

Abstract
Despite more than a decade of empirical work on the role of genetic polymorphisms in the serotonin system on behavior, the details across levels of analysis are not well understood. We describe a mathematical model of the genetic control of presynaptic serotonergic function that is based on control theory, implemented using systems of differential equations, and focused on better characterizing pathways from genes to behavior. We present the results of model validation tests that include the comparison of simulation outcomes with empirical data on genetic effects on brain response to affective stimuli and on impulsivity. Patterns of simulated neural firing were consistent with recent findings of additive effects of serotonin transporter and tryptophan hydroxylase-2 polymorphisms on brain activation. In addition, simulated levels of cerebral spinal fluid 5-hydroxyindoleacetic acid (CSF 5-HIAA) were negatively correlated with Barratt Impulsiveness Scale (Version 11) Total scores in college students (r = −.22, p = .002, N = 187), which is consistent with the well-established negative correlation between CSF 5-HIAA and impulsivity. The results of the validation tests suggest that the model captures important aspects of the genetic control of presynaptic serotonergic function and behavior via brain activation. The proposed model can be: (1) extended to include other system components, neurotransmitter systems, behaviors and environmental influences; (2) used to generate testable hypotheses.
Keywords: Cybernetic, Control system, Simulation, Genetic polymorphism
Introduction
Characterizing pathways from genes to behavior will be the primary task for generations of behavior geneticists. The Nature versus Nurture controversy that dominated the first two or three decades of behavior genetics no longer inflames passion. That genetic variation in a population is associated with individual differences in behavior in that population, for virtually all behaviors that psychologists find worthy of study, is no longer controversial (Gottesman and Hanson 2005; McClearn 2004). The task of characterizing the mechanisms by which genetic polymorphisms produce individual differences in behavior makes up the bulk of the fascinating, yet difficult, work that lies ahead for twenty-first century behavior geneticists (Green et al. 2008; Plomin et al. 2003). Such work cannot be easily done in isolation; it will require cross-disciplinary training and collaboration across disciplines. Because the effects of genes on phenotypes are likely to be dependent on both the environmental and genetic context, it is important to consider the dynamics of such interaction and co-action to understand pathways from genes to behavior (McClearn 2004).
As all roads once led to Rome, all pathways from genes to behavior lead to the brain. While there may be minor exceptions to this rule, as in the case of reflexes, for the vast majority of behaviors of interest to behavior geneticists the brain is the most important point of interest on the route. Such a statement may seem to be a platitude, but it is intended to orient behavior genetic analyses towards systematically elucidating relevant components and pathways in a systems approach. For those investigators interested in constructs such as self-regulation and emotion, and in disorders that arise by dysfunctional behavioral or emotional control the serotonin neurotransmitter system is a good candidate for a systems behavior genetic approach (Carver et al. 2008).
Serotonin system is a good candidate
Serotonin has been studied as a neurotransmitter for over 50 years, which has produced a sizeable corpus of data. A recent PubMed search of “serotonin” produced over 107,800 hits. Serotonin-containing neurons have been studied in the nervous systems of several species across taxa including aplysia (Glanzman 2008), lobsters (Kravitz 2000), fruit flies (Vomel and Wegener 2008), dogs (Vage and Lingaas 2008), mice (Murphy and Lesch 2008), monkies (Barr et al. 2003a, b), and humans (Canli and Lesch 2007; Gotlib et al. 2008; Haghighi et al. 2008; Rao et al. 2007; Williams et al. 2003). The power of convergent evidence can be brought to bear when studying associations between individual differences in the structure or function of the serotonin system and behavior across taxonomic units.
The projection systems of serotonergic neurons in the mammalian central nervous system are identified (Baumgarten and Grozdanovic 1999). Most serotonergic neurons have cell bodies in the raphe nuclei and project to many brain areas including the prefrontal cortex and the amygdala. The components of the serotonergic system are described and include at least 14 receptor subtypes (Hartig 1999), metabolic enzymes (Hotamisligil and Breakefield 1991; Veenstra-VanderWeele and Cook 2003), and a reuptake transporter (Lesch et al. 1993), among others. Genetic polymorphisms have been identified in each of the components that influence serotonergic function either in the structural genes, the regulatory regions or the introns.
Serotonin modulates a wide array of behaviors and behavioral states including, but not limited to, mood, arousal, impulsivity, feeding, motor behavior and aggression (Lucki 1998). Several behavioral disorders such as depression (Johnson 2004; Leonardo and Hen 2006; Levinson 2006), alcohol use disorders (Feinn et al. 2005; Johnson 2004) and obsessive compulsive disorder (Baumgarten and Grozdanovic 1998; El Mansari and Blier 2006) are linked to serotonergic dysfunction. Some of the effects of serotonergic dysfunction are manifest through altered brain development, which can be permanent and influence risk for subsequent development of behavioral disorders (Thompson and Stanwood 2008). Such developmental effects may be the result of exposure to early life stress and affect risk for adult onset disorders in a genotype dependent manner (Caspi et al. 2002, 2003); but also see (Risch et al. 2009).
Measures of serotonin function can be defined as traits with complex genetic architecture, in that they do not appear to be inherited in a simple, Mendelian fashion, and that complicating factors such as epistasis (Pezawas et al. 2008; Stoltenberg 2005), epigenesis (Philibert et al. 2007, 2008a, b), gender moderation and ethnic differences (Williams et al. 2003) influence these traits. Neuroscientists have long appreciated the context dependence and plasticity of neurotransmitter system function. Such complexities, however, limit the effectiveness of current behavior genetic methodologies and analytic strategies.
Epistasis should be expected between genes in neurotransmitter systems because the resulting proteins interact with each other to produce neural activity (Grigorenko 2003). In the serotonin system, there is some empirical evidence that the influence of particular polymorphisms is dependent on the genetic and environmental context in which they are found. For example, the S allele of a polymorphism in the 5-HT transporter regulatory region (5-HTTLPR) is associated with a reduction in volume of a region of the anterior cingulate except in those individuals who carry a brain derived neurotrophic factor (BDNF) Met allele (Pezawas et al. 2008). Another recent study described a complex interaction between 5-HTTLPR, monoamine oxidase A (MAOA) and catechol-O-methyl transferase (COMT) genotype, gender and stress response and susceptibility to major depression (Jabbi et al. 2007). These are just two examples presented as evidence for epistasis in the pathways from neurotransmitter genes to behavior via brain circuits. While it is true, as pointed out by a reviewer, that these and other specific empirical findings of statistical epistasis may eventually turn out to be false (Ioannidis 2005), the inclusion of physiological epistasis in models of neurotransmitter systems is likely to enhance their realism (Brodie 2000), and may, in fact, be necessary for understanding the systems.
Dynamic systems modeling
Progress in the characterization of pathways from genes to behaviors will require the development of new tools and methodologies and will benefit from the application of technologies not typically applied to problems in behavioral genetics. One approach that is well developed, but not in a behavior genetics context, is dynamic systems modeling. In this article, we present a case for the use of dynamic systems modeling in behavior genetics research by presenting and validating a model of neurotransmitter genes and function.
Dynamical system modeling, feedback, filtering and signal processing has been applied to biology or biological engineering for quite some time [for a review of concepts relating to modeling, properties of differential equations, stability and controllability issues see Stoltenberg and Nag (2007)]. Indeed, one of the pioneers of the field of biological modeling, Norbert Wiener, formalized the ideas of feedback and filtering in biology (Weiner 1965). He also developed the field of cybernetics which is a multidisciplinary study involving control systems applications, amongst other fields, to evolutionary biology, neuroscience and psychology. Various oscillators like Duffing Oscillator, Van der Pol oscillator and Lienard systems has been motivated and used to model cardiac and neural oscillatory behavior. Finally, dynamical system modeling in evolutionary biology was pioneered by J.B.S. Haldane where he argued that a systematic theory of natural selection must be a quantitative theory (Haldane 1924).
Dynamical system and control theory can contribute significantly to the modeling of biological subsystems by incorporating multiple parameters whose values may depend upon genetic polymorphisms. The advantage in modeling using differential equations and feedback control theory is generating hypotheses which can then be empirically tested. Moreover, medical advances can be made by perturbing certain parameters governing the dynamics of the subsystem to simulate the actions of pharmacological agents and studying the overall impact on system function prior to conducting costly and risky experiments. On the other hand, even though in principle detailed modeling of interconnected subsystems is possible, however, there could be multiple biochemical processes whose aggregate effect is currently understood but individually the processes are still unknown. In such cases, the dynamic system model is describing the average effects rather than modeling the detailed intricate subsystems that contribute to the aggregate effects. Also, in some cases since the model parameters are not known, the control theoretic model uses values on per unit basis; however, the output is topologically equivalent to the output of the system if the values of the model parameters were known. Thus, in the spirit of Henri Poincare’ we are interested in qualitative analysis and prediction using ordinary differential equations, rather than actual numerical values of the model output. This approach relieves the modeler from the responsibility of absolute accuracy in setting parameter values because the focus is on relative differences and qualitative outcomes.
We shall introduce two mathematical models as examples of the application of dynamical system or control theory to model biological systems. The first model we shall present is that of a bistable switch which has been studied in great detail in mathematical biology [see Cinquin and Demongeot (2002) for details]. The bistable switch is used to model cellular differentiation because it is a dynamic process in which differentiated cells can be in two or more states due to epigenetic regulation for example DNA histone acetylation or re-acetylation. Let us consider two proteins whose concentration are denoted by x1, x2. It is usually assumed that each of the proteins are undergoing exponential decay and they inhibit the synthesis of other proteins in the switch with cooperativity of repression c1, c2 and rate of synthesis s1, s2. Then the bistable switch can be modeled by the following autonomous differential equations:
| (1) |
| (2) |
The second model is the well known Predator and Prey model described by the set of Lotka–Volterra differential equations (see Gottman et al. 2002 or Perko 1991 for details). Let x1 represent the prey population and x2 represents the predator population. Then the well known Lotka–Volterra equations describing the two competitive species can be written as follows:
| (3) |
| (4) |
where a > 0 and d ≥ c > b > 0 are constant parameters. It is to be noted that the differential Eqs. 1–4 are nonlinear because of the presence of terms such as , , x1x2. These systems are usually referred to as planar systems and their properties can be understood by analyzing their phase portrait.
