

Abstract
Although the expression of cilia on chondrocytes was described over 40 years ago, the importance of this organelle in skeletal development and maintenance has only recently been recognized. Primary cilia are found on most mammalian cells and have been shown to play a role in chemosensation and mechanosensation. A growing number of human pleiotropic syndromes have been shown to be associated with ciliary or basal body dysfunction. Skeletal phenotypes, including alterations in limb patterning, endochondral bone formation, craniofacial development, and dentition, have been described in several of these syndromes. Additional insights into the potential roles and mechanisms of cilia action in the mammalian skeleton have been provided by research in model organisms including mouse and zebrafish. In this article we describe what is currently known about the localization of cilia in the skeleton as well as the roles and underlying molecular mechanisms of cilia in skeletal development.
A. Introduction
The primary cilium is an organelle that consists of a microtubule based axoneme covered by a specialized plasma membrane (Bisgrove and Yost, 2006; Davenport and Yoder, 2005; Pan et al., 2005; Satir and Christensen, 2007). A single, non-motile primary cilium is present on the surface of most eukaryotic cells. Primary cilia are generated through a process called Intraflagellar Transport (IFT) (Bisgrove and Yost, 2006; Davenport and Yoder, 2005; Pan et al., 2005; Satir and Christensen, 2007). Protein synthesis does not occur in cilia so the proteins that make up the structure must be transported into the cilium along microtubules. There are two IFT complexes which can be separated biochemically. Complex A mediates retrograde transport of cargoes from the tip to the base of the cilium and complex B mediates anterograde transport of specific cargoes from the base to the tip. Anterograde transport is driven by hetromeric kinesin 2 motors, which are composed of Kif3a and Kif3b motor subunits. Retrograde transport is mediated by dynein 1B. In addition, an assortment of accessory proteins are required for normal ciliary function.
It has been over 40 years since primary cilia were first identified on chondrocytes (Scherft and Daems, 1967). Ultrastructural studies showed that each chondrocyte has one cilium with the 9+0 microtubule pattern associated with non-motile primary cilia (Scherft and Daems, 1967; Wilsman and Fletcher, 1978). It was shown chondrocyte cilia vary in length from 1 to 4 microns and are about 0.2 microns in width (Poole et al., 2001; Scherft and Daems, 1967). The microtubules of cilia are enriched with detyrosinated and acetylated tubulin, allowing the use of antibodies to these modified forms of tubulin to immunolocalize cilia in a variety of cells including chondrocytes, perichondrial cells, and osteoblasts (Figure 1; Poole et al., 1997; Poole et al., 2001; Xiao et al., 2006, Song et al. 2007). Electron micrograhic studies showed that the chondrocyte cilium projects into the extracellular matrix (Poole et al., 1997). Electron, confocal, and tomographic microscopy indicate that chondrocyte cilia display various bending patterns potentially due to association of the cilium with the surrounding pericellular matrix (Jensen et al., 2004; Poole et al., 2001). Studies using flourescent microscopy suggest that cilia on chondrocytes have a specific orientation. Cilia on articular chondrocytes point away from the articular surface while cilia on columnar chondrocytes protrude from the center of the cell between the Golgi and nucleus (McGlashan et al., 2008; McGlashan et al., 2007; Song et al., 2007). The significance of this orientation is not clear. Future studies using mathematical models in combination with multiphoton microscopy will help clarify the orientation of cilia in different types of connective tissue (Ascenzi et al., 2007).
Figure 1. Localization of cilia on chondrocytes and perichondrial cells.
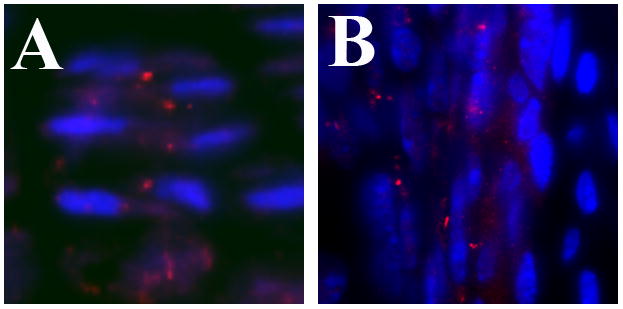
(A) Cilia, visualized by immunostaining with anti-acetylated α-tubulin antibodies (red), are aligned in the center of columns of proliferating chondrocytes in the growth plate of endochondral bones. Nuclei (blue) are located on alternate sides of the cells within a column. (B) In the perichondrium, cilia are present on both elongated and cuboidal cells adjacent to the cartilage anlagen (right side of image). Cilia were visualized by immunostaining with anti-Ift88 antibodies (red). Nuclei are blue.
Studies over the past decade have revealed an association between defects in the normal structure or function of the primary cilium and various congenital human conditions including polycystic kidney disease (PKD), situs-inversus, and retinal degeneration (Bisgrove and Yost, 2006; Davenport and Yoder, 2005; Pan et al., 2005). Pleiotrophic syndromes such as Jeune syndrome, Meckel syndrome, and Bardet-Biedl syndrome, which are associated with mutations in ciliary or basal body genes are characterized by various combinations of pathological changes including those in the skeleton (Table 1) (Bisgrove and Yost, 2006; Davenport and Yoder, 2005; Pan et al., 2005). The most common alterations in the skeleton associated with mutations in ciliary genes include polydactyly, shortened bones, and craniofacial dysmorphology. It has been shown that primary cilia are involved in transmitting both chemical and mechanical signals but the molecular mechanisms of these conditions are just now being determined.
Table 1.
Human cilopathies affecting the skeleton.
| Gene affected | OMIM# | Name | References |
|---|---|---|---|
| BBS | |||
| 209900 | Bardet-Biedl Syndrome (BBS) | Ansley et al. 2003; Tobin et al. 2008 | |
| CXORF5/Ofd1 | |||
| 311200 300209 |
Oral facial digital syndrome1 (OFD1) Simpson-Golabi-Behmel syndrome type 2 (SGBS2) |
Ferrante et al. 2006 | |
| EVC | |||
| 225500 | Ellis-van Creveld syndrome (EvC) | Ruiz-Perez et al. 2007 | |
| IFT80 | |||
| 611623 | Asphyxiating thoracic dystrophy 2 (ATD2); Juene’s asphyxiating thoracic dystorophy | Beales et al. 2007 | |
| MKS1 | |||
| 249000 | Meckels Syndrome type I (MKS1) | Kyttala et al. 2006 | |
| PKD1 | |||
| 17390 | Autosomal dominant polycyctic kidney disease (ADPKD) | Turco et al. 1993 | |
| RPGRIP1L/Ftm | |||
| 611561 611560 |
Meckels syndrome type 5 (MKS5) Joubert syndrome type 7 (JBTS7) |
Delous et al. 2007 |
To help in this regard, several mouse models have been generated and used to determine the role and mechanism of action for primary cilia in various aspects of skeletal development (Table 2). Furthermore, the zebrafish has emerged as another useful model to help uncover the molecular mechanisms of skeletal development. These animal models provide interesting insights into the how the cilium functions in the skeleton.
Table 2.
Animal models for IFT/cilia in the skeleton.
| Gene alteration | Strain name | References |
|---|---|---|
| Bbs | ||
| Null | Bbs4null/null; Bbs6null/null | Tobin et al. 2008 |
|
Zebrafish Knock down |
morpholino | Tayeh et al. 2008; Tobin et al. 2008 |
| Evc | ||
| Null allele | Evcnull/null | Ruiz-Perez et al. 2007 |
| Ftm/RPGRIP1L | ||
| Null allele | Ftm null/null | Vierkotten et al. 2007 |
| Ift80 | ||
|
Zebrafish Knock down |
morpholino | Beales et al. 2007 |
| Ift88/Tg737/Polaris | ||
| Hypomorphic allele | Ift88orpk | Zhang et al. 2003. |
| Haycraft et al. 2005 | ||
| McGlashan et al. 2007 | ||
| Ift88flexo | Liu et al. 2005 | |
| Null allele | Ift88tm1Rpw | Zhang et al. 2003. |
| Haycraft et al. 2005 | ||
| Liu et al. 2005 | ||
| Cre-LoxP conditional | Prx1Cre;Ift88LoxP/LoxP | Haycraft et al. 2007 |
| Col2aCre;Ift88LoxP/LoxP | Song et al. 2007 | |
| Kif3a | ||
| Cre-LoxP conditional | Prx1Cre;Kif3aLoxP/LoxP | Haycraft et al. 2007 |
| Kolpakova-Hart et al. 2007 | ||
| Col2aCre;Kif3aLoxP/LoxP | Song et al. 2007 | |
| Koyama et al. 2007 | ||
| Dermo1-Cre;Kif3aLoxP/LoxP | Kolpakova-Hart et al. 2007 | |
| Wnt1-Cre;Kif3aLoxP/LoxP | Kolpakova-Hart et al. 2007 | |
| Ofd1/CXORF5 | ||
| Cre-LoxP conditional | pCX-NLS-Cre;OfdLoxP/LoxP | Ferrante et al. 2006 |
| Pkd1 | ||
| Null allele | Pkd1null | Lu et al. 2001 |
| Strong hypomorphic/Null | Pkd1m1Bei | Xiao et al. 2006 |
| Truncation | Pkd1Δ17–21βgeo | Boulter et al. 2001 |
| Thm1 | ||
| Null allele | Thm1aln | Tram et al. 2008 |
B. Cilia are required for anterior-posterior limb patterning
The skeletal elements of the limb must be properly patterned along both the anterior-posterior and proximal-distal axes (for detailed review see Tickle, 2006). During outgrowth of the limb bud, the distal ectoderm forms a specialized epithelium called the apical ectodermal ridge (AER) that secretes factors including Fibroblast Growth Factors (Fgfs) essential for outgrowth of the limb. The anterior-posterior patterning of the limb is controlled mainly by a region of cells in the posterior mesenchyme known as the zone of polarizing activity (ZPA). Mice lacking the IFT protein Ift88 develop ectopic digits suggesting a role for cilia in the patterning of the mammalian digits. Indeed, cilia are expressed on both ectodermal and mesenchymal cells of the developing limb; however, only cilia in the mesoderm are required for proper limb patterning (Haycraft et al., 2005; Haycraft et al., 2007).
