

Abstract
The serotonin transporter (SERT) is the principal mechanism for terminating serotonin (5-HT) signals in the nervous system and is a site of action for a variety of psychoactive drugs including antidepressants, amphetamines, and cocaine. Here we show that human SERTs (hSERTs) and rat SERTs are capable of robust dopamine (DA) uptake through a process that differs mechanistically from 5-HT transport in several unanticipated ways. DA transport by hSERT has a higher maximum velocity than 5-HT transport, requires significantly higher Na+ and Cl− concentrations to sustain transport, is inhibited noncompetitively by 5-HT, and is more sensitive to SERT inhibitors, including selective serotonin reuptake inhibitors. We use a thiol-reactive methane thiosulfonate (MTS) reagent to modify a conformationally sensitive cysteine residue to demonstrate that hSERT spends more time in an outward facing conformation when transporting DA than when transporting 5-HT. Cotransfection of an inactive or an MTS-sensitive SERT with wild-type SERT subunits reveals an absence of cooperative interactions between subunits during DA but not 5-HT transport. To establish the physiological relevance of this mechanism for DA clearance, we show using in vivo high-speed chronoamperometry that SERT has the capacity to clear extracellularly applied DA in the hippocampal CA3 region of anesthetized rats. Together, these observations suggest the possibility that SERT serves as a DA transporter in vivo and highlight the idea that there can be distinct modes of transport of alternative physiological substrates by SERT.
Introduction
Neurotransmitter transporters terminate the actions of the biogenic amines serotonin (5-HT), dopamine (DA), and norepinephrine by clearing them from the extracellular space and are the primary sites of action for many psychostimulant and antidepressant drugs. Within the CNS, biogenic amine transporters are generally expressed in the cells that synthesize their cognate neurotransmitters (Hoffman et al., 1998). Although this correlation fits with the idea that transporters function to terminate signaling and to facilitate recycling of neurotransmitter molecules, transporters are not only found on nerve terminals near sites of vesicle release but also are located throughout the somatodendritic and axonal membranes (Hoffman et al., 1998; Tao-Cheng and Zhou, 1999; Schroeter et al., 2000). This distribution of carriers in regions outside the nerve terminal is relevant to “volume transmission,” a process in which biogenic amine neurotransmitters diffuse for micrometer distances before acting on receptors (Daws, 2009). This process, which occurs on a slower time scale and outside of conventional synapses, raises the possibility that neurotransmitters can be transported into other cell types by heterologous carriers (Bunin and Wightman, 1999; Zoli et al., 1999; Jansson et al., 2000; Mengual and Pickel, 2002). Thus, the question of how well transporters recognize alternative substrates has significance not only in drug action and pathological processes but also in the physiological context of volume transmission.
It is well established that both the norepinephrine transporter (NET) and the dopamine transporter (DAT) transport DA (Carboni and Silvagni, 2004). It was demonstrated that DAT could potentially transport 5-HT and that it could be coreleased with DA from striatal dopaminergic terminals (Stamford et al., 1990; Jackson and Wightman, 1995). It has also been established in striatal slice (Zhou et al., 2002; Mossner et al., 2006), synaptosomal (Norrholm et al., 2007) preparations, and in vivo (Callaghan et al., 2005; Zhou et al., 2005) that 5-HT can be accumulated in dopaminergic neurons through the DAT. This potential for cross talk between transporter substrates also exists for the serotonin transporter (SERT), which is expressed in several brain regions receiving dopaminergic input. Data from microdialysis studies in rats after extensive 6-hydroxydopamine denervation of striatal DA neurons suggest that 5-HT-producing neurons can take up DA by SERT (Kannari et al., 2006).
Current models for transport by SERT and other family members center on an alternating access model. The molecular details for the transport mechanism are not well understood, and whether SERT accumulates alternative substrates by a mechanism identical to 5-HT transport has not been examined. Here we show, using high-speed chronoamperometry, that SERT can clear DA when it is applied locally to the hippocampus in anesthetized rats. A more detailed investigation of DA transport by human SERT (hSERT) reveals that DA transport occurs through a process remarkably different from 5-HT uptake. DA transport by SERT is inhibited noncompetitively by 5-HT, has a different ionic dependence, and is more sensitive to SERT inhibitors. We also show that a DA-transporting hSERT spends less time in an inward facing conformation than a 5-HT-transporting hSERT and lacks the subunit cooperativity characteristic of 5-HT transport.
Materials and Methods
Animals.
Male Sprague Dawley rats (200–250 g; Sasco) were maintained on a 12 h light/dark cycle (6:00 A.M. to 6:00 P.M.) and housed four rats per cage with ad libitum water and rat chow. The vivarium and this research program operate in accordance with the United States Public Health Service Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication 85-23, revised 1985), the Animal Welfare Act, and other applicable federal, state, and local laws.
In vivo electrochemical measurement of 5-HT and DA clearance.