We shall introduce two mathematical models as examples of the application of dynamical system or control theory to model psychology systems. The first model is the well known model describing neurons [see Dunn et al. (2004) or Khalil (1996) for more details] as electrical circuits. Let xi be the activation level of ith neuron. The function σi : R → (−V, V), where R is the set of real numbers, is the ith sigmoidal function with asymptotic activation levels −V and V. Let Tij be the synaptic input from neurons j to neuron i and li represents a constant input usually referred to as static bias. Let τi be the time constant which determines how rapidly the activation rapidly reaches its steady state value. Then the n neurons circuit can be mathematically modeled as interconnected differential equations,
| (5) |
for i = 1…n.
The second model is a mathematical model describing marital interaction between husbands and wives [see Gottman et al. (2002) for more details]. This dynamical system model is different from the previous models (1)–(4), in the sense that the independent variable time takes on discrete value, that is, t = 1, 2, 3,…. Let H(t) and W(t) be the husband and wife’s behavior score at time t > 0. The influence function IHW(H(t)) is the influence of husband’s state at time t on wife’s state at time t + 1. Similarly, the influence function IWH(W(t)) is the influence of wife’s state at time t on husband’s state at time t + 1. Then the discrete dynamical system model describing the marital interaction between husband and wife is given as follows
| (6) |
| (7) |
The parameters a, b, c, and d are constants which are usually estimated for each person. The influence functions IHW(H(t)) or IWH(W(t)) are usually piecewise constant functions or piecewise linear and they depend on various issues/factors that influence the marriage.
From the above examples, and many more that can be found in various literature regarding system theoretic modeling of biological or psychological processes, one of the main points is that dynamic systems modeling of cognitive processes and that of neurons and neural networks, and brain circuits is well-developed, but that genetic variation at system components has not been modeled yet, with the exception of work from our own laboratory (Stoltenberg 2003, 2005, 2010; Stoltenberg and Nag 2007). These examples also show that the approach can be used to model systems at different levels of analysis. To advance in our understanding of pathways from genes to behaviors, we need to model the effects of genetic polymorphisms on parameters in such models.
Dynamic systems modeling of the serotonin system
Substantial empirical and theoretical evidence implicates variation in the function of the serotonin neurotransmitter system in the etiology of individual differences in behavioral control (Carver and Miller 2006). Candidate gene association studies have identified polymorphisms in genes that code for enzymes involved in serotonin metabolism as being associated with impulsive traits (Reuter et al. 2007; Stoltenberg et al. 2006) and with behavioral disorders characterized by deficient behavioral control (Bondy et al. 2006; Hill et al. 2002; Sheehan et al. 2005; Virkkunen et al. 1996). Genetic variants in the serotonin system are associated with brain volume differences and reactivity in areas associated with emotional processing and executive control (Canli et al. 2005, 2008; Herrmann et al. 2007; Pezawas et al. 2008). Clearly, the serotonin system is an important candidate system for understanding the biology of impulsivity.
To begin modeling the effects of genetic variation on the functioning of a neurotransmitter system, one must determine which components of the system to model. The components of interest should have known roles in controlling aspects of system function. From the perspective of a systems approach, one should strive to simplify nature while including sufficient complexity to manage the system (Ward 2002). Our primary focus has been on presynaptic control of serotonin function, which has kept us, for the moment, from modeling postsynaptic outcomes of serotonin function. Presynaptic control of serotonin function is sufficiently complex to merit such a focus and eventually we plan to extend the model to include postsynaptic effects.
The present model is an extension of an earlier, more basic one that included three presynaptic components: the 5-HT reuptake transporter and two autoreceptors (Stoltenberg 2005). Reuptake via the serotonin transporter (SERT) is the primary means by which the action of synaptic 5-HT is terminated. The 5-HT1A somatodendritic autoreceptor inhibits 5-HT neural firing and further 5-HT release when extracellular levels of 5-HT exceed a threshold. The 5-HT1B terminal autoreceptor adjusts the amount of 5-HT to be released. These three components contain common genetic polymorphisms, play a significant control in 5-HT presynaptic function and appear to epistatically interact (Stoltenberg 2005).
In this report, we describe our extensions and refinement of the dynamic system model of 5-HT presynaptic function. We added two metabolic enzymes to the model, (a) tryptophan hydroxylase, which is the rate-limiting enzyme in serotonin synthesis, and (b) monoamine oxidase A, which catalyzes the catabolism of serotonin. The addition of these two components provides a much more complete model of 5-HT function, one that includes most major aspects of the 5-HT “life cycle” in the brain. By including serotonin synthesis and metabolism, we will be able to simulate acute tryptophan depletion as well as cerebral spinal fluid levels of the 5-HT metabolite 5-hydroxyin-doleacetic acid (CSF 5-HIAA), both of which provide opportunities for further model validation.
In addition to determining which parameters to model, one must determine which outcomes to include. In the present case, we modeled aspects of central 5-HT function that were directly impacted by components of the model and we focused on those indices of 5-HT function that would provide avenues for comparison to measures in the empirical literature. Specifically, measures of extracellular (or synaptic) levels of 5-HT are often measured in mice using microdialysis techniques (de Groote et al. 2002; Stenfors and Ross 2004), measures of neural firing are measured using single unit recording (Sharp et al. 1997) or on a much grosser scale, functional magnetic resonance imaging (functional magnetic resonance imaging) (Hariri and Weinberger 2003), and measures of CSF 5-HIAA are obtained via lumbar puncture (Williams et al. 2003). In our model, we included the following outcome measures: (a) 5-HT cell firing rate; (b) extracellular (synaptic) 5-HT level; (c) intracellular 5-HT level; and (d) CSF 5-HIAA level.
Our model is based on the assumption that genetic polymorphisms in system components, as well as pharmacological action, produce alterations in aspects of neurotransmitter system function (e.g., neural firing rates, level of synaptic neurotransmitter, level of neurotransmitter metabolites in the cerebral spinal fluid) in an interactive fashion. Moreover, we assume that these variations in system function produce structural or functional differences in brain regions that influence the behaviors of interest. Whether these assumptions are supported by empirical findings at this point in time is immaterial to this modeling work. We assume that such genetic variation exists or alternatively that pharmacological intervention can produce the differences in function that we are modeling. Based on empirical evidence, epistasis, gene by environment interactions and gender moderation of neurotransmitter function are expected. In this study, we describe a control system model of presynaptic serotonin function and present results that support the validity of the model.
Methods
Model description
In the following sections we describe our control system model of presynaptic serotonin function, which consists of a series of differential equations. Several of the equations are modifications of our previous work and others are slight modifications of well known equations that model enzyme kinetics. In each case, a single parameter models genetically mediated functional variation from “low” to “high” across a range of values optimized to influence one or more of the outcome variables. Such parameter optimization is necessary in model development and is a first step in establishing the face validity of the model.
Intracellular and extracellular serotonin dynamics
In this section, we describe the equations used to model the action-potential mediated release of 5-HT into the synapse as well as 5-HT reuptake mediated by the serotonin transporter. Our model is based on known functions of 5-HT system components including autoreceptor mediated firing inhibition and release, refractory periods and reuptake (Baumgarten and Gothert 1999). To our knowledge, no standard differential equations exist for these functions. The equation that models extracellular 5-HT level simply describes the amount of extracellular 5-HT at a given time is a function of how much enters the synapse via release and exits via reuptake and diffusion. The equation that models intracellular 5-HT levels simply describes the amount of intracellular 5-HT at a given time is a function of how much enters the presynaptic neuron via synthesis and reuptake and how much exits the neuron via release and catabolism.
Let x1(t) be the amount of extracellular serotonin compartment at any time t > 0. Let γ be the reuptake rate parameter whose value is influenced by an individual’s 5-HTTLPR genotype. Let δ be the diffusion rate coefficient which characterizes the loss of serotonin from the extracellular serotonin compartment. The numerical value of δ is usually very small.
Let u(t) be the feedback control input to the extracellular serotonin compartment. The signal u(t) is a composite of output of the autoreceptors 5-HT1A, 5-HT1B the refractory period and the amount of intracellular serotonin x2(t) in the intracellular compartment (see Fig. 1). The refractory period is modeled by a sinusoidal function whose period can be modulated. All the control inputs from the autoreceptors 5-HT1A and 5-HT1B are non-zero during the positive part of the refractory period, thus allowing the neurons to fire. The autoreceptor 5-HT1A inhibits the firing of serotonin containing neurons if the level of extracellular serotonin x1(t) is greater than the set point value of serotonin x1th. Thus, the feedback u(t) contains the activation or inhibition function for set point regulation or homeostasis. The functioning of 5-HT1A has a stochastic variable rand which takes on random values in the interval (0,1) and there is a parameter aprob whose numerical value if equal to one implies normal functioning of the autoreceptor and any other value in the interval (0,1) will lead to stochastic inhibition to the firing rate (i.e., modeling inefficient firing inhibition). The autoreceptor 5-HT1B determines the amount of release of serotonin when activated. The autoreceptor 5-HT1B is modeled by two parameters Rmax which is the maximum amount of serotonin release in per unit from intracellular compartment and β a constant parameter (for a given genotype) which models the proportion of extracellular serotonin present at any time in the extracellular compartment such that Rmax – βx1(t) is the proportion of serotonin input to the synapse. This controller design is adopted so as to avoid excess serotonin in the synapse. Moreover, when the autoreceptor 5-HT1B is not working at all (e.g., “knocked out”) then β = 0 and when it is fully functioning then β = 1. Genetic or pharmacological variation can be modeled such that β ∈ (0, 1). Thus, the resultant control function of refractory period, 5-HT1A and 5-HT1B is mathematically modeled as follows,
| (8) |
where the parameter per modulates the refractory period.