The major signaling pathway implicated in digit patterning is the hedgehog (Hh) pathway. In addition to an important role in digit patterning, Hh signaling is involved in the development of many tissues in the body (for detailed review see Huangfu and Anderson, 2006). There are three mammalian Hh ligands, Sonic hedgehog (Shh), Indian hedgehog (Ihh) and Desert hedgehog (Dhh). Hh signaling is both positively and negatively regulated. In the absence of Hh ligand, Smothened (Smo) activity is repressed by the receptor Patched1 (Ptc1) or Patched2 (Ptc2). Upon ligand binding, Smo repression is relieved and the Gli transcription factors activate transcription of downstream target genes. There are three mammalian Gli proteins, Gli1, Gli2, and Gli3. Gli3 is processed to generate a potent transcriptional inhibtor (Gli3R) in the absence of Hh while Gli2 is a potent activator of transcription in the presence of ligand. Unlike Gli2 and Gli3, Gli1 is expressed only after pathway activation.
While Hh pathway regulation has been well characterized in Drosophila, there are significant differences in the mammalian signaling pathway, such as the requirement for the primary cilium in mammals, which are not fully understood. Recent work has shown that Ptc1 and Smo translocate out of and into the cilium, respectively, upon ligand binding while the Gli proteins localize to the tip of the cilium with SuFu, a regulator of Gli processing and activity (Figure 2; Corbit et al., 2005; Haycraft et al., 2005; Huangfu and Anderson, 2005; Rohatgi et al., 2007). Loss of cilia in Ift88 or Kif3a null mice or in the limb mesoderm only using Prx1-Cre results in extensive polydactyly with up to 8 unpatterned digits on each limb (Haycraft et al., 2005; Liu et al., 2005; Haycraft et al., 2007). Both activation and repression of Hh signaling are affected in Ift88 and Kif3a null embryos and Gli3 processing is disrupted (Haycraft et al., 2005; Liu et al., 2005). Even though localization of Shh mRNA is normal, expression of Ptc1 and Gli1, direct targets of Hh activity, are reduced indicating that the activation of Hh signaling is repressed in the limb field (Haycraft et al., 2007). Nevertheless, a block to activation of Shh signaling cannot account for the severe polydactyly observed. Shh-null mice have only one digit not polydactyly (Chiang et al., 2001). It was previously shown that mutations in Gli3 result in polydactyly (te Welscher et al., 2002; Wang et al., 2000). In the absence of Hh signaling, Gli3 is normally processed to a repressor form, suppressing transcription of Hh target genes. The expression pattern of two genes, Gremlin and Hand2, that are normally repressed in the anterior limb bud by Gli3 are expanded in the limbs of IFT/cilia mutants indicating that the loss of the Gli3 repressor and overall misexpression of Shh target genes results in polydactyly (Figure 2; Haycraft et al., 2005; Liu et al., 2005; Haycraft et al., 2007).
Figure 2. Mammalian Hh signal transduction and limb patterning.

(A) Hh signal transduction. In the absence of Hh ligand, Ptc1 localizes to the ciliary membrane. Full length Gli3 (Gli3FL) is found at the distal tip of the cilium and subsequent proteolytic processing results in increased amounts of Gli3 repressor (Gli3R). Gli3R is a potent transcriptional repressor resulting in repression of target gene transcription including Ptc1 and Gli1. Upon ligand binding, Ptc1 translocates out of the cilium and Smo is localized in the cilium membrane. Smo translocation to the cilium inhibits Gli3 processing leading to an increased Gli3FL:Gli3R ratio allowing transcription of target genes. In Ift88 mutants, the cilium is not formed and Gli3 processing is inefficient. As a result, target genes such as Gremlin and Hand2, which are repressed by Gli3R, are expressed while transcription of Gli1 and Ptc1 is not activated likely due to disruption of Gli2 function by an unknown mechanism. (B) Role of Shh in development of the mammalian limb. In the developing limb, Shh is expressed by cells in the ZPA (red). Activation of Shh signaling leads to decreased Gli3R levels in the posterior limb bud while the level of Gli3R remains high in the anterior limb bud. In cilia mutants, such as those lacking Ift88 or Kif3a, disruption of the normal gradient of Gli3FL:Gli3R in conjunction with loss of Gli2 activity across the limb bud results in polydactyly.
Recent work has identified a mouse mutant, Thm1aln, containing a mutation in an IFT complex A protein. In contrast to mice with mutations in Ift88, a complex B protein, only Hh pathway repression is disrupted resulting in ectopic expression of Ptc1 and Gli1 in the anterior limb bud (Tran et al., 2008). The reason for the differences in these two models is not fully understood, but anterograde trafficking of IFT proteins remains intact in Thm1aln mutants while both anterograde and retrograde transport are disrupted in Ift88 and Kif3a mutants as evidenced by complete loss of the ciliary axoneme.
As described above, cilia play an integral role in Shh signaling and therefore disruption of cilia results in defects in digit patterning. It is not surprising that a number of pleiotropic syndromes that have been shown to be due to mutations in cilia or basal body-localized proteins, for example Bardet-Biedl syndrome (BBS; OMIM #209900), include alterations in the number of digits among their clinical features. BBS features include cystic kidney dysplasia, obesity, hypogonadism and digit patterning alterations including post-axial polydactyly, syndactyly or brachydactyly. Mutations in at least one of 14 distinct genes (BBS1-BBS14) lead to BBS although the same genes have been linked to other pleiotropic syndromes including, McKusick-Kaufman syndrome (MKKS; OMIM #236700) and Meckel syndrome 1 (MKS1; OMIM #249000). Additionally, mutations in more than one BBS gene may lead to alterations in clinical presentation and patients have been identified with mutations in additional BBS genes leading to the hypothesis that BBS can develop through triallelic inheritance (Katsanis et al., 2001; Katsanis et al., 2002).
While digit patterning defects are seen in a majority of BBS patients, the two BBS mouse models (Bbs4 null and Bbs6 null) do not develop polydactyly or syndactyly (Fath et al., 2005). Pectoral fin development in the zebrafish shares similarities with mammalian limb development including expression of Shh in the posterior mesenchyme. In BBS morpholino-injected zebrafish embryos, the region of Shh expression was increased relative to control-injected embryos although no alterations in pectoral fin size were observed (Tayeh et al., 2008). These results suggest that the polydactyly seen in BBS patients may be due to alterations in Shh signaling. The proposed triallelic inheritance of BBS mentioned above may also play a role in the differences between mouse models and features observed in human patients since the mouse models examined contain mutations in only one BBS gene (Kulaga et al., 2004).
Syndactyly, brachydactyly or polysyndactyly are also seen in Oral-facial-digital syndrome I (OFD1; OMIM #311200). The gene responsible for OFD1 is chromosome open reading frame 5 (CXORF5). The protein contains a LIS1 homology domain, which is thought to regulate microtubule dynamics and is localized to the centrosome and basal body of primary cilia (Ferrante et al., 2001; Romio et al., 2004). Studies in mice have shown that cells lacking Ofd1 fail to form a primary cilium. In heterozygous female embryos one allele is randomly inactivated by X inactivation resulting in a chimeric embryo with normal and Ofd1 null cells, whereas males have a single allele (Ferrante et al., 2006). Female mice lacking one copy of the gene mutated in Ofd1 also show severe defects in patterning of the digits including development of seven to nine digits per limb. The neural tube of male embryos shows a reduction in the level of Ptc1 and Gli1 expression indicating a possible requirement for Ofd1 in activating Shh signaling. No change in Ptc1 or Gli1 expression was seen in the limb bud of heterozygous female embryos however regions of expanded Hox gene expression were detected in the anterior limb bud (Ferrante et al., 2006). It is unclear how loss of Ofd1 results in ectopic or expanded Hox gene expression and polydactyly, but it is hypothesized to be due to disruption of Gli3 function.
Postaxial polydactyly is also common in Ellis-van Creveld syndrome (EvC; OMIM#225500). EvC is a chondroectodermal dysplasia characterized by numerous skeletal and craniofacial abnormalities. Recently, one of the two proteins mutated in EvC was identified (EVC) and localized to the base of the cilium (Ruiz-Perez et al., 2007; Ruiz-Perez et al., 2003). EVC is not required for the formation or the maintenance of cilia structure but appears to be required for normal Hedghog signaling. In mouse, Evc expression was restricted to the skeleton and the developing face (Ruiz-Perez et al., 2007). Recently, mice lacking Evc were generated but no alterations in digit patterning were seen. The lack of digit defects in Evc null mice was attributed to either low levels of Evc expression during early stages of limb patterning or the lack of Gli3 processing defects in Evc mutants despite reduced Gli1 and Ptc1 expression in the developing bones (Ruiz-Perez et al., 2007).
