CAR1 and CAR2 LBP.
Because of the proximity of the SPTV insertion to the CAR LBP and the differences in ligand specificity of CAR2, we submitted the CAR1 structure (1XVP) and the CAR2 Phyre model for LBP detection. Figure 7C shows the superimposed CAR1 (red) and CAR2 (aqua) LBP representations generated by the PockerFinder algorithm with their respective ligands, CITCO (red) for CAR1 or DEHP (blue) for CAR2. Calculated pocket volumes for the CAR1 crystal structure and the Phyre CAR2 model were 722 and 662 Å3, respectively. The CAR1 PocketFinder calculated volume was similar to the reported pocket volume of 675 Å3 (Xu et al., 2004). Although the CAR1 and CAR2 predicted volumes were similar, the shapes of the pockets differed, as did the orientations of the respective ligands in the pockets (Figs. 7C–E).
The CAR3 sequence was also submitted for structure prediction and subsequent pocket detection (data not shown). For the CAR3 model, Ramachandran analysis showed that 86.3% of the residues were in favored regions, 11.2% were in allowed regions, and 2% were in outlier regions. One outlier was an arginine (R277), which was also a pre-proline residue, two residues away from the carboxy end of the APYLT insertion. The calculated pocket volume (701 Å3) of CAR3 was virtually identical to pocket predicted for CAR1 (model not shown), an anticipated result since the APLYT insertion lies between helices 8 and 9 and therefore not expected to affect the LBP.
DISCUSSION
The studies presented here provide novel insights into the CAR/PXR xenobiotic sensing systems and demonstrate that the variant CARs and PXR possess distinct ligand-selective activation profiles. Our previous observation that DEHP is a highly potent agonist of CAR2 (Dekeyser et al., 2009), coupled to our new findings, reveal that alternative splicing of CAR functions to enhance the receptors’ ability to distinguish a broad range of xenobiotics that otherwise possess similar chemical properties. We also identify BPA as a CAR1 and CAR3 agonist, with little effect on CAR2 activation. The finding that CAR1 and CAR3 have similar ligand-binding properties that are distinct from CAR2 supports the conceptual use of CAR3 as a tool for higher throughput prediction of CAR1 ligands, as suggested previously (Auerbach et al., 2005; Faucette et al., 2007). Furthermore, the ability of phthalates to activate PXR, albeit at high concentrations relative to CAR2, provides insight into the cross talk between the two receptors.
DEHP and DiNP induced expression of CYP2B6 and CYP3A4 proteins in primary human hepatocytes; however, they were not as potent as CITCO, which contrasts our in vitro studies. This discrepancy may be due to the extensive hepatic metabolism of phthalates in humans (Frederiksen et al., 2007; Wittassek and Angerer, 2008). Nevertheless, induction is likely mediated via CAR2 at low exposure concentrations and perhaps by both CAR2 and PXR at higher, e.g., 10μM concentrations. Although the induction response was relatively consistent among the donors, one donor exhibited lesser responsiveness. We previously observed interindividual variability in the hepatocyte induction of CYP2B6 and CYP3A4 messenger RNA (mRNA) by DEHP (Dekeyser et al., 2009), as well as to other prototypical inducers (Goyak et al., 2008). Differential xenobiotic responsiveness among humans likely results from a complex interplay of genetics, previous chemical exposures, and perhaps differences in the expression profiles of the xenobiotic receptors that mediate these responses.