Fig. 1.
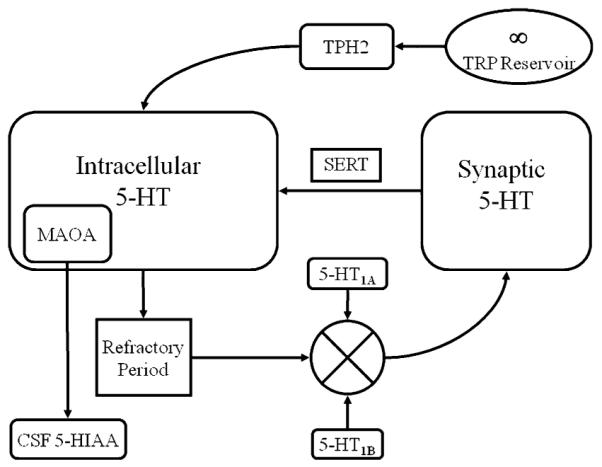
Flow diagram of presynaptic control elements in the serotonin system. Dietary tryptophan is converted to serotonin by TPH2 and stored in the intracellular compartment. Serotonin is released into the synapse as a function of a refractory period and input from two autoreceptors that inhibit firing (5-HT1A) or influence the amount of serotonin released (5-HT1B). Serotonin is removed from the synaptic space by the serotonin transporter (SERT) via reuptake. Serotonin is either repackaged for re-release or it is catabolized by MAOA and its by product 5-HIAA is transported to the cerebral spinal fluid
We also model the feedback input u(t) to the extracellular serotonin compartment by using (input–output) characteristic of the sigmoidal function such that there is a gradual build up of extracellular serotonin x1(t) with a growth rate of , which is fixed a priori in (the) model simulation. Thus, the feedback input to the synapse is modeled as a “gate” which has delay or time lag of for the serotonin to build up to the final state instead of a “light switch” [see Sontag (2005) for details regarding sigmoidal response] which is usually modeled by a step function. The input–output sigmoidal function is given by, .
Thus, the dynamics of extracellular serotonin component is given by,
| (9) |
The intracellular serotonin dynamics is modeled as follows,
| (10) |
where κ4 is a positive constant which is the coupling rate that causes metabolic breakdown of serotonin through the involvement of the enzyme monoamine oxidase A (MAOA), ρ(t) is a state variable which corresponds to the serotonin input to the intracellular serotonin compartment produced from dietary tryptophan catalyzed by a type of rate-limiting enzyme tryptophan hydroxylase which has been identified as TPH2 and u(t) is given by Eq. 8.
Serotonin synthesis
Equations describing enzyme kinetics are well known, and in this section and the next we describe our minor modifications of those equations to model genetically mediated functional variation in enzyme function. In both sets of equations we assume that genetic variation influences the rate at which the enzyme converts substrate to product.
We mathematically model the conversion of dietary tryptophan into serotonin by the application of the ratelimiting enzyme TPH2 by modifying the Michaelis–Menten kinetics model (see Edelstein-Keshet 1988 for more details). Let c(t) be the amount of tryptophan from diet in the synthesis chamber and v be the constant amount of tryptophan input from the reservoir (see Fig. 1). Let κ−1 be a positive coefficient indicating reverse reaction rate and κ1trp be a positive constant indicating forward reaction rate. Then the rate of change of tryptophan in the synthesis chamber is given by
| (11) |
where y1(t) is the amount of TRP-TPH2 complex. Let ε be a singular perturbation coefficient which is relatively small indicating a very slow, quasi-static change of the TRP-TPH2 complex and κ2trp a positive number modeling the synthesis rate coefficient. Then, the rate of change of the TRP-TPH2 complex is given by
| (12) |
Finally, the rate of production of serotonin by catalysis of TRP-TPH2 complex is given by
| (13) |
where κ3 is a positive constant which is designed for asymptotic stabilization.
Serotonin catabolism
We shall model the metabolic breakdown of serotonin by the enzyme MAOA by applying very similar technique as was discussed in the context of conversion of dietary tryptophan into serotonin. Let λ(t) be the amount of serotonin involved in the metabolic breakdown in the catabolic chamber (see Fig. 1) and z1(t) be the 5-HT-MAOA complex. Let κ1maoa is a positive constant indicating forward reaction rate. Then, we have the following,
| (14) |
Let κ2maoa be a positive number modeling the metabolic rate coefficient. Then, the rate of change of the 5-HT-MAOA complex is given by,
| (15) |
Finally, the rate of production of CSF 5-HIAA is given by
| (16) |
where, p(t) is the amount of CSF 5-HIAA at any time t > 0.
Firing rate
An important outcome measure for mathematical models of neurons is the number of action potentials in a given time period. Such an outcome is a representation of neural activity, which can then be compared to empirical observations of single unit recordings or more gross measures of brain activation such as EEG or MRI. In our model, we are measuring the number of times that the system “fires” or releases 5-HT in a fixed number of time steps (i.e., 200, with a dt = .01). So, in this study, the maximum number of times that a system could fire is 20,000 (i.e., 200/.01).
We integrate Eqs. 8–16 using the Runge–Kutta-4 routine of Berkeley Madonna 8.0.1 with a pre-determined step size of Δt > 0. To measure the firing rate we define the following discrete dynamical equation,
| (17) |
and FIRERATE(t) is a discrete variable which measures the firing rate of the neuron. The discrete dynamical Eq. 17 is initialized by FIRERATE(0) = 0 and FIRE(0) = 0.
Software
To solve the systems of differential equations, we used Berkeley Madonna 8.0.1 (Zahnley et al. 2001). The code for the model and the basic model parameters are included in the Appendix.
Model validation
To test the validity of the model, we conducted three separate tests. The first validity test examined the face validity of the model by comparing parameter plots to empirical or theoretical expectations of parameter variation for the five primary model components (a) synthesis [tryptophan hydroxylase], κ1trp, (b) release threshold [5-HT1A], aprob, (c) release quantity [5-HT1B], β, (d) reuptake [5-HTTLPR], γ, and (e) catabolism [MAOA], κ1maoa. Each parameter was varied across a range of values that represented “low” to “high” function. For each parameter, plots were generated for each outcome of interest (a) firing rate, (b) synaptic 5-HT level, (c) intracellular 5-HT level and, (d) CSF 5-HIAA level. It should be noted that we optimized each parameter so that varying it across the range of low to high function produced noticeable changes on at least one outcome variable. Such an approach is necessary at the early stages of model development because the appropriate scale for parameters is not obvious a priori. Therefore, these tests of face validity indicate simply that when the parameter of interest is varied across a given range of values it influences a given outcome in a direction that fits with a general understanding of how the system functions.
The second validity test examined criterion validity of the model by comparing model output to empirical reports of brain activity for individuals with different genotypes for TPH2 and 5-HTTLPR (Canli et al. 2008). We simulated firing rate data for nine groups defined by combinations of “low”, “medium” and “high” levels of the synthesis (TPH2) and reuptake parameters (5-HTTLPR). This test is an attempt to determine whether the model is capable of producing a pattern of output that is similar to a recent finding that identifies an association between genotype and patterns of brain activation. If the model produces output patterns that are generally similar to empirical findings it suggests that the model is capturing some important aspects of 5-HT system function.
The third validity test examined criterion validity of the model by comparing a model outcome to an observed measure of impulsivity in groups of subjects defined by their THP2 intron-8 (rs1386483) and MAOA (u-VNTR) genotypes. Subjects completed a questionnaire measure of impulsivity and provided buccal cells for DNA extraction and PCR-based genotyping. We simulated CSF 5-HIAA data for four groups defined by “low” and “high” levels of synthesis (TPH2) and catabolism (MAOA) parameters. Individuals were assigned simulated CSF 5-HIAA values based on their joint genotypes (“low” synthesis = TPH2 C/C; “high” synthesis = TPH2 T/_; “low” catabolism = MAOA short [i.e., presence of 3-repeat]; “high” catabolism = MAOA long). We chose to examine simulated CSF 5-HIAA level with impulsivity because of the well documented negative correlation between them (Soderstrom et al. 2001; Westergaard et al. 2003). We focused on TPH2 and MAOA because in the face validity tests both the synthesis and catabolism parameters influenced CSF 5-HIAA level. This is an attempt to determine whether the model is capable of predicting an individual’s self-reported impulsivity based solely on their genotype at two loci. An important aspect of a systems behavior genetic approach is to improve our ability to predict behavior based on an individual’s genotype. We do not have observed levels of CSF 5-HIAA in this dataset. It would be desirable to first test whether our simulated levels of CSF 5-HIAA determined by genotype were associated with actual CSF 5-HIAA levels. However, lumbar punctures were outside of the scope of this study. Therefore, in an exploratory analysis, we compared simulated CSF 5-HIAA with observed impulsivity scores as a way to examine the potential of the model to predict behavior.
Subjects
Subjects who provided data for the third validation tests were recruited for another study to examine potential associations between polymorphisms in serotonergic genes and individual differences in impulsivity and health-risk behaviors (Stoltenberg et al. 2008). Subjects (N = 200) were recruited from the student population at a small midwestern university and were paid $5 for their participation. The sample was mostly Caucasian (95%), female (62%), and ranged in age from 18 to 47 (M = 22.67, SD = 5.65). The Institutional Review Board approved the study and all subjects provided informed consent.
Measures
Barratt Impulsiveness Scale (version 11). The Barratt Impulsiveness Scale, Version 11 (BIS-11) is a widely used 30-item questionnaire that assesses levels of impulsivity. Higher scores on the BIS-11 denote higher levels of impulsivity. Internal consistency of BIS-11 Total score for college students is acceptable (Cronbach’s α = 0.82, (Patton et al. 1995)).