Similarly, polydactyly is also prevalent in Meckel-Gruber syndrome (MKS1; OMIM #249000). Meckel-Gruber syndrome is an autosomal lethal disorder due to mutations in one of several distinct loci including MKS1, which was shown to be expressed in the developing digits in mice (Kyttala et al., 2006). Meckels type 5 (OMIM#611561) is a form of Meckel-Gruber syndrome resulting from mutations in Retinitis Pigmentosa GTPase Regulator-interacting-protein 1-like (RPGRIP1L) (Delous et al., 2007). Fantom (Ftm) encodes the mouse homolog of RPGRIP1L and mice lacking Ftm develop ectopic digits (Vierkotten et al., 2007). Similar to what is seen in Ift88 mutants, Gli3 processing is disrupted in Ftm mutant embryos and excess full length Gli3 is present. Unlike Ift88 mutants, Ptc1 expression is present although at reduced levels in the developing limbs.
Reduced numbers of cilia are seen by SEM on the node and by immunostaining in the developing limb of Ftm mutant embryos suggesting that Ftm is required for proper cilia formation or maintenance, although the defects are not as severe as in Ift88 or Kif3a mutants.
C. A role for IFT/primary cilia in endochondral bone formation and development of the post-natal growth plate
Most of the bones in the body develop through a process called endochondral bone formation (Cancedda et al., 1995; Colnot, 2005; van der Eerden et al., 2003). Embryonic mesenchymal cells condense at the sites where the skeletal elements will form. Condensed mesenchymal cells then undergo differentiation to become chondrocytes forming the cartilaginous anlagen of the future bone. Proliferation allows for the initial expansion of the skeletal element. Chondrocytes then differentiate toward their terminally differentiated form, the hypertrophic chondrocyte, also allowing for an increase in size of the skeletal element. Hypertrophic chondrocytes do not proliferate but they synthesize a unique extra-cellular matrix that allows mineralization of the matrix. The hypertrophic cells also secrete factors that attract vasculature to the bone anlagen and promote the differentiation of osteoblasts from cells in the adjacent perichondrium leading to the development of the bone collar. Finally, hypertrophic chondrocytes undergo apoptosis and the mineralized matrix is replaced with osteoblasts forming trabecular bone.
As described above, there are three members of the vertebrate Hh family, of these Ihh is the most important regulator of endochondral bone formation. Ihh is expressed in cells that are committed to become hypertrophic and it regulates proliferation of chondrocytes, the rate of hypertrophic differentiation, as well as the formation of the bone collar (St-Jacques et al., 1999). Regulation of proliferation is thought to be a direct effect of Ihh on chondrocytes while hypertrophic differentiation is mediated indirectly (Long et al., 2001). Ihh acts in a negative feedback loop with Parathyroid Hormone related Protein (PTHrP) and other factors to sense and regulate the rate of chondrocyte differentiation and thus limit hypertrophic differentiation (Lanske et al., 1996; Vortkamp et al., 1996).
Since Ihh is a major regulator of endochondral bone formation and it has been shown that cilia are required to mediate both the activator and repressor functions of Hh signaling (Haycraft et al., 2005; Liu et al., 2005) it makes sense that mice and humans with mutations in ciliary genes often present with defects in the development of the skeleton. Recently, it was demonstrated that at least two human syndromes that include significant defects in endochondral bone formation are associated with mutations in genes that code for ciliary proteins. First, Juene’s asphyxiating thoracic dystrophy (ATD2; OMIM #611177) was associated with a missense mutation in one of the WD40 domains of IFT80. IFT80 is localized to the basal body and the ciliary axoneme and is part of IFT complex B (Beales et al., 2007). The missense mutation resulted in missing or shortened cilia. Although a connection to Hedghog signaling could not be examined in human tissues, knock down of IFT80 in zebrafish using morpholino technology resulted in skeletal defects as well as renal cysts (Beales et al., 2007). Some of the skeletal defects resembled those seen in Shh depleted fish. Furthermore, Ptc1 expression was down-regulated in the IFT80 morphants suggesting alterations in Hedgehog signaling (Beales et al., 2007).
As mentioned above, EvC is characterized by numerous skeletal and craniofacial abnormalities. One of the proteins mutated in EvC is localized to the base of the cilium and is required for normal Hedgehog signaling (Ruiz-Perez et al., 2007). In the growth plate, Evc was expressed in the perichondrium as well as in resting and proliferating chondrocytes (Ruiz-Perez et al., 2007). Expression was not detected in late prehypertophic or hypertrophic cells. Disruption of Evc in mice resulted in a variety of skeletal and craniofacial abnormalities as well as alterations in the teeth and nails. The phenotypes were similar to that seen in EvC patients (Ruiz-Perez et al., 2007). Alterations in chondrocyte proliferation or early differentiation were not observed; however, accelerated hypertrophic differentiation was observed in the Evc-null growth plates. Defects in the growth plate were further associated with diminished Ihh signaling as measured by a reduction in the expression of Ptc1 and Gli1; however, processing of Gli3 to the repressor form was normal. The results suggest that Evc is specifically involved in Hh-dependent activation of gene expression. This is in contrast to studies in which cilia themselves are absent and both activator and repressor functions of Hh are affected (Haycraft et al., 2005; Haycraft et al., 2007; Liu et al., 2005; Song et al., 2007). Phenotypic differences in Evc-null mice and mice with conditional mutations in Kif3a or Ift88 (see below) can in part be accounted for by intact Gli3 function in the Evc mutant mice.
Mutations in Pkd1 are associated with autosomal dominant Polycystic Kidney Disease (ADPKD; OMIN#173900). Skeletal malformations have been detected in unusual patients with this form of PKD (Turco et al., 1993). Mice with mutations in Pkd1 demonstrate defects in embryonic endochondral bone formation as well as osteopenia in adults (Boulter et al., 2001; Lu et al., 2001; Xiao et al., 2006). Pkd1 mRNA is expressed in early condensing mesenchyme and later in prechondrogenic tissue (Guillaume et al., 1999). As skeletal development progresses, it becomes enriched in the perichondrium and invertervertebral disc. Pkd1 mutant mice demonstrate shortened long bones as well as defects in the formation of the vertebrae (spina bifida occulta) (Lu et al., 2001). A delay in hypertrophic differentiation, vascularization, and skeletal mineralization were also noted (Boulter et al., 2001; Lu et al., 2001; Xiao et al., 2006). The molecular mechanism accounting for the defects seen in endochondral bone formation in Pkd1 mutant mice is not known.
Mice with mutations in other ciliary genes also demonstrate alterations in endochondral bone formation resulting in shortening of the bones in the limbs (Haycraft et al., 2007; McGlashan et al., 2007; Song et al., 2007; Zhang et al., 2003). Mice with conditional deletion of Ift88 or Kif3a in early limb mesenchyme (Prx1-Cre;Ift88loxP/loxP or Prx1-Cre;Kif3aloxP/loxP) have defects in embryonic endonchondral bone formation including accelerated hypertrophic differentiation as well as a delay in vascularization of the primary ossification center resulting in the persistence of hypertrophic cells in the growth plate (Haycraft et al., 2007). Defects were observed as early as 15.5 days of gestation. Mice in which Kif3a was deleted using Dermo1-Cre had a similar, although milder, phenotype to the Prx1-Cre mutants (Kolpakova-Hart et al., 2007). The phenotypes resembled those seen in mice with germline mutations in Ihh (St-Jacques et al., 1999). Ptc1 and Gli1 mRNA were dramatically reduced in the long bones of Prx1-Cre;Ift88loxP/loxP and Prx1-Cre;Kif3aloxP/loxPmice relative to controls, supporting of a role for IFT/cilia in mediating Ihh signaling during embryonic endochondral bone formation (Figure 3A). Expression of Ptc1 and Gli1 was reduced in both chondrocytes and perichondrial cells suggesting Ihh signaling was disrupted in both compartments. Somewhat suprisingly, PTHrP expression was maintained; perhaps due to the loss of Gli3 repressor function (Hilton et al., 2005; Koziel et al., 2005). In contrast to the Prx1-Cre and Dermo1-Cre mutant mice, mice in which Ift88 or Kif3a were deleted using Col2a-Cre (Col2a-Cre;Ift88loxP/loxP or Col2a-Cre;Kif3aloxP/loxP) did not demonstrate alterations in embryonic endochondral bone formation (Song et al., 2007). Immunoflourescent staining confirmed that cilia were missing in chondrocytes by E15.5 days. Furthermore, Ptc1 expression was dramatically reduced in embryonic chondrocytes but expression was maintained in the perichondrium (Figure 3A). The differences in the phenotype between Prx1-Cre and Col2a-Cre mutants were likely due to the expression pattern of Col2a-Cre, which targets chondrocytes but does not efficiently target cells in the perichondrium (Ovchinnikov et al., 2000). Prx1-Cre and Dermo1-Cre target the perichondrium and cartilage (Logan et al., 2002; Yu et al., 2003). The combined results suggest an important role for perichondrial IFT/cilia in regulating embryonic endochondral bone formation possibly by mediating signaling by Ihh. There is prior evidence indicating the importance of perichondrial Ihh signaling in the regulation of hypertrophic differentiation that supports this model (Alvarez et al., 2002; Colnot, 2005; Long et al., 2001; Vortkamp et al., 1996).