It is noteworthy that DEHP- and DiNP-mediated induction of both CYP2B6 and CYP3A4 proteins was apparent even at the lowest concentration tested (100nM). Data from our transcriptional transactivation experiments indicated that even lower DEHP concentrations, e.g., 10nM, are capable of activating CAR2 (Fig. 6B). Human studies have reported mean DEHP blood levels of 4.8μM in humans undergoing dialysis (Pollack et al., 1985). Additionally, a study of Swedish mothers (Hogberg et al., 2008) found significant levels of DEHP in serum (median 1.2nM, mean 15nM) and maternal milk (mean 43nM). Thus, vulnerable populations appear exposed to DEHP at concentrations that likely activate CAR2 and/or PXR. DEHP and DiNP are extensively metabolized, and based on urinary excretion of DEHP and DiNP metabolites, even “normal healthy” humans are likely exposed to low but significant levels of DEHP and DiNP (Meeker et al., 2009; Silva et al., 2006), levels that may activate CAR2. Furthermore, Silva et al. (2006) suggest that human DiNP exposures may be underestimated because DiNP is an isomeric mixture that is extensively metabolized and only a few metabolites are measured when assessing human exposures. Although DEHP appears more potent than DiNP by measures of both CYP450 induction and transcriptional transactivation activity, it should be noted that as a mixture of 9-carbon isomers, it is possible that only select DiNP isomers contribute to the activation responses noted. This aspect will require further study; however, it is likely that most human exposures are to DiNP isomer mixtures.
Although we did not test BPA in hepatocytes, our transactivation assays demonstrated that BPA activated CAR1 and CAR3 at concentrations of 10μM. To our knowledge, this is the first report of BPA activation of human CAR variants. However, the EC50 of BPA for estrogenic activity in MCF-7 cells is 0.63μM (Kitamura et al., 2005). In addition, BPA also mediates other pathways that do not involve nuclear receptors at subnanomolar doses (Welshons et al., 2006); thus, our prediction is that CAR is not involved in BPA-induced effects at lower doses. Another interesting observation was that NP had little or no activity with human CAR variants or human PXR. These latter results are in contrast to a report that NP activates mouse and human CAR and human PXR (Hernandez et al., 2007). The differences noted may result from the NP preparations used. Our study used a 99% pure preparation of 4-N-NP, whereas the Hernandez study used technical grade 4-NP, which is an 85% para-isomer mix (Hernandez et al., 2007).
We performed mutagenesis studies to investigate potential mechanisms of how CAR2’s SPTV insertion contributes to ligand specificity. Mutation of the S233 residue greatly alters the receptor’s response to DEHP and DiNP. Replacing S233 with the smaller more hydrophobic alanine residue (CAR2A) may have created a more hydrophobic LBP environment for interacting with the alkyl chains of DEHP and DiNP, and thus, increased the response to these compounds. In contrast, substitution of S233 with aspartic acid, which has a slightly larger volume than serine and is negatively charged, may have created a less favorable LBP environment for DEHP and DiNP, resulting in a markedly decreased response. The differences in DEHP and DiNP response among the CAR2 mutants were not nearly as pronounced with the pan-CAR ligand, CITCO. Taken together, these results suggest that the naturally occurring SPTV insertion of CAR2 alters the nature of the receptor’s LBP and creates a more favorable binding site conformation for DEHP and DiNP.
The differences in activation of the CAR variants by phthalates and BPA are important observations regarding the diversity of CAR and led us to further investigate the nature of the CAR LBP using CAR2 and CAR3 structural models. The human CAR1 ligand-binding domain has been characterized in the presence of two agonists, CITCO and 5β-preganedione (Xu et al., 2004). These studies show that helices 2, 3, 4, 5, 6, 7, and 10 and β-strands 3 and 4 surround the LBP of CAR1. Furthermore, amino acids in helices 3, 5, 6, and 7 are important for ligand binding. The SPTV insertion between helices 6 and 7 thus likely alters the spatial orientation of this region, resulting in altered ligand specificity. Although the overall volume of the modeled CAR2 LBP was similar, the results suggested that the CAR2 SPTV leads to an altered LBP conformation in comparison to CAR1 and CAR3. Specifically, the CAR2 insertion is predicted to alter the conformation of helix 6, allowing for an expanded pocket proximal to that region.