Genotyping
DNA was extracted for genotyping from buccal cells (DNeasy Blood & Tissue Kit®, QIAGEN®). Gene amplification was performed on an GeneAmp® PCR System 9700 (Applied Biosystems), and a FOTO/Analyst Digital Imaging System (FOTODYNE®) was used to document gels stained with Ethidium Bromide and visualized under ultraviolet light.
The TPH2 intron-8 (rs1386483) polymorphism was amplified using forward primer: 5′-GCT GGC TCT GAA CGT GTA TTT TG-3′, and reverse primer: 5′-TTT GGC TGA TTT TCC TAA TTA AT-3′ (note this primer was designed with a mismatched base pair A to generate an Ssp1 restriction site, (Stoltenberg et al. 2006). PCR conditions were 60 s at 95°C, 45 s at 52°C, and 45 s at 72°C for 30 cycles. To ensure complete digestion, PCR products were digested (in same tube used for PCR) overnight with 3–5 units of Ssp1 and then 8 μl of each digest was separated by electrophoresis in 3.5% agarose gels.
The MAOA u-VNTR was amplified using forward primer 5′-ACA GCC TGA CCG TGG AGA AG-3′, and reverse primer 5′-GAA CGG ACG CTC CAT TCG GA-3′ (Huang et al. 2004). PCR reaction conditions were 60 s at 95°C, 45 s at 55°C, and 90 s at 72°C for 32 cycles. Amplification products were separated on 3.0% agarose gels.
Results
Face validity
Parameter plots are shown in Fig. 2 across a range of synthesis rates to model variation in TPH2 activity (from 1.0 to 2.0) for (a) firing rate, (b) synaptic 5-HT level, (c) intracellular 5-HT level and (d) CSF 5-HIAA level. As synthesis rate increased, firing rates diminished. No substantial change in Synaptic level of 5-HT was observed. Dramatic increases for both Intracellular 5-HT and CSF 5-HIAA levels were observed.
Fig. 2.

Parameter plots of synthesis rate (i.e., TPH2) showing simulated a firing rate, b synaptic 5-HT level, c intracellular 5-HT level, and d CSF 5-HIAA level
Parameter plots are shown in Fig. 3 across a range of inhibition probabilities to model variation in capacity of the 5-HT1A receptor to inhibit firing (from 0.9 to 1.0) for (a) firing rate, (b) synaptic 5-HT level, (c) intracellular 5-HT level and (d) CSF 5-HIAA level. As the probability of firing inhibition increased, firing rates drastically diminished. With the exception of a slight initial decrease, no substantial change in Synaptic level of 5-HT was observed. No change was observed for both Intracellular 5-HT and CSF 5-HIAA levels.
Fig. 3.

Parameter plots of probability of inhibition (i.e., 5-HT1A) showing simulated a firing rate, b synaptic 5-HT level, c intracellular 5-HT level, and d CSF 5-HIAA level
Parameter plots are shown in Fig. 4 across a range of release parameters to model variation in capacity of the 5-HT1B receptor to control release amount (from 0.0 to 1.0) for (a) firing rate, (b) synaptic 5-HT level, (c) intracellular 5-HT level and (d) CSF 5-HIAA level. As the release parameter increased, firing rates increased. No substantial change in was observed for Synaptic level of 5-HT, Intracellular 5-HT level and CSF 5-HIAA level.
Fig. 4.

Parameter plots of release amount (i.e., 5-HT1B) showing simulated a firing rate, b synaptic 5-HT level, c intracellular 5-HT level, and d CSF 5-HIAA level
Parameter plots are shown in Fig. 5 across a range of reuptake rates to model variation in capacity of the 5-HT transporter to remove 5-HT from the synapse (from 0.0 to 1.0) for (a) firing rate, (b) synaptic 5-HT level, (c) intracellular 5-HT level and (d) CSF 5-HIAA level. As reuptake rate increased, firing rates increased. A dramatic decrease in Synaptic level of 5-HT was observed. No change was observed for both Intracellular 5-HT and CSF 5-HIAA levels.
Fig. 5.

Parameter plots of reuptake rate (i.e., SERT) showing simulated a firing rate, b synaptic 5-HT level, c intracellular 5-HT level, and d CSF 5-HIAA level
Parameter plots are shown in Fig. 6 across a range of catabolism rates to model variation in capacity of MAOA to degrade 5-HT (from 1.0 to 2.0) for (a) firing rate, (b) synaptic 5-HT level, (c) intracellular 5-HT level and (d) CSF 5-HIAA level. As catabolism rates increased, no change was observed for firing rates, Synaptic or Intracellular levels of 5-HT. A substantial increase in CSF 5-HIAA levels was observed as catabolism rates increased.
Fig. 6.

Parameter plots of catabolism rate (i.e.,. MOAA) showing simulated a firing rate, b synaptic 5-HT level, c intracellular 5-HT level, and d CSF 5-HIAA level
Simulated firing rate by TPH2 and 5-HTTLPR genotype
Figure 7a shows simulated firing rates for nine genotypes defined by level of function for the synthesis (TPH2) and reuptake (5-HTTLPR) parameters. Synthesis rates (TPH2) were modeled as High (TT, 2.0), Medium (TG, 1.5) and Low (GG, 1.0). Reuptake rates (5-HTTLPR) were modeled as High (LL, 1.0), Medium (LS, 0.5) and Low (SS, 0.0). The lowest firing rates are observed in the Low reuptake condition, with very little difference observed due to synthesis rates when coupled with Low reuptake. As reuptake rate increases to Medium or High, firing rates also increase. In addition, within Medium or High reuptake groups, firing rate is highest for the Low synthesis conditions and lowest for the High synthesis conditions with the Medium synthesis condition intermediate. The non-parallel lines shown in Fig. 7a indicate epistatic interaction between TPH2 and 5-HTTLPR on firing rate.
Fig. 7.

Simulated firing rate of serotonergic neurons across groups defined by 5-HTTLPR and TPH2 genotypes shown in a separate genotype groups, and b pooled groups after Canli et al. (2008) [i.e., L + G = L/L, G/G; L + T = L/L, T/_; S + G = S/_, G/G; S + T = S/_, T/_]. The pattern of effects shown in panel B is consistent with the additive effect reported by Canli et al. (2008) although the underlying pattern shown in panel A (i.e., the raw scores before grouping) suggests epistasis not additivity
Figure 7b shows the nine genotypes combined into the three groups that were compared with respect to brain activation in response to emotional stimuli (Canli et al. 2008). Grouping the genotypes in that way produced a graph that suggests an additive relationship for TPH2 and 5-HTTLPR on firing rate. Our results suggest that the model captures the relationship between TPH2 and 5-HTTLPR genotypes on firing rate such that higher rates of 5-HT firing are associated with lower levels of brain activation in response to affective stimuli. In other words, higher rates of 5-HT firing produce a higher level of constraint on the amygdala. Additionally, our simulation results suggest that TPH2 and 5-HTTLPR may actually epistatically interact to affect brain activation in response to emotional stimuli and that the grouping used by Canli et al. (2008) may have obscured the interaction.
Simulated CSF 5-HIAA and impulsivity
TPH2 allele frequencies were calculated by pooling across sex. Because the MAOA u-VNTR is X-linked, allele frequencies were analyzed separately by sex. For TPH2 (intron-8; rs1386483): C (142 bp) = 0.73, T (123 bp) = 0.27 (N = 189); for MAOA u-VNTR: for males, 3-repeat (321 bp) = 0.42, 4-repeat (351 bp) = 0.57, 5-repeat (381 bp) = 0.01 (N = 69); for females, 3-repeat = 0.37, 4-repeat = 0.61, 5-repeat = 0.02 (N = 124). All markers were in H–W equilibrium. Due to small sample sizes in some cells and because of documented activity differences associated with MAOA u-VNTR genotype (Deckert et al. 1999) we grouped the sample into four genotype categories defined by TPH2 (T/_ and C/C) and MAOA (Short = 3-repeat homo- or hemizygotes, Long = all others). Analyses were conducted using data from the 187 individuals that had both genotypes and BIS-11 scores available.
The mean score for the observed BIS-11 Total was 62.86 (SD = 9.83, N = 187). The mean level for the simulated CSF 5-HIAA was 2.68 (SD = 1.08, N = 187). Z-scores of the observed BIS-11 Total score and the simulated CSF 5-HIAA level were significantly correlated (r = −0.22, p = .002). Figure 8 shows observed mean BIS-11 Total scores and simulated mean CSF 5-HIAA levels for groups defined by observed TPH2 and MAOA genotypes.
Fig. 8.
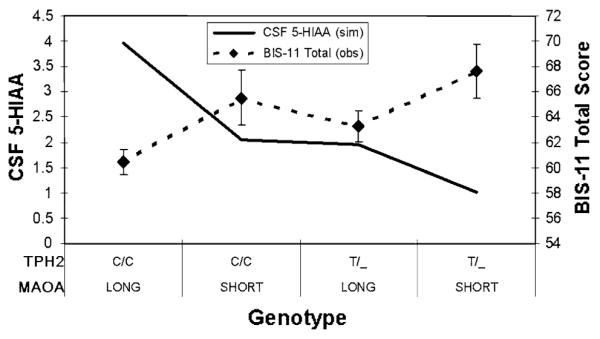
Mean observed BIS-11 Total scores and simulated CSF 5-HIAA levels for subjects grouped by observed MAOA and TPH2 genotypes
Discussion
We modeled the effects of genetic polymorphisms in controlling components of presynaptic serotonin neurotransmitter function with a control system model consisting of differential equations and difference Eqs. 8–17 and tested the validity of the model by comparing its output with both published empirical reports and with observed genetic and questionnaire data. Basic validation tests, such as observing increases in simulated extracellular 5-HT levels when the reuptake parameter is reduced, suggest that the model captures aspects of 5-HT function. Further validation of the model was obtained by comparing simulation results to a published empirical report of the association between TPH2 and 5-HTTLPR genotypes on brain activity in response to observing facial expressions of fear (Canli et al. 2008). Interestingly, our results suggest that the genotype grouping used by Canli et al. (2008) may have limited their capacity to detect potential epistatic interaction between TPH2 and 5-HTTLPR. Furthermore, the model was validated by simulating the CSF 5-HIAA levels of individuals based on their TPH2 and MAOA genotypes and observing a negative correlation between the simulated CSF 5-HIAA and observed impulsivity scores as measured by the Barratt Impulsiveness Scale. At this juncture, it is important to note that further validation tests of the model should be conducted, but the validation tests presented here provide important, albeit indirect, evidence that the model captures important aspects of the influence of genetic polymorphisms on 5-HT system function. More direct validation tests with both human and non-human animal models and direct measures of 5-HT function are needed.