Figure 3. A role for primary cilia in endochondral bone formation and the post natal growth plate.
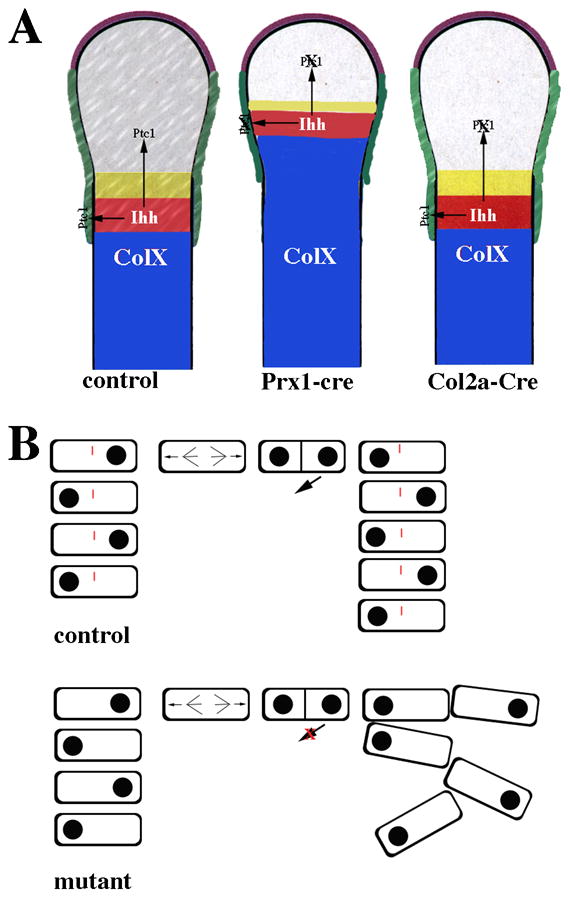
(A) Primary cilia regulate Ihh signaling during embryonic endochondral bone formation. Ihh is expressed in chondrocytes that are committed to hypertrophic differentiation (red area). Ihh normally acts on both prehypertrophic chondrocytes (gray and yellow areas) and the perichondrium (green). Ptc1, a downstream target of Ihh signaling, was used to determine how depletion of cilia affects Ihh signaling during endochondral bone formation. In normal control mice, cilia are present on chondrocytes as well as cells in the perichondrium and Ptc1 expression is detected in both cell types. In mice in which ciliogenesis is disrupted in Prx1-Cre expressing cells, cilia are absent from both chondrocytes and cells in the perichondrium. Disruption of ciliogenesis in the cartilage and in the perichondrium resulted in accelerated hypertrophic differentiation (blue area) similar to that seen in the Ihh-null mice. Ptc1 expression was dramatically reduced in both chondrocytes and cells of the perichondrium confirming that Ihh signaling was disrupted. In contrast, when ciliogenesis was disrupted using Col2a-Cre, which only targets the chondrocytes, hypertrophic differentiation was normal. Ptc1 expression was reduced in chondrocytes but maintained in the perichondrium. (B) Primary cilia mediate chondrocyte rotation in the post-natal growth plate. In normal control mice, flat cells in the growth plate have polarity and are aligned in columns parallel to the long-axis of the bone. Cell division occurs perpendicular to the long axis. One cell then migrates under the other to form the columns. This process is called chondrocyte rotation. Cilia are present on chondrocytes in the columns and are required for normal chondrocyte rotation. In cilia depleted growth plates, chondrocytes still divide perpendicular to the long axis of the bone and polarity is maintained; however, the orientation of the cells one to another is altered and the cells are not maintained in columns.
Although Col2a-Cre;Ift88loxP/loxP and Col2a-Cre;Kif3aloxP/loxP mice did not demonstrate defects in embryonic endochondral bone formation, the mice demonstrated post-natal dwarfism due to a progressive loss of the growth plate (Song et al., 2007). After birth, the growth plate allows for continued longitudinal growth of the bone through continued proliferation, differentiation, and replacement by bone. The cells in the growth plate are aligned into columns representing a continuum of differentiation. The cells are maintained in columns through a process that has recently been referred to as rotation. During cell division, chondrocytes divide perpendicular to the long axis of the bone. This is followed by a series of cell shape changes and movements that result in the positioning of flat cells one on top of the other (Figure 3B; (Dodds, 1930). Very little is known about this process. The first alterations seen in the growth plate of Col2a-Cre;Kif3aloxP/loxP mice were at post natal day 7 (P7) and included a decrease in the length of the proliferation zone associated with reduced cell proliferation. By P10 days, the columnar organization of the growth plate was severely altered. In addition to accelerated hypertrophy, alterations in the localization of activated focal adhesion kinase (FAK) and disorganization of the actin cytoskeleton suggested defects in the process of chondrocyte rotation (Figure 3B). The phenotype of Col2a-Cre;Ift88loxP/loxP and Col2a-Cre;Kif3aloxP/loxP mice had some similarities to mice with conditional deletion of Ihh induced in post-natal cartilage (Col2a-CreER;IhhloxP/loxP) (Maeda et al., 2007). Nevertheless, alterations in Ptc1 expression were not detected by RT-PCR or in situ hybridization suggesting the phenotype was independent of Ihh signaling. It was proposed that defects in rotation could be the result of alterations in cell adhesion and shape mediated by mechanical stimulation (Aszodi et al., 2003; Jensen et al., 2004; Park et al., 2006).
It was previously suggested that chondrocyte cilia transmit mechanical forces through their interaction with the surrounding ECM (Poole et al., 1985; Jensen et al., 2004). It is known in other cell types that cilia are involved in transmitting both chemical and mechanical signals. The role of cilia in transmitting mechanical signals is most well characterized in the kidney where the protein products for genes associated with polycycstic kidney disease (PKD) are localized to cilia (Nauli and Zhou, 2004; Yoder et al., 2002). Autosomal dominant PKD results from mutations in Pkd1 or Pkd2, which encode polycystin 1 (Pc1) and polycystin 2 (Pc2). Pc1 and Pc2 are integral membrane proteins that form a regulatory protein and a Ca+2 channel in the primary cilium of renal epithelial cells. Cilia on renal cells are sensitive to fluid shear stress and respond with changes in the concentration of cytosolic Ca+2 (Praetorius and Spring, 2001). Cells without cilia or cells in which the function of Pc1 or Pc2 has been blocked do not respond to mechanical stimulation (Praetorius et al., 2003; Praetorius and Spring, 2003a; Praetorius and Spring, 2003b). It is possible that mechanical forces are transmitted through the cartilage extracellular matrix to chondrocytes by bending of the cilium (Jensen et al., 2004). Using electron microscopy is has been shown that the chondrocyte cilium projects into the extracellular matrix and is tightly associated with the Golgi apparatus (Poole et al., 1997). A combination of addditional imaging techniques including confocal, and tomographic microscopy indicate that chondrocyte cilia display various bending patterns in association with the surrounding pericellular matrix (Jensen et al., 2004; Poole et al., 2001). Some of the bending patterns fit with a model of shear stress while others suggested deflection by the matrix. Furthermore, the close anatomical association of the cilium with the microtubule cytoskeleton, Golgi apparatus, and microtubule organizing centers within the cell support a model of direct signaling between the matrix, the cilium, and the inside of the cell (Poole et al., 1997). Cilia were dramatically depleted from the cartilage of Col2a-Cre;Kif3aloxP/loxP mice by E15.5 days, nevertheless, alterations in the organization of the growth plate were not observed until seven to ten days after birth, a time when the young mice are starting to become subjected to significant mechanical load. Alterations in the perception of mechanical load as a result of the loss of cilia could lead to alterations in the growth plate and articular cartilage. Previously, it was shown that when young rats are subjected to unloading, alterations in the growth plate are observed including a reduction in the height of the growth plate due to reduced number of cells in the proliferative zone. Older rats were less responsive suggesting that there is a window of development that requires appropriate loading (Yu et al., 2003; Zerath et al., 1997).
The molecular mechanism whereby cilia potentially mediate mechanical signals in cartilage is not known. It has been shown that integrins are present on the chondrocyte cilium and integrin dependent signaling cascades have been decribed in chondrocyte mechanotransduction (McGlashan et al., 2006; Millward-Sadler and Salter, 2004; Praetorius et al., 2004). Furthermore, deletion of chondrocyte β1 integrin results in defects in rotation that include alterations in cell shape and orientation with some similarities to that seen in post-natal mice with disrupted cilia (Aszodi et al., 2003). In support of this model, deletion of Pkhd1, a ciliary protein mutated in autosomal recessive PKD, in cultured renal epithelial cells resulted in alterations in cell adhesion and activation of FAK supporting a link between primary cilia and integrin mediated cell-ECM interactions in the kidney (Mai et al., 2005).