In silico comparisons of CITCO in the CAR1 structure with that of docked DEHP within the CAR2 pocket suggest these compounds exploit respective differences of their chemistries as well as their LBP. DEHP is primarily a hydrophobic molecule that appears to interact with CAR2 through hydrophobic interactions. One alkyl side chain of DEHP appears to project into the expanded region of the CAR2 pocket, which is primarily bounded by hydrophobic amino acids F217, I226, and F247. In the CAR1 structure, the CITCO imidazothiazole moiety extends into a more polar region of the pocket, enclosed by N165, C202, H203, and Y326 (Xu et al., 2004). In the CAR2-predicted pocket, DEHP does not appear to utilize this portion of the LBP. Although the LBPs are displayed to emphasize the differences (Figs. 7C–E), an interesting similarity is that the phenyl ring of DEHP is almost superimposed with the para-chlorophenyl ring of CITCO (not shown), lending enhanced credibility to the modeling predictions. However, efforts are required and ongoing to obtain the actual crystal structure of CAR2.
Initial concerns regarding phthalates focused on peroxisome proliferation demonstrated in rodents through PPAR activation; concerns that have since waned as humans are relatively refractory to peroxisome proliferation (Rusyn et al., 2006). More recently, concern has shifted toward possible anti-androgenic endocrine–disrupting effects of phthalates (Kavlock et al., 2006). It is well established that both CYP2B6 and CYP3A4 are induced by CAR and PXR and that these enzymes are involved in testosterone metabolism (Imaoka et al., 1996). The potential impact of phthalates on androgen metabolism through the activation of CAR and PXR will require additional investigation. Furthermore, the emerging regulatory role of CAR in energy metabolism and insulin sensitivity (Dong et al., 2009; Gao et al., 2009; Konno et al., 2008) also suggests intriguing areas for future phthalate research.
In comparison to the essentially infinite number of small molecules to which humans are exposed, the number of xenosensing receptors is relatively small. The current report demonstrates that alternatively spliced human CAR gene transcripts increase CAR’s capacity to serve as a selective xenosensor. Although mRNA for these splice variants has been detected as substantial levels in human liver (Dekeyser et al., 2009; Jinno et al., 2004; Ross et al., 2010), it is noteworthy that most rodent species, due to the absence of a canonical splice acceptor site, appear incapable of producing the CAR2 variant. These differences, when combined with the known species differences in the receptors’ ligand specificity, suggest that rodent models alone may not be adequate for the study of human CAR activation responses (Dekeyser et al., 2009). Finally, the observation that phthalates extensively interact with the human CAR and PXR receptors and that CAR2 is extraordinarily sensitive to both DEHP and DiNP provides new direction for investigating the toxicological implications of human exposure to these ubiquitous environmental agents.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (R01 GM066411 to C.J.O.).
Supplementary Material
Acknowledgments
The authors would like to express special thanks to Dr Stephen Strom for his support and assistance with human hepatocyte procurement.
References
- Arnold KA, Eichelbaum M, Burk O. Alternative splicing affects the function and tissue-specific expression of the human constitutive androstane receptor. Nucl. Recept. 2004;2:1. doi: 10.1186/1478-1336-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach SS, Dekeyser JG, Stoner MA, Omiecinski CJ. CAR2 displays unique ligand binding and RXRalpha heterodimerization characteristics. Drug Metab. Dispos. 2007;35:428–439. doi: 10.1124/dmd.106.012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach SS, Ramsden R, Stoner MA, Verlinde C, Hassett C, Omiecinski CJ. Alternatively spliced isoforms of the human constitutive androstane receptor. Nucleic Acids Res. 2003;31:3194–3207. doi: 10.1093/nar/gkg419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach SS, Stoner MA, Su S, Omiecinski CJ. Retinoid X receptor-{alpha}-dependent transactivation by a naturally occurring structural variant of human constitutive androstane receptor (NR1I3) Mol. Pharmacol. 