Reducing the synthesis parameter (from 2.0 to 1.0) in our face validity tests produced an increase in the rate of neural firing and significant decreases in intracellular 5-HT and CSF 5-HIAA (see Fig. 2). A change in the rate of neural firing is consistent with the interpretation that vesicles are less than full when the rate of 5-HT synthesis is reduced, which then requires more firing events to achieve sufficient synaptic 5-HT levels. A reduction in intracellular 5-HT is consistent with a recent study that found serotonergic neurons in the raphe nucleus to be devoid of 5-HT in mice with the TPH2 gene knocked out (Gutknecht et al. 2008). Our parameter range did not model a complete knockout of TPH2, but our results are consistent in the sense that a reduction of synthesis produced a reduction in intracellular levels of 5-HT. We did not see a reduction in extracellular 5-HT, which is likely due to the fact that we did not model a complete knockout of synthesis and that even when intracellular levels are relatively low they are sufficient to maintain sufficient 5-HT in the extracellular space. Our finding, that reducing synthesis also reduces CSF 5-HIAA levels is not consistent with a recent study that found no association between variants of a TPH2 promoter single nucleotide polymorphism (SNP) rs4131347 (−C8347G) and CSF 5-HIAA (Mann et al. 2008). It is not clear, however, whether this particular SNP is associated with significant differences in 5-HT synthesis. Our findings suggest that significant changes in 5-HT synthesis are likely to be associated with CSF 5-HIAA levels.
The primary effect of reducing the probability of inhibition, which models a reduction in 5-HT1A autoreceptor function, is an increase in neural firing rate (see Fig. 3). Consistent with our earlier work (Stoltenberg 2005), our present findings for modeling the 5-HT1A autoreceptor correspond to work with 5-HT1A knockout mice that show a significant increase in 5-HT neural firing rates (He et al. 2001; Parsons et al. 2001; Richer et al. 2002) and little to no change in extracellular 5-HT levels (He et al. 2001; Parsons et al. 2001; Richer et al. 2002).
Varying the release parameter, which models variation in the efficiency of the 5-HT1B terminal autoreceptor to adjust the amount of 5-HT released in response to an action potential, affected only firing rate in our study (see Fig. 4). Reducing the release parameter from its baseline of 1.0–0.0 produced a substantial decrease in neural firing rate because without the capacity to reduce the amount of 5-HT released from the maximum results in excess 5-HT in the synapse, which then results in firing inhibition via the 5-HT1A autoreceptor. The firing rate of 5-HT containing neurons in the dorsal raphe is reduced in 5-HT1B knockouts relative to wild type (Evrard et al. 1999).
Varying the reuptake rate parameter in the present study modeled variation that could result from genetic polymorphisms, such as 5-HTTLPR, or by pharmacological intervention, such as selective serotonin reuptake inhibitors. Reducing reuptake from its baseline value of 1.0–0.0 increased the level of extracellular 5-HT and reduced the firing rate (see Fig. 5). Both of these effects are seen in mice, whose reuptake rate is manipulated by knocking out the 5-HTT structural gene or by administration of SSRI (de Groote et al. 2002; Evrard et al. 2002; Gobbi et al. 2001; Mannoury la Cour et al. 2001). We did not observe changes in levels of intracellular 5-HT or in CSF 5-HIAA. However, there is evidence that 5-HTTLPR genotype may be associated with differences CSF 5-HIAA levels (van der Stelt et al. 2004; Williams et al. 2003). Our model does not capture this aspect of 5-HT function, which suggests that further refinements to the model are necessary.
Varying the catabolic parameter in the present study modeled variation that could result from genetic polymorphism, such as the MAOA u-VNTR, or pharmacological intervention, such as a monoamine oxidase inhibitor. Such variation in the model produced changes in only CSF 5-HIAA level (see Fig. 6). There is evidence that the MAOA u-VNTR genotype affects CSF 5-HIAA level, but that the outcome may depend on gender (Jonsson et al. 2000). We will extend the model to include gender so that we can better describe the function of the system.
For the most part, the model produces output that has substantial face validity. That is not to say, however, that the model cannot be improved. It does appear to capture important aspects of 5-HT system function.
In our tests of criterion validity, we compared the output of the model to specific conditions of interest. In the first case, we found that the pattern of firing rates across groups defined by High, Medium and Low rates of both synthesis and reuptake was consistent with the constraining role that 5-HT neurons play on amygdala activation. That is, higher 5-HT firing rates should be associated with lower rates of amygdala activation. We found that the group characterized by Low synthesis and High reuptake (L + G, see Fig. 7) had the highest firing rate and the group characterized by High Synthesis and Low reuptake had the lowest firing rate (S + T) and the remaining group (L + T and S + G) was intermediate. This pattern can be interpreted in the context of the findings of recent amygdala activation patterns in response to facial expressions of fear where the L + G group had the lowest activation, the S + T group had the highest activation and the remaining group was intermediate (Canli et al. 2008). Our results suggest the testable hypothesis that the G allele of the −703 G/T TPH2 polymorphism is associated with low synthesis rates and the T allele is associated with high synthesis rates. We were unable to locate any extant studies to support this prediction. The results of this criterion validity test should provide a level of confidence in the model.
In addition, we preformed another criterion validity test in which we compared the simulated level of CSF 5-HIAA for groups of subjects defined by TPH2 intron-8 and MAOA u-VNTR genotypes to observed scores on the Barratt Impulsiveness Scale (Version 11; see Fig. 8). The significant negative correlation between simulated CSF 5-HIAA scores and the impulsivity scores is consistent with the negative correlation in rhesus macaques between CSF 5-HIAA and rate of long (i.e., risky) leaps through the tree canopy (Westergaard et al. 2003) and outward directed aggression (i.e., impulsive aggression) in humans (Soderstrom et al. 2001). Because of the indirect nature of the evidence, this test should be considered to be exploratory. It would be highly desirable to directly measure CSF 5-HIAA in such a study.
Although serotonin is known to play a role in many behaviors and behavioral disorders, it is difficult to directly measure serotonin function in the human brain. Levels of extracellular serotonin are relatively low and indirect measures, such as whole blood serotonin levels, may not be good indicators of central serotonin function. No analog for single unit recording to measure firing rates of serotonergic neurons or microdialysis of extracellular serotonin levels is available for use in humans. Several indirect measures of serotonin function have been used in humans such as whole blood or plasma serotonin level, binding of receptors or transporters in blood platelets, CSF 5-HIAA, and fMRI. Each of these indirect measures of serotonin function has both advantages and limitations. Ideally, a measure of serotonin function would be valid, reliable, simple to measure, non-invasive and inexpensive. If this control system model could provide valid simulated measures of central 5-HT function by using genotypes, which can be obtained using non-invasive and relatively inexpensive techniques, it would be useful tool in both the research laboratory and the clinic for better understanding and treating disorders that result from 5-HT system dysfunction.
Control system models, such as the one presented here may be useful in behavior-genetic analyses because they provide a platform on which to develop a more rich understanding of how genetic variation may contribute to individual differences in the function of neurotransmitter systems and brain circuits. Candidate gene association studies are commonly used to investigate heredity-behavior relations in humans in a hypothesis driven manner. The case–control approach can be used to study genetic associations with disorders or other dichotomous traits. A regression-based approach can be used to study associations between genetic polymorphisms and the expression of dimensional traits. Although candidate gene association studies are widely used, they have been criticized for a paucity of replication across studies (Sullivan 2007). Population stratification has long been a focus of criticism of case–control designs (Hamer and Sirota 2000) although it may not be as critical as once thought, especially if studies are carefully designed (Hutchison et al. 2004).
Using an approach to the study of heredity-behavior relations that incorporates a dynamical systems modeling is consistent with the recommendations provided by Hutchison et al. (2004) to maximize effect sizes and improve the explanatory power of candidate gene association studies. Their first recommendation is to use a design that controls for third variables. Our proposed model explicitly identifies components of the 5-HT system that are known to influence its function. While the risk for confounding variables exists in any model, a theoretically and empirically derived dynamic systems model seeks to include the factors that are known to significantly influence function thereby reducing the number of unknown third variables. This is especially true when one compares the dynamic systems approach to a candidate gene analysis of a single polymorphism. Hutchison et al.’s (2004) second recommendation is to use continuous rather than dichotomous outcome measures. Such an approach fits well within the framework of a dynamic systems approach. Their third recommendation is to use an endophenotypes because such traits will be simpler genetically than will diagnoses. In addition, they suggest using quasi-experimental designs that include assigning individuals to groups based on their genotypes in a pretest posttest design. The model that we describe can be used to identify genotype combinations that produce the most divergent 5-HT function outcomes. One could screen a large sample of individuals to obtain groups of individuals with the target genotypes and then phenotype those individuals in detail. By using this approach with wellvalidated, reliable, continuous psychological or physiological measures one could maximize effect sizes and minimize the number of individuals that would be studied intensely. Hutchison et al.’s (2004) fourth recommendation was to specify a theoretically motivated mediational model. Our system dynamic approach clearly fits with this recommendation in that our model is based on theoretical and empirical relations among 5-HT system components. Finally, Hutchison et al. (2004) recommend that researchers provide effect size estimates and increase sample sizes of studies. Our approach, while not specifically addressing this final recommendation, is not conflict with it. It is our position that an approach to studying heredity-behavior relations that includes a dynamic systems approach to candidate gene association studies would conform to the recommendations of Hutchison et al. (2004) and thereby increase the reliability of candidate gene association studies.