Growth plates with some similarities to those in the long bones are present at the base of skull. Synchondroses consist of two mirror image growth plates that are important for development and growth of the cranial base. The synchondroses in Col2a-Cre;Kif3aloxP/loxP mice have been characterized at post-natal stages and compared to mice with inducible and conditional deletion of Ihh (Col2a-CreER;IhhloxP/loxP) (Koyama et al., 2007). Similar to what was seen in the post-natal long bone, the synchondroses were disorganized with significant reduction in chondrocyte proliferation. Histologically hypertrophic cells were present but expression of Col10a mRNA was dramatically reduced indicating a delay in complete hypertrophic differentiation. Ptc1 and Gli1 expression were reduced in chondrocytes suggesting Hh signaling was disrupted in the cranial base growth plates. Nevertheless, the cranial base in mice deficient in Ihh only minimally resembled the cranial base of mice in which cilia were disrupted suggesting IFT/cilia also have unique Ihh-independent roles in synchondroisis development (Koyama et al., 2007).
Mice harboring the Ift88orpk mutation also demonstrate short limbs (McGlashan et al., 2007). Minor alterations in the shape and organization of growth plate chondrocytes were detected by P4. In contrast to Col2a-Cre;Ift88loxP/loxP and Col2a-Cre;Kif3aloxP/loxP mice, Ift88orpk mutants demonstrated a delay in hypertrophic differentiation as measured by a reduction in the expression domain of Type X collagen protein with continued expression of Type II collagen. The differences in the Ift88orpk and Col2a-Cre models may reflect differences between systemic effects of germline mutation versus conditional deletion or the effect of a hypomorphic versus a null allele.
Recently, it was shown that another IFT protein, IFT46, is localized to cilia and preferentially expressed in early hypertrophic chondrocytes (Gouttenoire et al., 2007). IFT46 was initially identified as a BMP-responsive gene. BMP is a known mediator of chondrocyte differentiation. Knock down of IFT46 using siRNA technology in cultured chondrocytes stimulated the expression of genes that are primarily expressed by prehypertrophic columnar chondrocytes. The results suggest that IFT46 regulates specific aspects of growth plate biology. Since BMP regulated expression of IFT46, it is possible that BMP mediates the function of cilia in early hypertrophic cells.
D. Primary cilia in articular cartilage
Articular cartilage differs from the growth plate in that it is maintained as a mature resting cartilage. It is required for normal joint function but has limited capacity for repair. Osteoarthritis is a leading cause of disability in the world so understanding how articular cartilage is maintained is of primary importance. Mechanical load is a critical factor in maintaining articular cartilage but how the load is sensed is not known. Since it is likely that primary cilia function as mechanosensors, understanding the role of primary cilia in articular cartilage is of interest. Recently, the fate of primary cilia on articular chondrocytes during the progression of bovine osteoarthritis was investigated (McGlashan et al., 2008). Cilia were present on a subset of cells within all the layers, superficial, middle and deep, of normal articular cartilage. The incidence of cilia was greater in the deep zone cartilage. Cilia in normal cartilage also had a distinct orientation. As seen by others using mouse tissue, cilia in the superficial layer were oriented away from the articular surface (McGlashan et al., 2007; Poole et al., 1985; Wilsman et al., 1980). The majority of cilia in the middle and deep zones also projected away from the surface. Primary cilia were present during all stages of osteoarthritis examined; however, the proportion of ciliated cells within each zone increased except for that in the deep zone, which was not significantly different from the normal controls. In contrast to normal cartilage, cilia within cell clusters at the eroding osteoarthritic surface projected toward the center of the cluster. The majority of cilia in middle and deep zone osteoarthritic cartilage projected away from the articular surface similar to normal cartilage. The significance of this orientation remains unclear.
A biological role for primary cilia in maintaining articular chondrocyte function is suggested in mice with mutations in ciliary genes. Alterations in the shape of cells in the superficial layers of the articular cartilage were described in Ift88orpk mice (McGlashan et al., 2007). In addition, the cartilage in these mice demonstrated reduced toluidine blue and Type II collagen staining suggesting defects in the maintenance of the cartilage phenotype. Col2a-Cre;Kif3aloxP/loxP mice had alterations in the shape of the joints and alterations in the organization of the articular cartilage but expression of aggrecan was maintained up to P30 days (Song et al., 2007). Dermo-1Cre;Kif3aloxP/loxP mice had severely misshapen joint surfaces that were thought to be due to early patterning defects in the articular cartilage (Kolpakova-Hart et al., 2007). Severe systemic defects in the Ift88orpk mouse and perinatal lethality in Dermo-1Cre;Kif3aloxP/loxP mice preclude comprehensive studies on osteoarthritis. Progression of osteoarthritis in the Col2a-Cre;Kif3aloxP/loxP line has not yet been addressed.
E. A role for primary cilia in the development of the bone collar
As the chondrocytes in the developing skeleton begin to undergo hypertrophy, mesenchymal cells in the perichondrium begin to differentiate into osteoblasts and secrete osteoid, which will mineralize to form the mature bone (Day and Yang, 2008). Ihh signaling induces expression of Wnt ligands in the perichondrium and canonical Wnt signaling which is essential to drive cells to differentiate along the osteoblast lineage. In the absence of Wnt signaling, perichondrial cells differentiate into chondrocytes or adipocytes.
Wnt signaling is essential for development of many tissues (Clevers, 2006; Veeman et al., 2003). In the mammalian genome there are at least 19 Wnt ligands which bind to a subset of seven pass membrane receptors, the Frizzled proteins. Wnt signaling is further subdivided into two general categories, canonical and non-canonical. In canonical Wnt signaling, the absence of ligand results in β-catenin phosphorylation by glycogen synthase kinase-3β (GSK3β) and targetting for proteosome-mediated degradation. Upon ligand binding, dishevelled inhibits GSK3β activity leading to stabilization of β-catenin and its subsequent translocation to the nucleus. In the nucleus, β-catenin interacts with members of the LEF/TCF family to activate transcription of target genes. In contrast to canonical Wnt signaling, non-canonical signaling does not require β-catenin. Non-canonical signaling is not as well characterized at the molecular level as canonical Wnt signaling, but has been shown to be required for morphological movements such as convergent extension in the inner ear in mice and the body axis in Xenopus and zebrafish. Convergent extension movements during embryonic development require the planar cell polarity (PCP) signaling pathway, which leads to cytoskeletal alterations through RhoA and JNK activation.
Cilia have been implicated in regulating both canonical and non-canonical Wnt signaling (Beales et al., 2007; Corbit et al., 2008; Gerdes et al., 2007; Simons et al., 2005). Overexpression of Inversin, which localizes to cilia and is required for left-right axis formation in mice, leads to disruption of canonical Wnt signaling (Simons et al., 2005). In contrast, cells lacking Kif3a show enhanced canonical Wnt signaling due to constitutive phosphorylation of disheveled (Corbit et al., 2008). These data have led to the hypothesis that the cilium and basal body may regulate the switch between canonical and non-canonical Wnt signaling. Defects in PCP have also been shown in BBS morpholino-injected zebrafish embryos resulting in shortening of the body along the anterior posterior axis (Gerdes et al., 2007; Tayeh et al., 2008; Tobin et al., 2008).
The bone collar fails to develop in Prx1-Cre;Ift88/Kif3aloxP/loxP mutant mice, but it is not affected in the limbs of Col2a-Cre;Kif3aloxP/loxP mice suggesting cilia on the perichondrial cells are essential for bone collar formation and the defects are not due to loss of cilia on the chondrocytes (Haycraft et al., 2007; Song et al., 2007). In these mice and Dermo1-Cre;Kif3aloxP/loxP mutant mice, ectopic cartilage is seen along the diaphysis of the forming bone and Ihh signaling is lost as indicated by lack of Ptc1 or Gli1 expression (Haycraft et al., 2007; Kolpakova-Hart et al., 2007). While loss of Ihh signaling has been shown to result in defects in bone collar formation, it does not result in ectopic cartilage (St-Jacques et al., 1999). In contrast, disruption of β-catenin in the perichondrium results in a similar phenotype as seen in Prx1-Cre;Ift88/Kif3aloxP/loxP mutant mice and suggests that canonical Wnt signaling is disrupted in the perichondrium (Hilton et al., 2005; Hu et al., 2005). One function of Ihh is to induce expression of Wnt ligands. It is not known whether the similarity to conditional deletion of β-catenin is due to a requirement for cilia in canonical Wnt signaling in the perichondrial cells or loss of Wnt ligand expression downstream of Ihh signaling.
While the bone collar surrounding the long bones of Col2a-Cre;Kif3aloxP/loxP mutant mice develop normally, there are defects in bone formation in the synchondroses of the skull including excessive intramembraneous ossification and ectopic cartilage formation (Koyama et al., 2007). In these mutants, Gli1 and Ptc1 expression is present in the perichondrium and may be expanded where cilia formation is not disrupted. In agreement with expanded Ihh signaling, syndecan 3 expression, which has been proposed to restrict Ihh signaling in the chondrocytes, is absent providing a mechanism for increased Ptc1 and Gli1 expression in the adjacent perichondrium (Koyama et al., 2007).
F. Primary cilia in the maintenance of bone
Mature osteoblasts secrete osteoid that is subsequently mineralized to form bone. As bone formation progresses, a subset of osteoblasts become trapped in the bone and differentiate into osteocytes. Osteocyte cell bodies reside within the lacunae of the bone and they extend processes through the canaliculi where they communicate through gap junctions. Canonical Wnt signaling has been implicated in the control of postnatal bone formation since activating mutations in the Frizzled co-receptor Lrp5 lead to increased bone mass in adults (reviewed in Bonewald and Johnson, 2008; Macsai et al., 2008).