Oxidation currents from exogenously applied 5-HT and DA were recorded in brain regions of urethane-anesthetized rats using high-speed chronoamperometric methodology, as described previously (Cass et al., 1993; Cass and Gerhardt, 1995; Zahniser et al., 1999; Gulley et al., 2006). The stereotaxic coordinates used for placement of the Nafion-coated carbon fiber electrode–micropipette assemblies (calculated from bregma) were −4.1 mm (anteroposterior), ±3.3 mm (lateromedial), and −3.6 to −3.8 mm (dorsoventral) for the CA3 hippocampal region and +1.5, ±2.2, and −4.0 to −4.3 mm for the dorsal striatum (Paxinos and Watson, 2007). Solutions containing either 100 μm 5-HT or 200 μm DA and 154 mm NaCl/100 μm ascorbic acid, pH 7.4, were pressure ejected once every 5 min. Two recording modes were used: for 5-HT, the “delayed pulse” mode was used to retain electrode sensitivity, whereas for DA, the “fast–slow” mode was used (Luthman et al., 1997). After two reproducible baseline signals were obtained (<10% variation in maximal signal amplitude), either saline (1 ml/kg, i.p.) or citalopram (10 mg/kg, i.p.) was injected and oxidation currents were recorded at 5 min intervals for 1 h after injection. Clearance time was calculated from these currents as the time for the signal to increase to its maximal value and to decrease by 80% (T80).
Site-directed mutagenesis.
cDNA encoding hSERT was reverse transcribed and amplified using RNA extracted from human placenta and inserted into the mammalian expression vector pcDNA3.1(+) (Invitrogen). The predicted amino acid sequence of the hSERT clone was identical to that reported previously (Entrez Protein Database accession number NP_001036). hSERT Y95F, C109A, Y95F/D98G, Y95F/C109A, Y95F/C109A/I172C, Y95F/C109A/I179C, and Y95F/C109A/Q332C were created using the Quickchange Mutagenesis kit (Stratagene) according to the manufacturer's recommendations. The entire coding regions of mutated constructs were sequenced using an ABI PRISM 310 genetic analyzer (Applied Biosystems).
Cell culture and transfections.
COS-7 cells were maintained in DMEM supplemented with 10% fetal calf serum, 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C and 5% CO2 in a humidified atmosphere. For transfections, 0.3 μg of plasmid and 0.9 μl of Fugene6 (Roche Molecular Biochemicals) were used per square centimeter of plating area. Appropriate amounts of plasmid and Fugene6 were mixed with DMEM according to the manufacturer's recommendations. COS-7 cells were trypsinized, suspended in growth media, combined with the plasmid/Fugene6 mixture, and transferred into growth plates to give an initial cell confluence of 70–80% after cell adhesion. White clear-bottomed 96-well plates (Corning) were used for uptake experiments.
Serotonin and DA uptake assays in COS-7 cells.
All uptake assays comparing 5-HT and DA uptake and the response to different treatments were performed in parallel on the same batch of transfected cells. Uptake assays were performed 40–64 h after transfection. The media were removed, and the cells were washed with PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.4) containing 0.1 mm CaCl2 and 1 mm MgCl2 (PBSCM). After washing, cells were incubated at room temperature for 10 min in PBSCM, including 50 μm ascorbic acid, 10 mm d-glucose, and 5 μm of the catechol-O-methyltransferase inhibitor RO 41-0960 (PBSCM-AGR) containing 12 (in duplicate) increasing concentrations of [3H]5-HT or [3H]DA diluted with unlabeled 5-HT (total 5-HT concentration, 0.1–21 μm) or DA (total DA concentration, 0.5–51 μm) for Km and Vmax determinations. Alternatively, after initial washing, the cells were incubated at room temperature for 10 min with a constant concentration of radiolabeled substrate (100 nm [3H]5-HT or 200 nm [3H]DA) and increasing concentrations of unlabeled substrate (0.01–150 μm 5-HT or 0.2–1000 μm DA). Washing with PBSCM terminated the uptake. For uptake in 48-well plates at 37°C, COS-7 cells were transfected in 48-well plates with cDNA encoding hSERT wild type (WT) 48 h before performing the uptake assay. The cells were then washed in PBSCM and incubated with 200 nm [3H]5-HT or 200 nm [3H]DA and increasing concentrations of unlabeled 5-HT (0.7–1000 μm 5-HT) or DA (8–1000 μm DA) for 10 min at 37°C. Incubations were terminated with three washes, and the plates were prepared for scintillation counting. Specific uptake was determined as the difference in radioactivity between cells transfected with SERT-containing constructs and mock-transfected cells. Counts from inhibition of a fixed radiolabeled substrate concentration were converted to total moles transported by multiplying the number of counts by the ratio of ([radiolabeled substrate] + [unlabeled substrate])/[radiolabeled substrate] and by dividing this product by the product of the tracer's specific activity and the efficiency of the scintillation counter.