There are some limitations to the present model. The model does not include a number of factors known to influence serotonin function such as gender (Weiss et al. 2005; Williams et al. 2003), and stress (Barr et al. 2003a, b; Caspi et al. 2003; Konno et al. 2007). Not all effects reported in the empirical literature are not reflected in the simulation output such as the approximately 30% reduction in CSF 5-HIAA that is seen when reuptake is reduced (Stenfors and Ross 2004) although this might be due to relatively course scaling that might not detect a change of that magnitude. Alternatively, it may be that our current model might not yet fully capture the complex relations among aspects of 5-HT function. No postsynaptic effects or interactions with other neurotransmitter systems are included in the present model. These and other shortcomings of the model do not detract from the demonstrated validity of the model to capture specific aspects of 5-HT function, but underscore the real complexity inherent in pathways from genes to behavior.
Undoubtedly there are alternative approaches to modeling the influence of genetic variation on presynaptic 5-HT function. The control system approach that we have chosen has a long history, is well understood and provides a powerful and versatile platform with which to model complex systems. Our approach is non-proprietary and we encourage other investigators to adopt a control system approach to studying relations among genes and behaviors. However, developing equations and selecting parameters to model a real system is fraught with peril. To model the dynamics of a system one is forced to make decisions regarding the precise direction and magnitude of relationships among variables to a degree not required in static diagrams describing systems. Where practical, we have indicated the factors that influenced our decisions about the model. Other investigators may choose to model specific processes or parameters differently. Our decisions were based on empirical data when it was available or on theoretical considerations with respect to neurotransmitter system function. Parameter values were often chosen so that parameter changes across components would be of similar relative magnitude so that artifacts of scale would not be produced. In the end, models such as this require validation in well controlled studies in biological systems. We acknowledge that our work has not yet progressed to the stage where we can be fully confident that it fully captures the behavior of the serotonin system and we continue to work toward that end.
A long-term goal of this research program is to use genotypes and other patient characteristics in a control system model to provide information on system function that might lead to improved diagnoses and treatments. In other words, we hope that modeling efforts such as this might facilitate the development of personalized medicine (Stoltenberg 2010).
The present findings add to the substantial literature on the effects of genetic polymorphisms on 5-HT function and individual differences in behavior. Specifically, we presented a system of differential equations that appears to capture important aspects of the joint influence of genetic polymorphisms in the 5-HT system on the system’s function. This work may help to orient investigators toward a systems approach for the next stage in behavior-genetic analysis that will be focused on better characterizing pathways from genes to behavior.
Conclusions
Control system modeling is an approach that provides a solid platform on which to build a systems view of pathways from genes to behavior. Our results provide validation for this approach and suggest several testable hypotheses. The techniques of control system modeling enable investigators to explore the complex genetic architectures that are likely to underlie the behaviors and behavioral disorders of greatest interest to psychologists.
Acknowledgments
This work was partially supported by a three year research grant from the National Institute of Mental Health (NIMH) grant no. 1R15MH077654-01A. Also, the first author in this work was partially supported by NIH grant no. 2 P20 RR016479 from the INBRE program of the National Center for Research Resources. The second author will also like to thank the Department of Mathematics for their generous support and also for the Berkeley Madonna Software.
Appendix
Berkeley Madonna Code to simulate differential and difference Eqs. 8–17:
METHOD RK4
STARTTIME = 0
STOPTIME = 200
DT = 0.01
d/dt(x1) = −(g + d) * x1 + u * (1 − exp(−2 * x2))/(1 + exp(−2 * x2))
INIT x1 = 0
d/dt(x2) = g * x1 − k4 * x2 + r − u
INIT x2 = 1
d/dt(c) = −c + ((k_1/k1trp) + c) * y1 + v
INIT c = 1
d/dt(y1) = (c − ((k_1 + k2trp)/k1trp + c) * y1)/e
INIT y1 = 1
d/dt(r) = −k3 * r + k2trp * y1
INIT r = 1
d/dt(l) = −l + ((k_1/k1maoa) + l) * z1 + k4 * x2
INIT l = 1
d/dt(z1) = (l −((k_1 + k2maoa)/k1maoa + l) * z1)/e
INIT z1 = 0
d/dt(p) = −k3 * p + k2maoa * z1
INIT p = 0
rand = RANDOM(0, 1)
u = IF (SIN(TIME/PER) > 0) THEN
(IF (rand > 1 − aprob) AND ((x1 − x1th) >0)
THEN 0 ELSE (Rmax − b * x1) * x2) ELSE 0
INIT Fire Rate = 0
INIT Fire = 0
FireRate(t + Dt) = FireRate + Fire
Next Fire = IF u > 0 THEN 1 ELSE 0
See Table 1.
Table 1.
Model parameter values
| Parameter | Baseline | Face validity | Criterion validity 1 | Criterion validity 2 |
|---|---|---|---|---|
| γ | 1.0 | 0.0–1.0 | SS = 0.0 (low) LS = 0.5 (medium) LL = 1.0 (high) |
1.0 |
| aprob | 1.0 | 0.9–1.0 | 1.0 | 1.0 |
| β | 1.0 | 0.0–1.0 | 1.0 | 1.0 |
| κ1trp | 1.0 | 1.0–2.0 | GG = 1.0 (low) TG = 1.5 (medium) TT = 2.0 (high) |
TT & TC = 1.0 (low) CC = 2.0 (high) |
| κ1maoa | 1.0 | 1.0–2.0 | 1.0 | Short = 1.0 (low) Long = 2.0 (high) |
Note: This table presents parameter values for the baseline and validity tests reported in the present study. For the Face validity tests, we ran parameter plots of 10 runs across the range of values indicated. For Criterion validity test 1, we examined firing rates over nine conditions resulting from “low”, “medium” and “high” levels of reuptake (γ) and synthesis (κ1trp). For Criterion validity test 2, we examined CSF 5-HIAA levels (p) over four conditions resulting from “low”, and “high” levels of catabolism (κ1maoa) and synthesis (κ1trp). Genotypes representing different levels of synthesis do not match because for Criterion validity 1 we were comparing to the −703 G/T (rs4570625) polymorphism reported by Canli et al. (2008), whereas for Criterion validity 2 we were utilizing observed genotypes for the intron-8 (rs1386483) polymorphism
Contributor Information
Scott F. Stoltenberg, Department of Psychology, University of Nebraska-Lincoln, 238 Burnett Hall, Lincoln, NE 68588-0308, USA
Parthasarathi Nag, Department of Mathematics, Black Hills State University, Spearfish, SD 57799-9127, USA.
REFERENCES
- Barr CS, Newman TK, Becker ML, Champoux M, Lesch KP, Suomi SJ, Goldman D, Higley JD. Serotonin transporter gene variation is associated with alcohol sensitivity in rhesus macaques exposed to early-life stress. Alcohol Clin Exp Res. 2003a;27(5):812–817. doi: 10.1097/01.ALC.0000067976.62827.ED. [DOI] [PubMed] [Google Scholar]
- Barr CS, Newman TK, Becker ML, Parker CC, Champoux M, Lesch KP, Goldman D, Suomi SJ, Higley JD. The utility of the non-human primate; model for studying gene by environment interactions in behavioral research. Genes Brain Behav. 2003b;2(6):336–340. doi: 10.1046/j.1601-1848.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- Baumgarten HG, Gothert M. Serotonergic neurons and 5-HT receptors in the CNS. Springer; Berlin: 1999. p. 767. [Google Scholar]
- Baumgarten HG, Grozdanovic Z. Role of serotonin in obsessive-compulsive disorder. Br J Psychiatry. 1998;35(Suppl):13–20. [PubMed] [Google Scholar]
- Baumgarten HG, Grozdanovic Z. Anatomy of central serotonergic projection systems. In: Baumgarten HG, Gothert M, editors. Serotonergic neurons and 5-HT receptors in the CNS. Springer; Berlin: 1999. [Google Scholar]
- Bondy B, Buettner A, Zill P. Genetics of suicide. Mol Psychiatry. 2006;11(4):336–351. doi: 10.1038/sj.mp.4001803. [DOI] [PubMed] [Google Scholar]
- Brodie ED., III . Why evolutionary genetics does not always add up. In: Wolf JB, Brodie ED III, Wade MJ, editors. Epistasis and the evolutionary process. Oxford University Press, Inc.; New York: 2000. [Google Scholar]
- Canli T, Lesch KP. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci. 2007;10(9):1103–1109. doi: 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- Canli T, Congdon E, Gutknecht L, Constable RT, Lesch KP. Amygdala responsiveness is modulated by tryptophan hydroxylase-2 gene variation. J Neural Transm. 2005;112(11):1479–1485. doi: 10.1007/s00702-005-0391-4. [DOI] [PubMed] [Google Scholar]