Throughout life, the bones of the mature skeleton are remodeled in response to many stimuli including mechanical loading. Loss of mechanical stimulation leads to loss of bone mass while increased loading leads to formation of bone. Intriguingly, both Pc1 and Pc2 are localized to the primary cilium of cultured osteoblasts and osteocytes suggesting a role for the cilium in mechanosensation in the bone as well as the kidney as described above (Nauli and Zhou, 2004; Xiao et al., 2008). In mice, loss of Pc1 results in delayed vascular invasion and a thin bone collar. Specifically, Pkd1 null samples show decreased Runx2-II expression as well as decreases in expression of other osteoblast/osteocyte specific genes such as osteocalcin and osterix (Xiao et al., 2006). Pkd1 null osteoblasts cultured in vitro also had lower basal intracellular calcium than wild type controls.
Despite the decrease in Runx2-II expression, no change in Runx2-I expression was detected (Xiao et al., 2006). Runx2-II is required for terminal osteoblast differentiation while Runx2-I is essential to early stages of osteoblast differentiation (Ducy et al., 1997; Xiao et al., 2005; Xiao et al., 2004). This selective decrease in Runx2-II expression may explain the Pkd1 bone phenotype. Indeed, Pkd1 null bones resemble those in Runx2-II mutant mice.
In addition to the embryonic defects in Pkd1 null mice, heterozygous Pkd1 adult mice show a 9% reduction in bone mineral density at 12 weeks of age (Xiao et al., 2006). Both trabecular bone volume and cortical bone thickness were affected suggesting a general requirement for Pkd1 in postnatal bone maintenance. More recent work has shown that mice lacking one allele of Pkd1 and one allele of Runx2-II have a more pronounced phenotype than either heterozygote alone (Xiao et al., 2008). While Pkd2 expression has been shown both in cultured osteoblasts and osteocytes by in situ hybridization, the role of Pc2 in the skeleton remains unknown (Markowitz et al., 1999; Xiao et al., 2006). However, loss of the Pc2-coupling domain of Pc1 abolished Runx2-II expression in vitro suggesting that Pc2 also plays a role in development and maintenance of the mammalian skeleton. Pc1 activation of Runx2-II transcription was also shown to require Pc1-mediated intracellular calcium increases resulting in increased binding of the transcription factor NFI to the Runx2-II promoter region (Xiao et al., 2008). How these defects are related to the proposed mechanosensation by Pc1 and Pc2 awaits further investigation.
In contrast to the requirement for intracellular calcium signaling via Pc1 shown for Runx2-II expression, other work has demonstrated that mechanosensation in bone cells occurs through a calcium-independent mechanism (Malone et al., 2007). MC3T3-E1 osteoblasts were subjected to dynamic flow and the cilium was observed to deflect in response to flow suggesting that it is positioned to receive and respond to mechanical stimuli in vivo. Mechanical stimulation has been shown to upregulate expression of osteopontin and synthesis of prostaglandin E2 (Malone et al., 2007). In MC3T3-E1 or MLO-Y4 cells treated with Ift88-directed siRNA, no change in prostaglandin E2 release was detected. Similarly, osteopontin mRNA levels did not increase in Ift88-siRNA treated cells. Calcium influx in response to flow has been shown and requires Pc2 in kidney epithelia(Praetorius and Spring, 2001). In contrast, correlation between calcium influx in response to fluid flow and expression of a cilium was observed in MC3T3-E1 or MLO-Y4 cells suggesting that cilia-mediated mechanosensation is calcium independent (Malone et al., 2007). Treatment of cells with gadolinium chloride to disrupt Pc2 function had no effect on calcium influx in response to flow indicating that calcium influx in response to flow in bone cells occurs through a different mechanism than in kidney epithelia.
The apparent discrepancies between the two proposed roles for cilia and Pc1 in the skeleton have yet to be investigated although it the results are not mutually exclusive. The work performed in vivo and with cultured cells did not examine the response of the cells to dynamic flow while the experiments performed under flow conditions did not investigate the potential role of Pc1 in the responses observed nor did the authors examine Runx2-II expression. It should be noted that alterations in bone mineral density have not been reported in patients with ADPKD. Additional experiments both in vivo and in vitro are necessary to address the role of cilia and Pc1 and Pc2 in osteoblast function and mechanosensation.
G. Primary cilia in craniofacial development
Many human ciliopathies are characterized by defects in craniofacial development. The face develops from the branchial arches through a series of very complicated morphogenetic events (Chai and Maxson, 2006). The bones of the face are derived from cranial neural crest and cranial mesoderm. In addition to malformations in the digits as described above, OFD1 is characterized by malformations in the craniofacial region including clefts in the jaw, tongue, and upper lip. Female mice in which CXORF5/Ofd1 was deleted using a ubiquitously expressed Cre demonstrated craniofacial malformations including a severe cleft palate and short snout (Ferrante et al., 2006). Males died early in gestation with defects in left-right patterning. The molecular mechanism of the craniofacial defects was not examined; however, alterations in Hh signaling were noted in the neural tube and alteration in Hox gene expression were detected in early limb buds (Ferrante et al., 2006).
One of the characteristics of Meckels type 5 is cleft lip and palate. Meckels type 5 and Joubert type 7 (OMIN#611560) are allelic and, as described above, result from mutations in RPGRIP1L (Delous 2007). A mouse homolog of this protein is called Fantom (Ftm). Null mutations of Ftm in mouse result in, among many defects, hypoplastic lower jaw and cleft lip with similarities to the human condition (Delous 2007, Vierkotten et al 2007). RPGRIP1L and Ftm are both localized to the basal body but do not appear to disrupt the formation or maintenance of cilia stucture (Delous et al., 2007; Vierkotten et al., 2007). Molecular mechanisms underlying the craniofacial defects have not been examined but alterations in Hh signaling were detected in mouse embryonic fibroblast cultured from mutant mice suggesting Hh signaling could be involved.
Craniofacial dymorphology has also been observed in some patients with BBS. Characteristic facial dysmorphology include displacement of the nose, mid face hypoplasia, and mild mandibular retrognatia. Bbs4-null and Bbs6-null mice demonstrated a short snout due to premaxillary and mandibular hypoplasia (Tobin et al., 2008). Using morpholino technology, fish in which various BBS proteins were depleted demonstrated defects in craniofacial development that were considered similar to those observed in humans and mice. Specifically, the mandibles were reduced and the branchial arch derivates were hypoplastic. Bbs8 depleted fish had the most severe phenotype including partial cyclopia and in some cases the mandibles were completely missing (Tobin et al., 2008). Many of the bones in the face are derived from neural crest cells and it has been shown that migrating neural crest cells bear primary cilia (Tobin et al., 2008). Using the zebrafish model is was suggested that the craniofacial defects observed in BBS mutant fish were due to defects in migration of neural crest cells. It was shown that the neural crest proliferated and was specified and maintained. Further experiments with the zebrafish suggested that non-canonical Wnt, PCP, signaling was involved. Induction of non-canonical Wnt signaling using either a truncated form of Dishevelled (Dvl) or a membrane targeted form of Dvl partially rescued the phenotype in Bbs8 depleted fish (Tobin et al., 2008). Although Hh signaling was also disrupted, it was concluded that the defect in neural crest migration was primarily due to a defect in PCP signaling since a drug that blocks Hh signaling, cyclopamine, did not have any effect on migration of cranial neural crest (Tobin et al., 2008).
Craniofacial abnormalities including cleft palate and supernumerary teeth have been described in Ift88orpk mice (Zhang et al., 2003). A more severe craniofacial phenotype including severe frontonasal dysplasia and shortening of the lower jaw as well as profound cleft secondary palate was documented in mice with conditional deletion of Kif3a in Wnt1 expressing neural crest cells (Kolpakova-Hart et al., 2007). Patterning defects in the midline of the face including midfacial clefting, missing tongue and missing incisors were also observed. Both endochondral and intramembranous bones of the head and face, which are primarily derived from neural crest, were affected. Since Wnt1-Cre expression was restricted to the neural crest, as expected structures derived from cephalic mesoderm were not affected. Similarities in Wnt1-Cre;Kif3aloxP/loxP mice to the craniofacial phenotypes seen in mice with conditional deletion of Smo in Wnt1 expressing cells suggested Hh signaling was altered in the absence of Kif3a (Jeong et al., 2004). In support of this model, Gli1 mRNA was limited to the epithelial structures and was absent from the mesenchyme in mutant mice whereas it was expressed in both the epithelial and mesenchymal components of the midface region in control mice (Kolpakova-Hart et al., 2007). The results suggested that Hh signaling was disrupted in the neural crest derived mesenchyme leading to some of the defects observed. However, mid-facial clefting, like that seen in the Wnt1-Cre;Kif3aloxP/loxP mutants, was not observed in Smo mutants suggesting defects in midline patterning were due to disruptions in the Hh-independent repressor function of Gli3 although alterations in other signaling pathways, for example, PCP, could not be excluded (Kolpakova-Hart et al., 2007). Potential effects on neural crest migration were not specficially examined. Together, craniofacial defects in humans and mice with alterations in cilia-related genes indicate an important role for primary cilia and intraflagellar transport in craniofacial development.