2005;68:1239–1253. doi: 10.1124/mol.105.013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol. Cell. Biol. 1994;14:1544–1551. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonefeld-Jorgensen EC, Long M, Hofmeister MV, Vinggaard AM. Endocrine-disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: new data and a brief review. Environ. Health Perspect. 2007;115(Suppl. 1):69–76. doi: 10.1289/ehp.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Tompkins LM, Li L, Li H, Kim G, Zheng Y, Wang H. A single amino acid controls the functional switch of human constitutive androstane receptor (CAR) 1 to the xenobiotic-sensitive splicing variant CAR3. J. Pharmacol. Exp. Ther. 2010;332:106–115. doi: 10.1124/jpet.109.159210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekeyser JG, Stagliano MC, Auerbach SS, Prabu KS, Jones AD, Omiecinski CJ. Di(2-ethylhexyl) phthalate is a highly potent agonist for the human constitutive androstane receptor splice variant, CAR2. Mol. Pharmacol. 2009;75:1005–1013. doi: 10.1124/mol.108.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr. Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, Wakil SJ, Stevens RD, Ilkayeva O, Newgard CB, Chan L, et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18831–18836. doi: 10.1073/pnas.0909731106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 2007;320:72–80. doi: 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Tzameli I, Choi HS, Chen J, Simha D, Seol W, Evans RM, Moore DD. Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature. 1998;395:612–615. doi: 10.1038/26996. [DOI] [PubMed] [Google Scholar]
- Frederiksen H, Skakkebaek NE, Andersson AM. Metabolism of phthalates in humans. Mol. Nutr. Food Res. 2007;51:899–911. doi: 10.1002/mnfr.200600243. [DOI] [PubMed] [Google Scholar]
- Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 2009;284:25984–25992. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyak KM, Johnson MC, Strom SC, Omiecinski CJ. Expression profiling of interindividual variability following xenobiotic exposures in primary human hepatocyte cultures. Toxicol. Appl. Pharmacol. 2008;231:216–224. doi: 10.1016/j.taap.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 2003;278:45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- Hernandez JP, Huang W, Chapman LM, Chua S, Moore DD, Baldwin WS. The environmental estrogen, nonylphenol, activates the constitutive androstane receptor. Toxicol. Sci. 2007;98:416–426. doi: 10.1093/toxsci/kfm107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogberg J, Hanberg A, Berglund M, Skerfving S, Remberger M, Calafat AM, Filipsson AF, Jansson B, Johansson N, Appelgren M, et al. Phthalate diesters and their metabolites in human breast milk, blood or serum, and urine as biomarkers of exposure in vulnerable populations. Environ. Health Perspect. 2008;116:334–339. doi: 10.1289/ehp.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaoka S, Yamada T, Hiroi T, Hayashi K, Sakaki T, Yabusaki Y, Funae Y. Multiple forms of human P450 expressed in Saccharomyces cerevisiae. Systematic characterization and comparison with those of the rat. Biochem. Pharmacol. 1996;51:1041–1050. doi: 10.1016/0006-2952(96)00052-4. [DOI] [PubMed] [Google Scholar]
- Jinno H, Tanaka-Kagawa T, Hanioka N, Ishida S, Saeki M, Soyama A, Itoda M, Nishimura T, Saito Y, Ozawa S, et al. Identification of novel alternative splice variants of human constitutive androstane receptor and characterization of their expression in the liver. Mol. Pharmacol. 2004;65:496–502. doi: 10.1124/mol.65.3.496. [DOI] [PubMed] [Google Scholar]
- Kavlock R, Barr D, Boekelheide K, Breslin W, Breysse P, Chapin R, Gaido K, Hodgson E, Marcus M, Shea K, et al. NTP-CERHR expert panel update on the reproductive and developmental toxicity of di(2-ethylhexyl) phthalate. Reprod. Toxicol. 2006;22:291–399. doi: 10.1016/j.reprotox.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Suzuki T, Sanoh S, Kohta R, Jinno N, Sugihara K, Yoshihara S, Fujimoto N, Watanabe H, Ohta S. Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds. Toxicol. Sci. 2005;84:249–259. doi: 10.1093/toxsci/kfi074. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol. Cell. Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno Y, Negishi M, Kodama S. The roles of nuclear receptors CAR and PXR in hepatic energy metabolism. Drug Metab. Pharmacokinet. 2008;23:8–13. doi: 10.2133/dmpk.23.8. [DOI] [PubMed] [Google Scholar]