- Canli T, Congdon E, Constable R Todd, Lesch KP. Additive effects of serotonin transporter and tryptophan hydroxylase-2 gene variation on neural correlates of affective processing. Biol Psychol. 2008;79(1):118–125. doi: 10.1016/j.biopsycho.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Carver CS, Miller CJ. Relations of serotonin function to personality: current views and a key methodological issue. Psychiatry Res. 2006;144(1):1–15. doi: 10.1016/j.psychres.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Carver CS, Johnson SL, Joormann J. Serotonergic function, two-mode models of self-regulation, and vulnerability to depression: what depression has in common with impulsive aggression. Psychol Bull. 2008;134(6):912–943. doi: 10.1037/a0013740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297(5582):851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Cinquin O, Demongeot J. Positive and negative feedback: striking a balance between necessary antagonists. J Theor Biol. 2002;216(2):229–241. doi: 10.1006/jtbi.2002.2544. [DOI] [PubMed] [Google Scholar]
- de Groote L, Olivier B, Westenberg HG. Extracellular serotonin in the prefrontal cortex is limited through terminal 5-HT(1B) autoreceptors: a microdialysis study in knockout mice. Psychopharmacology (Berl) 2002;162(4):419–424. doi: 10.1007/s00213-002-1117-z. [DOI] [PubMed] [Google Scholar]
- Deckert J, Catalano M, Syagailo YV, Bosi M, Okladnova O, Di Bella D, Nothen MM, Maffei P, Franke P, Fritze J, Maier W, Propping P, Beckmann H, Bellodi L, Lesch KP. Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Hum Mol Genet. 1999;8(4):621–624. doi: 10.1093/hmg/8.4.621. [DOI] [PubMed] [Google Scholar]
- Dunn NA, Lockery SR, Pierce-Shimomura JT, Conery JS. A neural network model of chemotaxis predicts functions of synaptic connections in the nematode caenorhabditis elegans. J Comput Neurosci. 2004;17:137–147. doi: 10.1023/B:JCNS.0000037679.42570.d5. [DOI] [PubMed] [Google Scholar]
- Edelstein-Keshet L. Mathematical models in biology. McGraw-Hill; New York: 1988. [Google Scholar]
- El Mansari M, Blier P. Mechanisms of action of current and potential pharmacotherapies of obsessive-compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(3):362–373. doi: 10.1016/j.pnpbp.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Evrard A, Laporte AM, Chastanet M, Hen R, Hamon M, Adrien J. 5-HT1A and 5-HT1B receptors control the firing of serotoninergic neurons in the dorsal raphe nucleus of the mouse: studies in 5-HT1B knock-out mice. Eur J Neurosci. 1999;11(11):3823–3831. doi: 10.1046/j.1460-9568.1999.00800.x. [DOI] [PubMed] [Google Scholar]
- Evrard A, Malagie I, Laporte AM, Boni C, Hanoun N, Trillat AC, Seif I, De Maeyer E, Gardier A, Hamon M, Adrien J. Altered regulation of the 5-HT system in the brain of MAO-A knock-out mice. Eur J Neurosci. 2002;15(5):841–851. doi: 10.1046/j.1460-9568.2002.01917.x. [DOI] [PubMed] [Google Scholar]
- Feinn R, Nellissery M, Kranzler HR. Meta-analysis of the association of a functional serotonin transporter promoterpolymorphism with alcohol dependence. Am J Med Genet B Neuropsychiatr Genet. 2005;133(1):79–84. doi: 10.1002/ajmg.b.30132. [DOI] [PubMed] [Google Scholar]
- Glanzman DL. New tricks for an old slug: the critical role of postsynaptic mechanisms in learning and memory in Aplysia. Prog Brain Res. 2008;169:277–292. doi: 10.1016/S0079-6123(07)00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbi G, Murphy DL, Lesch K, Blier P. Modifications of the serotonergic system in mice lacking serotonin transporters: an in vivo electrophysiological study. J Pharmacol Exp Ther. 2001;296(3):987–995. [PubMed] [Google Scholar]
- Gotlib IH, Joormann J, Minor KL, Hallmayer J. HPA axis reactivity: a mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biol Psychiatry. 2008;63(9):847–851. doi: 10.1016/j.biopsych.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Hanson DR. Human development: biological and genetic processes. Annu Rev Psychol. 2005;56:263–286. doi: 10.1146/annurev.psych.56.091103.070208. [DOI] [PubMed] [Google Scholar]
- Gottman JM, Murray JD, Swanson CC, Tyson R, Swanson KR. The mathematics of marriage: dynamic nonlinear models. The MIT Press; Cambridge: 2002. [Google Scholar]
- Green AE, Munafo MR, Deyoung CG, Fossella JA, Fan J, Gray JR. Using genetic data in cognitive neuroscience: from growing pains to genuine insights. Nat Rev Neurosci. 2008;9(9):710–720. doi: 10.1038/nrn2461. [DOI] [PubMed] [Google Scholar]
- Grigorenko EL. Epistasis and the genetics of complex traits. In: Plomin R, deFries JC, Craig IW, McGuffin P, editors. Behavioral genetics in the postgenomic era. American Psychological Association; Washington: 2003. pp. 247–266. [Google Scholar]
- Gutknecht L, Waider J, Kraft S, Kriegebaum C, Holtmann B, Reif A, Schmitt A, Lesch KP. Deficiency of brain 5-HT synthesis but serotonergic neuron formation in Tph2 knockout mice. J Neural Transm. 2008;115(8):1127–1132. doi: 10.1007/s00702-008-0096-6. [DOI] [PubMed] [Google Scholar]
- Haghighi F, Bach-Mizrachi H, Huang YY, Arango V, Shi S, Dwork AJ, Rosoklija G, Sheng HT, Morozova I, Ju J, Russo JJ, Mann JJ. Genetic architecture of the human tryptophan hydroxylase 2 gene: existence of neural isoforms and relevance for major depression. Mol Psychiatry. 2008;13(8):813–820. doi: 10.1038/sj.mp.4002127. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. A mathematical theory of natural and artificial selection. Trans Camb Phil Soc. 1924;23:19–41. [Google Scholar]
- Hamer D, Sirota L. Beware the chopsticks gene. Mol Psychiatry. 2000;5(1):11–13. doi: 10.1038/sj.mp.4000662. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Weinberger DR. Imaging genomics. Br Med Bull. 2003;65:259–270. doi: 10.1093/bmb/65.1.259. [DOI] [PubMed] [Google Scholar]
- Hartig PR. Molecular biology and transductional characteristics of 5-HT receptors. In: Baumgarten HG, Gothert M, editors. Serotonergic neurons and 5-HT receptors in the CNS. Springer; Berlin: 1999. pp. 175–212. [Google Scholar]
- He M, Sibille E, Benjamin D, Toth M, Shippenberg T. Differential effects of 5-HT1A receptor deletion upon basal and fluoxetine-evoked 5-HT concentrations as revealed by in vivo microdialysis. Brain Res. 2001;902(1):11–17. doi: 10.1016/s0006-8993(01)02271-5. [DOI] [PubMed] [Google Scholar]
- Herrmann MJ, Huter T, Muller F, Muhlberger A, Pauli P, Reif A, Renner T, Canli T, Fallgatter AJ, Lesch KP. Additive effects of serotonin transporter and tryptophan hydroxylase-2 gene variation on emotional processing. Cereb Cortex. 2007;17(5):1160–1163. doi: 10.1093/cercor/bhl026. [DOI] [PubMed] [Google Scholar]
- Hill EM, Stoltenberg SF, Bullard KH, Li S, Zucker RA, Burmeister M. Antisocial alcoholism and serotonin-related polymorphisms: association tests. Psychiatr Genet. 2002;12(3):143–153. doi: 10.1097/00041444-200209000-00005. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Breakefield XO. Human monoamine oxidase A gene determines levels of enzyme activity. Am J Hum Genet. 1991;49(2):383–392. [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Cate SP, Battistuzzi C, Oquendo MA, Brent D, Mann JJ. An association between a functional polymorphism in the monoamine oxidase a gene promoter, impulsive traits and early abuse experiences. Neuropsychopharmacology. 2004;29(8):1498–1505. doi: 10.1038/sj.npp.1300455. [DOI] [PubMed] [Google Scholar]
- Hutchison KE, Stallings M, McGeary J, Bryan A. Population stratification in the candidate gene study: fatal threat or red herring? Psychol Bull. 2004;130(1):66–79. doi: 10.1037/0033-2909.130.1.66. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP. Why most published research findings are false. PLoS Med. 2005;2(8):e124. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbi M, Korf J, Kema IP, Hartman C, van der Pompe G, Minderaa RB, Ormel J, den Boer JA. Convergent genetic modulation of the endocrine stress response involves polymorphic variations of 5-HTT, COMT and MAOA. Mol Psychiatry. 2007;12(5):483–490. doi: 10.1038/sj.mp.4001975. [DOI] [PubMed] [Google Scholar]
- Johnson BA. Role of the serotonergic system in the neurobiology of alcoholism: implications for treatment. CNS Drugs. 2004;18(15):1105–1118. doi: 10.2165/00023210-200418150-00005. [DOI] [PubMed] [Google Scholar]
- Jonsson EG, Norton N, Gustavsson JP, Oreland L, Owen MJ, Sedvall GC. A promoter polymorphism in the monoamine oxidase A gene and its relationships to monoamine metabolite concentrations in CSF of healthy volunteers. J Psychiatr Res. 2000;34(3):239–244. doi: 10.1016/s0022-3956(00)00013-3. [DOI] [PubMed] [Google Scholar]
- Khalil HK. Nonlinear systems. Prentice Hall; Upper Saddle River: 1996. [Google Scholar]
- Konno K, Matsumoto M, Togashi H, Yamaguchi T, Izumi T, Watanabe M, Iwanaga T, Yoshioka M. Early postnatal stress affects the serotonergic function in the median raphe nuclei of adult rats. Brain Res. 2007;1172:60–66. doi: 10.1016/j.brainres.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kravitz EA. Serotonin and aggression: insights gained from a lobster model system and speculations on the role of amine neurons in a complex behavior. J Comp Physiol [A] 2000;186(3):221–238. doi: 10.1007/s003590050423. [DOI] [PubMed] [Google Scholar]
- Leonardo ED, Hen R. Genetics of affective and anxiety disorders. Annu Rev Psychol. 2006;57:117–137. doi: 10.1146/annurev.psych.57.102904.190118. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Wolozin BL, Murphy DL, Reiderer P. Primary structure of the human platelet serotonin uptake site: identity with the brain serotonin transporter. J Neurochem. 1993;60(6):2319–2322. doi: 10.1111/j.1471-4159.1993.tb03522.x. [DOI] [PubMed] [Google Scholar]