H. Primary cilia in tooth development
Like the skeleton, the teeth are mineralized tissues and therefore share some similarities with the bones. Although they do not erupt through the gingiva until after birth in both mice and humans, teeth begin to develop during embryogenesis from the oral ectoderm and neural crest-derived mesenchyme (reviewed in Miletich and Sharpe, 2003). The oral ectoderm first thickens, then invaginates as tooth development begins. Underlying neural crest cells condense around the invaginating ectoderm and reciprocal signaling interactions elaborate the structure through several stages as development progresses. As in many tissues, both Hh and Wnt signaling play important roles at several stages in tooth development including patterning and morphogenesis. The mature tooth is composed of enamel secreted by epithelial-derived ameloblasts overlaying mineralized dentin secreted by neural crest-derived mesenchyme cells. While the odontoblasts are retained within the pulp cavity following eruption, the ameloblasts are lost. The dental pulp is the inner cavity of the tooth and contains many cell types including vasculature, mesenchymal stem cells and nerves. There are significant differences in tooth development and patterning between mice and humans. While mice develop a single set of permanent dentition consisting of incisors separated from molars by a toothless diastema, humans develop two sets of dentition, deciduous and permanent. The deciduous teeth are shed during childhood and replaced by the permanent dentition that is retained. Humans also develop additional tooth types between the incisors and molars including the canines and premolars. Despite these differences, the mouse has proven to be an invaluable research model to study tooth development.
Cilia have been observed on both ameloblasts and odontoblasts in vivo using electron microscopy (Magloire et al., 2004; Sasano, 1986). On odontoblasts, cilia are hypothesized to play a role in mechanotransduction although this has yet to be investigated (Magloire et al., 2004). Ameloblasts showed dynamic expression of cilia throughout their differentiation in rats and were predicted to be present on all post-mitotic amelobalsts based on their prevelance in serial sections (Sasano, 1986).
In agreement with a role for cilia in tooth patterning, Ift88orpk mutants develop an ectopic molar mesial to the first molar although the mechanism for this has yet to be reported (Zhang et al., 2003). As mentioned above, Wnt1-Cre;Kif3aloxP/loxP mutant mice lack incisors but this may be due to midline defects and may not reflect a requirement for cilia in the initiation of incisor development (Kolpakova-Hart et al., 2007).
Defects in tooth number and morphology have also been reported in several ciliopathies but the molecular mechanism underlying the defects has not been investigated. Natal teeth, the eruption of teeth through the gingiva prior to birth, are a common feature of EvC. Additionally, EvC patients often present with missing permanent teeth (hypodontia or oligodontia) and malformed, often pointed, cusps. In agreement with a role for Evc in tooth morphogenesis, the first molar of Evc null mice is often smaller that that found in controls (Ruiz-Perez et al., 2007). Significant tooth defects are also observed in patients with OFD1. The maxillary canines are often malpositioned and supernumary deciduous teeth are frequent including the canines and premolars. In addition to defects in the number of teeth, canines are often misshapen. Alterations in the dimensions of the pulp cavity have also been reported in a small number of BBS patients but there has been no investigation of the underlying cause and no defects in tooth morphogenesis have been reported in BBS mutant mice. Overall the role of the cilium in the formation of the mammalian dentition has not been examined in detail and awaits further investigation, but defects in Shh and Wnt signaling may be involved based on the phenotype observed in other tissues.
I. Summary
Defects in the primary cilium were first shown to be involved in the development of cysts in the kidney less than 10 years ago (reviewed in Bisgrove and Yost, 2006; Davenport and Yoder, 2005; Pan et al., 2005; Satir and Christensen, 2007). Since that time, the expression and function of cilia has become an area of intense research. Mutations in proteins localized to the primary cilium or basal body lead to a diverse group of pleiotropic syndromes including BBS, MKS, EvC, and OFD1, which show defects in the skeleton including patterning of the digits, endochondral bone formation, craniofacial development and the dentition.
Cilia have been shown to play a role in signaling pathways important in the development of the skeleton including Hh and Wnt (Bisgrove and Yost, 2006; Pan et al., 2005). Many of the defects in digit patterning are attributed to loss of activation or repression of Shh signaling. In the developing bones, Ihh signaling is also disrupted although the potential role for cilia in Wnt signaling in the perichondrium either through induction of Wnt ligand expression or canonical signaling distinct from that described in other tissues is still under investigation (Haycraft et al., 2007; Koyama et al., 2007).
Despite the fact the both bone cells and kidney epithelia respond to fluid flow and express primary cilia as well as the mechanosensitive Pc1 and Pc2 proteins, there are distinct differences in the function of this organelle in the two cell types (Malone et al., 2007; Nauli and Zhou, 2004; Praetorius et al., 2003). Fluid flow in canine kidney epithelia results in Pc2 mediated calcium influx while no link to calcium influx and cilia is seen in bone cells. In cartilage, cilia are in direct contact with the extracellular matrix but the potential function of Pc1 and Pc2 in the cartilage has not been determined. This highlights the importance of studying the function of the cilium and cilia-localized proteins in multiple cells types throughout development and postnatally since cell types may respond differently.
The various roles of cilia in the formation and maintenance of the mammalian skeleton is an emerging area of research and will provide important insight into the growing number of roles cilia play throughout the body.
Acknowledgments
We would like to thank the members of our labs and S. R. McGlashan for helpful discussions during preparation of this manuscript.
References
- Alvarez J, Sohn P, Zeng X, Doetschman T, Robbins DJ, Serra R. Development. 2002;129:1913–24. doi: 10.1242/dev.129.8.1913. [DOI] [PubMed] [Google Scholar]
- Ascenzi MG, Lenox M, Farnum C. J Struct Biol. 2007;158:293–306. doi: 10.1016/j.jsb.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszodi A, Hunziker EB, Brakebusch C, Fassler R. Genes Dev. 2003;17:2465–79. doi: 10.1101/gad.277003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ. Nat Genet. 2007;39:727–9. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- Bisgrove BW, Yost HJ. Development. 2006;133:4131–43. doi: 10.1242/dev.02595. [DOI] [PubMed] [Google Scholar]
- Bonewald LF, Johnson ML. Bone. 2008;42:606–15. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R. Proc Natl Acad Sci U S A. 2001;98:12174–9. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancedda R, Cancedda FD, Castagnola P. International Review of Cytology. 1995;159:265–358. doi: 10.1016/s0074-7696(08)62109-9. [DOI] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr Dev Dyn. 2006;235:2353–75. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Harris MP, Simandl BK, Li Y, Beachy PA, Fallon JF. Dev Biol. 2001;236:421–35. doi: 10.1006/dbio.2001.0346. [DOI] [PubMed] [Google Scholar]
- Clevers H. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Colnot C. J Cell Biochem. 2005;95:688–97. doi: 10.1002/jcb.20449. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Nature. 2005;437:1018–21. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF. Nat Cell Biol. 2008;10:70–6. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- Davenport JR, Yoder BK. Am J Physiol Renal Physiol. 2005;289:F1159–69. doi: 10.1152/ajprenal.00118.2005. [DOI] [PubMed] [Google Scholar]
- Day TF, Yang Y. J Bone Joint Surg Am. 2008;90(Suppl 1):19–24. doi: 10.2106/JBJS.G.01174. [DOI] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U, Schneider-Maunoury S, Attie-Bitach T, Saunier S. Nat Genet. 2007;39:875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Dodds GS. Anatomical Record. 1930;46:385–399. [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Cell. 1997;89:747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Fath MA, Mullins RF, Searby C, Nishimura DY, Wei J, Rahmouni K, Davis RE, Tayeh MK, Andrews M, Yang B, Sigmund CD, Stone EM, Sheffield VC. Hum Mol Genet. 2005;14:1109–18. doi: 10.1093/hmg/ddi123. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O, Bernard L, Malcolm S, Winter R, Ballabio A, Franco B. Am J Hum Genet. 2001;68:569–76. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B. Nat Genet. 2006;38:112–7. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, Badano JL, Katsanis N. Nat Genet. 2007;39:1350–60. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- Gouttenoire J, Valcourt U, Bougault C, Aubert-Foucher E, Arnaud E, Giraud L, Mallein-Gerin F. J Biol Chem. 2007;282:30960–73. doi: 10.1074/jbc.M705730200. [DOI] [PubMed] [Google Scholar]