- Lamba JK, Lamba V, Yasuda K, Lin YS, Assem M, Thompson E, Strom S, Schuetz E. Expression of constitutive androstane receptor splice variants in human tissues and their functional consequences. J. Pharmacol. Exp. Ther. 2004;311:811–821. doi: 10.1124/jpet.104.069310. [DOI] [PubMed] [Google Scholar]
- Laurie AT, Jackson RM. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003;278:17277–17283. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002;62:638–646. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- Masson D, Qatanani M, Sberna AL, Xiao R, Pais de Barros JP, Grober J, Deckert V, Athias A, Gambert P, Lagrost L, et al. Activation of the constitutive androstane receptor decreases HDL in wild-type and human apoA-I transgenic mice. J. Lipid Res. 2008;49:1682–1691. doi: 10.1194/jlr.M700374-JLR200. [DOI] [PubMed] [Google Scholar]
- Meeker JD, Calafat AM, Hauser R. Urinary metabolites of di(2-ethylhexyl) phthalate are associated with decreased steroid hormone levels in adult men. J. Androl. 2009;30:287–297. doi: 10.2164/jandrol.108.006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitro N, Vargas L, Romeo R, Koder A, Saez E. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 2007;581:1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi JM, Gerbal-Chaloin S, Drocourt L, Maurel P, Vilarem MJ. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. Biochim. Biophys. Acta. 2003;1619:243–253. doi: 10.1016/s0304-4165(02)00483-x. [DOI] [PubMed] [Google Scholar]
- Pollack GM, Buchanan JF, Slaughter RL, Kohli RK, Shen DD. Circulating concentrations of di(2-ethylhexyl) phthalate and its de-esterified phthalic acid products following plasticizer exposure in patients receiving hemodialysis. Toxicol. Appl. Pharmacol. 1985;79:257–267. doi: 10.1016/0041-008x(85)90347-3. [DOI] [PubMed] [Google Scholar]
- Ross J, Plummer SM, Rode A, Scheer N, Bower CC, Vogel O, Henderson CJ, Wolf CR, Elcombe CR. Human constitutive androstane receptor (CAR) and pregnane X receptor (PXR) support the hypertrophic but not the hyperplastic response to the murine nongenotoxic hepatocarcinogens phenobarbital and chlordane in vivo. Toxicol. Sci. 2010;116:452–466. doi: 10.1093/toxsci/kfq118. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit. Rev. Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur RS, Wu Y, Bramlett KS, Wang M, Yao S, Perkins D, Totten M, Searfoss G, III, Ryan TP, Su EW, et al. Alternative splicing within the ligand binding domain of the human constitutive androstane receptor. Mol. Genet. Metab. 2003;80:216–226. doi: 10.1016/j.ymgme.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Schettler T. Human exposure to phthalates via consumer products. Int. J. Androl. 2006;29:134–139. doi: 10.1111/j.1365-2605.2005.00567.x. [DOI] [PubMed] [Google Scholar]
- Silva MJ, Reidy JA, Preau JL, Jr, Needham LL, Calafat AM. Oxidative metabolites of diisononyl phthalate as biomarkers for human exposure assessment. Environ. Health Perspect. 2006;114:1158–1161. doi: 10.1289/ehp.8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J. Biol. Chem. 1999;274:6043–6046. doi: 10.1074/jbc.274.10.6043. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, Lehmann JM, Negishi M. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol. Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- Wei P, Zhang J, Dowhan DH, Han Y, Moore DD. Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response. Pharmacogenomics J. 2002;2:117–126. doi: 10.1038/sj.tpj.6500087. [DOI] [PubMed] [Google Scholar]
- Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology. 2006;147(Suppl. 6):S56–S69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]
- Wittassek M, Angerer J. Phthalates: metabolism and exposure. Int. J. Androl. 2008;31:131–138. doi: 10.1111/j.1365-2605.2007.00837.x. [DOI] [PubMed] [Google Scholar]
- Xie W, Yeuh MF, Radominska-Pandya A, Saini SP, Negishi Y, Bottroff BS, Cabrera GY, Tukey RH, Evans RM. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4150–4155. doi: 10.1073/pnas.0438010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005;28:249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, et al. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol. Cell. 2004;16:919–928. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.