- Levinson DF. The genetics of depression: a review. Biol Psychiatry. 2006;60(2):84–92. doi: 10.1016/j.biopsych.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Lucki I. The spectrum of behaviors influenced by serotonin. Biol Psychiatry. 1998;44(3):151–162. doi: 10.1016/s0006-3223(98)00139-5. [DOI] [PubMed] [Google Scholar]
- Mann JJ, Currier D, Murphy L, Huang YY, Galfalvy H, Brent D, Greenhill L, Oquendo M. No association between a TPH2 promoter polymorphism and mood disorders or monoamine turnover. J Affect Disord. 2008;106(1–2):117–121. doi: 10.1016/j.jad.2007.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannoury la Cour C, Boni C, Hanoun N, Lesch KP, Hamon M, Lanfumey L. Functional consequences of 5-HT transporter gene disruption on 5-HT(1a) receptor-mediated regulation of dorsal raphe and hippocampal cell activity. J Neurosci. 2001;21(6):2178–2185. doi: 10.1523/JNEUROSCI.21-06-02178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClearn GE. Nature and nurture: interaction and coaction. Am J Med Genet B Neuropsychiatr Genet. 2004;124B(1):124–130. doi: 10.1002/ajmg.b.20044. [DOI] [PubMed] [Google Scholar]
- Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci. 2008;9(2):85–96. doi: 10.1038/nrn2284. [DOI] [PubMed] [Google Scholar]
- Parsons LH, Kerr TM, Tecott LH. 5-HT(1A) receptor mutant mice exhibit enhanced tonic, stress-induced and fluoxetine-induced serotonergic neurotransmission. J Neurochem. 2001;77(2):607–617. doi: 10.1046/j.1471-4159.2001.00254.x. [DOI] [PubMed] [Google Scholar]
- Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt impulsiveness scale. J Clin Psychol. 1995;51(6):768–774. doi: 10.1002/1097-4679(199511)51:6<768::aid-jclp2270510607>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Perko L. Differential equations and dynamical systems. Springer; New York: 1991. [Google Scholar]
- Pezawas L, Meyer-Lindenberg A, Goldman AL, Verchinski BA, Chen G, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR. Evidence of biologic epistasis between BDNF and SLC6A4 and implications for depression. Mol Psychiatry. 2008;13(7):709–716. doi: 10.1038/mp.2008.32. [DOI] [PubMed] [Google Scholar]
- Philibert R, Madan A, Andersen A, Cadoret R, Packer H, Sandhu H. Serotonin transporter mRNA levels are associated with the methylation of an upstream CpG island. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(1):101–105. doi: 10.1002/ajmg.b.30414. [DOI] [PubMed] [Google Scholar]
- Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A. MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet. 2008a;147B(5):565–570. doi: 10.1002/ajmg.b.30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Sandhu H, Hollenbeck N, Gunter T, Adams W, Madan A. The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am J Med Genet B Neuropsychiatr Genet. 2008b;147B(5):543–549. doi: 10.1002/ajmg.b.30657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, deFries JC, Craig IW, McGuffin P. Behavioral genomics. In: Plomin R, deFries JC, Craig IW, McGuffin P, editors. Behavioral genetics in the postgenomic era. American Psychological Association; Washington: 2003. pp. 531–540. [Google Scholar]
- Rao H, Gillihan SJ, Wang J, Korczykowski M, Sankoorikal GM, Kaercher KA, Brodkin ES, Detre JA, Farah MJ. Genetic variation in serotonin transporter alters resting brain function in healthy individuals. Biol Psychiatry. 2007;62(6):600–606. doi: 10.1016/j.biopsych.2006.11.028. [DOI] [PubMed] [Google Scholar]
- Reuter M, Ott U, Vaitl D, Hennig J. Impaired executive control is associated with a variation in the promoter region of the tryptophan hydroxylase 2 gene. J Cogn Neurosci. 2007;19(3):401–408. doi: 10.1162/jocn.2007.19.3.401. [DOI] [PubMed] [Google Scholar]
- Richer M, Hen R, Blier P. Modification of serotonin neuron properties in mice lacking 5-HT1A receptors. Eur J Pharmacol. 2002;435(2–3):195–203. doi: 10.1016/s0014-2999(01)01607-7. [DOI] [PubMed] [Google Scholar]
- Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. Jama. 2009;301(23):2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp T, Umbers V, Gartside SE. Effect of a selective 5-HT reuptake inhibitor in combination with 5-HT1A and 5-HT1B receptor antagonists on extracellular 5-HT in rat frontal cortex in vivo. Br J Pharmacol. 1997;121(5):941–946. doi: 10.1038/sj.bjp.0701235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan K, Lowe N, Kirley A, Mullins C, Fitzgerald M, Gill M, Hawi Z. Tryptophan hydroxylase 2 (TPH2) gene variants associated with ADHD. Mol Psychiatry. 2005;10(10):944–949. doi: 10.1038/sj.mp.4001698. [DOI] [PubMed] [Google Scholar]
- Soderstrom H, Blennow K, Manhem A, Forsman A. CSF studies in violent offenders. I. 5-HIAA as a negative and HVA as a positive predictor of psychopathy. J Neural Transm. 2001;108(7):869–878. doi: 10.1007/s007020170036. [DOI] [PubMed] [Google Scholar]
- Sontag ED. Molecular systems biology and control. Eur J Control. 2005;11(4–5):396–435. [Google Scholar]
- Stenfors C, Ross SB. Changes in extracellular 5-HIAA concentrations as measured by in vivo microdialysis technique in relation to changes in 5-HT release. Psychopharmacology (Berl) 2004;172(2):119–128. doi: 10.1007/s00213-003-1736-z. [DOI] [PubMed] [Google Scholar]
- Stoltenberg SF. Serotonergic agents and alcoholism treatment: a simulation. Alcohol Clin Exp Res. 2003;27(12):1853–1859. doi: 10.1097/01.ALC.0000098876.94384.0A. [DOI] [PubMed] [Google Scholar]
- Stoltenberg SF. Epistasis among presynaptic serotonergic system components. Behav Genet. 2005;35(2):199–209. doi: 10.1007/s10519-004-1019-4. [DOI] [PubMed] [Google Scholar]
- Stoltenberg SF. Dynamic and systems-based models for evaluating hypotheses related to predicting treatment response. In: Johnson BA, editor. Addictive disorders and substance abuse. Springer; New York: 2010. [Google Scholar]
- Stoltenberg SF, Nag P. Applying control system modelling to understand how genetic variation influences serotonin function and behavior. In: Lassau JA, editor. Neural synapse research trends. Nova Science Publishers, Inc.; New York: 2007. pp. 133–171. [Google Scholar]
- Stoltenberg SF, Glass JM, Chermack ST, Flynn HA, Li S, Weston ME, Burmeister M. Possible association between response inhibition and a variant in the brain-expressed tryptophan hydroxylase-2 gene. Psychiatr Genet. 2006;16(1):35–38. doi: 10.1097/01.ypg.0000176528.30362.34. [DOI] [PubMed] [Google Scholar]
- Stoltenberg SF, Batien BD, Birgenheir DG. Does gender moderate associations among impulsivity and health-risk behaviors? Addict Behav. 2008;33(2):252–265. doi: 10.1016/j.addbeh.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF. Spurious genetic associations. Biol Psychiatry. 2007;61(10):1121–1126. doi: 10.1016/j.biopsych.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Thompson BL, Stanwood GD. Pleiotropic effects of neurotransmission during development: modulators of modularity. J Autism Dev Disord. 2008;39(2):260–268. doi: 10.1007/s10803-008-0624-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vage J, Lingaas F. Single nucleotide polymorphisms (SNPs) in coding regions of canine dopamine- and serotonin-related genes. BMC Genet. 2008;9:10. doi: 10.1186/1471-2156-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt HM, Broersen LM, Olivier B, Westenberg HG. Effects of dietary tryptophan variations on extracellular serotonin in the dorsal hippocampus of rats. Psychopharmacology (Berl) 2004;172(2):137–144. doi: 10.1007/s00213-003-1632-6. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Cook EH., Jr Knockout mouse points to second form of tryptophan hydroxylase. Mol Interv. 2003;3(2):72–75. 50. doi: 10.1124/mi.3.2.72. [DOI] [PubMed] [Google Scholar]
- Virkkunen M, Goldman D, Linnoila M. Serotonin in alcoholic violent offenders. Ciba Found Symp 194168–194177; 1996. pp. 177–182. discussion. [DOI] [PubMed] [Google Scholar]
- Vomel M, Wegener C. Neuroarchitecture of aminergic systems in the larval ventral ganglion of Drosophila melanogaster. PLoS ONE. 2008;3(3):e1848. doi: 10.1371/journal.pone.0001848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LM. Dynamical cognitive science. The MIT Press; Cambridge: 2002. [Google Scholar]
- Weiner N. Cybernetics, second edition: or the control and communication in the animal and the machine. The MIT Press; Cambridge: 1965. [Google Scholar]
- Weiss LA, Abney M, Cook EH, Jr, Ober C. Sex-specific genetic architecture of whole blood serotonin levels. Am J Hum Genet. 2005;76(1):33–41. doi: 10.1086/426697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergaard GC, Suomi SJ, Chavanne TJ, Houser L, Hurley A, Cleveland A, Snoy PJ, Higley JD. Physiological correlates of aggression and impulsivity in free-ranging female primates. Neuropsychopharmacology. 2003;28(6):1045–1055. doi: 10.1038/sj.npp.1300171. [DOI] [PubMed] [Google Scholar]
- Williams RB, Marchuk DA, Gadde KM, Barefoot JC, Grichnik K, Helms MJ, Kuhn CM, Lewis JG, Schanberg SM, Stafford-Smith M, Suarez EC, Clary GL, Svenson IK, Siegler IC. Serotonin-related gene polymorphisms and central nervous system serotonin function. Neuropsychopharmacology. 2003;28(3):533–541. doi: 10.1038/sj.npp.1300054. [DOI] [PubMed] [Google Scholar]
- Zahnley T, Macey R, Oster G. Berkeley Madonna. Version 8.0.1. University of California; Berkeley: 2001. [Computer software] [Google Scholar]