- Guillaume R, D’Agati V, Daoust M, Trudel M. Dev Dyn. 1999;214:337–48. doi: 10.1002/(SICI)1097-0177(199904)214:4<337::AID-AJA6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Development. 2007;134:307–16. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- Hilton MJ, Tu X, Cook J, Hu H, Long F. Development. 2005;132:4339–51. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Development. 2005;132:49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Proc Natl Acad Sci U S A. 2005;102:11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Development. 2006;133:3–14. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- Jensen CG, Poole CA, McGlashan SR, Marko M, Issa ZI, Vujcich KV, Bowser SS. Cell Biol Int. 2004;28:101–10. doi: 10.1016/j.cellbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Genes Dev. 2004;18:937–51. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL, Lupski JR. Science. 2001;293:2256–9. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- Katsanis N, Eichers ER, Ansley SJ, Lewis RA, Kayserili H, Hoskins BE, Scambler PJ, Beales PL, Lupski JR. Am J Hum Genet. 2002;71:22–9. doi: 10.1086/341031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpakova-Hart E, Jinnin M, Hou B, Fukai N, Olsen BR. Dev Biol. 2007;309:273–84. doi: 10.1016/j.ydbio.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, Pacifici M. Development. 2007;134:2159–69. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel L, Wuelling M, Schneider S, Vortkamp A. Development. 2005;132:5249–60. doi: 10.1242/dev.02097. [DOI] [PubMed] [Google Scholar]
- Kulaga HM, Leitch CC, Eichers ER, Badano JL, Lesemann A, Hoskins BE, Lupski JR, Beales PL, Reed RR, Katsanis N. Nat Genet. 2004;36:994–8. doi: 10.1038/ng1418. [DOI] [PubMed] [Google Scholar]
- Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M. Nat Genet. 2006;38:155–7. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- Lanske B, Karapalis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LHK, Ho C, Mulligan RC, Abou-Samra A-B, Juppner H, Segre GV, Kronenberg HM. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Development. 2005;132:3103–11. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Development. 2001;128:5099–108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Lu W, Shen X, Pavlova A, Lakkis M, Ward CJ, Pritchard L, Harris PC, Genest DR, Perez-Atayde AR, Zhou J. Hum Mol Genet. 2001;10:2385–96. doi: 10.1093/hmg/10.21.2385. [DOI] [PubMed] [Google Scholar]
- Macsai CE, Foster BK, Xian CJ. J Cell Physiol. 2008;215:578–87. doi: 10.1002/jcp.21342. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS, Mackem S, Lanske B. Proc Natl Acad Sci U S A. 2007;104:6382–7. doi: 10.1073/pnas.0608449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magloire H, Couble ML, Romeas A, Bleicher F. Cell Biol Int. 2004;28:93–9. doi: 10.1016/j.cellbi.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Mai W, Chen D, Ding T, Kim I, Park S, Cho SY, Chu JS, Liang D, Wang N, Wu D, Li S, Zhao P, Zent R, Wu G. Mol Biol Cell. 2005;16:4398–409. doi: 10.1091/mbc.E04-11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, Jacobs CR. Proc Natl Acad Sci U S A. 2007;104:13325–30. doi: 10.1073/pnas.0700636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz GS, Cai Y, Li L, Wu G, Ward LC, Somlo S, D’Agati VD. Am J Physiol. 1999;277:F17–25. doi: 10.1152/ajprenal.1999.277.1.F17. [DOI] [PubMed] [Google Scholar]
- McGlashan SR, Cluett EC, Jensen CG, Poole CA. Dev Dyn. 2008;237:2013–2020. doi: 10.1002/dvdy.21501. [DOI] [PubMed] [Google Scholar]
- McGlashan SR, Haycraft CJ, Jensen CG, Yoder BK, Poole CA. Matrix Biol. 2007;26:234–46. doi: 10.1016/j.matbio.2006.12.003. [DOI] [PubMed] [Google Scholar]
- McGlashan SR, Jensen CG, Poole CA. J Histochem Cytochem. 2006;54:1005–14. doi: 10.1369/jhc.5A6866.2006. [DOI] [PubMed] [Google Scholar]
- Miletich I, Sharpe PT. Hum Mol Genet. 2003;12(Spec No 1):R69–73. doi: 10.1093/hmg/ddg085. [DOI] [PubMed] [Google Scholar]
- Millward-Sadler SJ, Salter DM. Ann Biomed Eng. 2004;32:435–46. doi: 10.1023/b:abme.0000017538.72511.48. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Zhou J. Bioessays. 2004;26:844–56. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Genesis. 2000;26:145–6. [PubMed] [Google Scholar]
- Pan J, Wang Q, Snell WJ. Lab Invest. 2005;85:452–63. doi: 10.1038/labinvest.3700253. [DOI] [PubMed] [Google Scholar]
- Park TJ, Haigo SL, Wallingford JB. Nat Genet. 2006;38:303–11. doi: 10.1038/ng1753. [DOI] [PubMed] [Google Scholar]
- Poole CA, Flint MH, Beaumont BW. Cell Motil. 1985;5:175–93. doi: 10.1002/cm.970050302. [DOI] [PubMed] [Google Scholar]
- Poole CA, Jensen CG, Snyder JA, Gray CG, Hermanutz VL, Wheatley DN. Cell Biol Int. 1997;21:483–94. doi: 10.1006/cbir.1997.0177. [DOI] [PubMed] [Google Scholar]
- Poole CA, Zhang ZJ, Ross JM. J Anat. 2001;199:393–405. doi: 10.1046/j.1469-7580.2001.19940393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius HA, Frokiaer J, Nielsen S, Spring KR. J Membr Biol. 2003;191:193–200. doi: 10.1007/s00232-002-1055-z. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Praetorius J, Nielsen S, Frokiaer J, Spring KR. Am J Physiol Renal Physiol. 2004;287:F969–78. doi: 10.1152/ajprenal.00096.2004. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. J Membr Biol. 2001;184:71–9. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. J Membr Biol. 2003a;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Curr Opin Nephrol Hypertens. 2003b;12:517–20. doi: 10.1097/00041552-200309000-00006. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Science. 2007;317:372–6. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. J Am Soc Nephrol. 2004;15:2556–68. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H, Goodship JA. Development. 2007;134:2903–12. doi: 10.1242/dev.007542. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, Hamel B, Gibbs JL, Young ID, Wright MJ, Goodship JA. Am J Hum Genet. 2003;72:728–32. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasano Y. Arch Histol Jpn. 1986;49:437–48. doi: 10.1679/aohc.49.437. [DOI] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- Scherft JP, Daems WT. J Ultrastruct Res. 1967;19:546–55. doi: 10.1016/s0022-5320(67)80080-7. [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Nat Genet. 2005;37:537–43. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Haycraft CJ, Seo HS, Yoder BK, Serra R. Dev Biol. 2007;305:202–16. doi: 10.1016/j.ydbio.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Genes Dev. 1999;13:2072–86. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayeh MK, Yen HJ, Beck JS, Searby CC, Westfall TA, Griesbach H, Sheffield VC, Slusarski DC. Hum Mol Genet. 2008;17:1956–67. doi: 10.1093/hmg/ddn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Welscher P, Zuniga A, Kuijper S, Drenth T, Goedemans HJ, Meijlink F, Zeller R. Science. 2002;298:827–30. doi: 10.1126/science.1075620. [DOI] [PubMed] [Google Scholar]
- Tickle C. Nat Rev Mol Cell Biol. 2006;7:45–53. doi: 10.1038/nrm1830. [DOI] [PubMed] [Google Scholar]
- Tobin JL, Di Franco M, Eichers E, May-Simera H, Garcia M, Yan J, Quinlan R, Justice MJ, Hennekam RC, Briscoe J, Tada M, Mayor R, Burns AJ, Lupski JR, Hammond P, Beales PL. Proc Natl Acad Sci U S A. 2008;105:6714–9. doi: 10.1073/pnas.0707057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR. Nat Genet. 2008;40:403–10. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco AE, Padovani EM, Chiaffoni GP, Peissel B, Rossetti S, Marcolongo A, Gammaro L, Maschio G, Pignatti PF. J Med Genet. 1993;30:419–22. doi: 10.1136/jmg.30.5.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Eerden BC, Karperien M, Wit JM. Endocr Rev. 2003;24:782–801. doi: 10.1210/er.2002-0033. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Axelrod JD, Moon RT. Dev Cell. 2003;5:367–77. doi: 10.1016/s1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- Vierkotten J, Dildrop R, Peters T, Wang B, Ruther U. Development. 2007;134:2569–77. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kroneberg HM, Tabin CJ. Science. 1996;273:613–621. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Cell. 2000;100:423–34. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wilsman NJ, Farnum CE, Reed-Aksamit DK. Anat Rec. 1980;197:355–61. doi: 10.1002/ar.1091970309. [DOI] [PubMed] [Google Scholar]
- Wilsman NJ, Fletcher TF. Anatomical Record. 1978;190:871–889. doi: 10.1002/ar.1091900408. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Awad HA, Liu S, Mahlios J, Zhang S, Guilak F, Mayo MS, Quarles LD. Dev Biol. 2005;283:345–56. doi: 10.1016/j.ydbio.2005.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zhang S, Magenheimer BS, Luo J, Quarles LD. J Biol Chem. 2008;283:12624–34. doi: 10.1074/jbc.M710407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, Dallas SL, Maser R, Calvet JP, Bonewald L, Quarles LD. J Biol Chem. 2006;281:30884–95. doi: 10.1074/jbc.M604772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao ZS, Hjelmeland AB, Quarles LD. J Biol Chem. 2004;279:20307–13. doi: 10.1074/jbc.M401109200. [DOI] [PubMed] [Google Scholar]
- Yoder BK, Hou X, Guay-Woodford LM. J Am Soc Nephrol. 2002;13:2508–16. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Development. 2003;130:3063–74. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zerath E, Holy X, Mouillon JM, Farbos B, Machwate M, Andre C, Renault S, Marie PJ. Life Sci. 1997;61:2397–406. doi: 10.1016/s0024-3205(97)00957-0. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK. Dev Dyn. 2003;227:78–90. doi: 10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]