For IC50 determinations, following the initial washing step, cells were incubated for 10 min with 12 increasing concentrations of drug (in duplicate) to reach equilibrium. Uptake was then initiated by the addition of a solution of [3H]5-HT or [3H]DA to give a final [3H]5-HT concentration of 100 nm or [3H]DA concentration of 200 nm. Drugs that are transporter substrates were not preincubated with the cells but added concomitantly with [3H]5-HT or [3H]DA. The uptake was allowed to continue for 10 min at room temperature and was terminated by two washes with PBSCM. All washing steps were performed using an automated plate washer. In ion substitution experiments, Na+ or Cl− ions was substituted in a HEPES-buffered saline solution with N-methyl-d-glucamine-Cl or gluconate salts, respectively. In chloride substitution experiments, gluconate salts were also used for magnesium, potassium, and calcium. In several instances, we took advantage of the high DA affinity of the Y95F mutant, which we showed to be otherwise identical to hSERT WT in all other measured features, except apparent DA transport affinity and inhibitor affinity, and used it in constructs with other mutations to elevate DA uptake activities at low DA concentrations.
For uptake experiments with [2-(trimethylammonium)ethyl] methane-thiosulfonate (MTSET)-sensitive Y95F/C109A/I179C, COS-7 cells were transfected as described above, and 40–64 h after transfection, the cells were washed in PBSCM and incubated for 10 min at room temperature with either PBSCM-AGR alone or PBSCM-AGR containing 10 mm MTSET. Cells were then washed with PBSCM and assayed for uptake with either 150 nm [3H]5-HT or 300 nm [3H]DA for 10 min at room temperature as described above, and uptake was terminated by washing twice with PBSCM.
To study the effect of substrates and ligands on reactivity of hSERT Y95F/C109A/Q332C mutant with MTSET, COS-7 cells were transfected as described. Forty to sixty-four hours later, cells were washed with PBSCM and incubated with substrate or ligand in PBSCM-AGR for 10 min at room temperature. Varying concentrations of MTSET were then added from a stock solution prepared in DMSO. After a 10 min incubation, the cells were washed five times in PBSCM-AGR and assayed for uptake with 250 nm [3H]5-HT for 10 min at room temperature as described above, and uptake was terminated by washing twice with PBSCM. The rate of reactivity was calculated after determining the concentration of MTSET that results in 50% of maximal inactivation (Rudnick, 2003).
For uptake experiments with (2-aminoethyl)methanethiosulfonate (MTSEA)-sensitive hSERT Y95F/C109A/I172C and MTSEA-insensitive hSERT Y95F/C109A, plasmids containing each construct were mixed at various ratios, while keeping a constant total DNA amount, and transfected into COS-7 cells as described above. Forty to sixty-four hours after transfection, cells were washed in PBSCM and incubated for 10 min with either PBSCM-AGR alone or PBSCM-AGR containing 0.25 mm MTSEA. Cells were then washed with PBSCM and assayed for uptake with either 150 nm [3H]5-HT or 300 nm [3H]DA for 10 min as described above. COS-7 cells transfected with varying ratios of Y95F and Y95F/D98G plasmids were washed with PBSCM 40–64 h after transfection and assayed for uptake with either 150 nm [3H]5-HT or 300 nm [3H]DA for 10 min as described above. All steps were performed at room temperature.
For all experiments with transiently transfected COS-7 cells, the cells were solubilized in scintillant after the uptake assay (30% Scintisafe; Thermo Fisher Scientific) and plates were counted in a PerkinElmer 1450 MicroBeta Jet plate counter. Specific uptake was determined as the difference between uptake counts from cells transfected with SERT-containing constructs and mock-transfected cells. Assuming Michaelis-Menten kinetics, the data were plotted and analyzed by the single-site nonlinear least-squares curve fit (GraphPad Prism).
Efflux from [3H]5-HT or [3H]DA preloaded COS-7 cells.
COS-7 cells were transfected and plated as described above. Forty-eight hours after transfection, cells were washed with PBSCM and incubated for 45 min with PBSCM-AGR containing [3H]5-HT or [3H]DA at room temperature to load the cells with equal concentrations of [3H]5-HT or [3H]DA. Cells were then washed every 5 min in PBSCM-AGR over a 45 min time period and efflux initiated by addition of 80 μl of PBSCM-AGR containing the appropriate drug to be tested. At specified time points, 40 μl of the PBSCM-AGR solution was transferred to another plate and prepared for scintillation counting to determine the amount of [3H]5-HT or [3H]DA efflux. At the end of the experiment, the cells were solubilized in scintillation fluid to determine the amount of [3H]5-HT or [3H]DA remaining in the cells after the efflux experiment, which was then used to calculate the total loading of the cells.
Radioligand binding assays.
HEK-293 cells stably transfected with hSERT WT were plated in 96-well plates, washed with ice-cold PBSCM 24 h after plating, and incubated with 0.2–0.5 nm [125I]RTI-55 with either a mixture of a fixed concentration of 5-HT plus a series of DA concentrations or a mixture of a fixed DA concentration plus a series of 5-HT concentrations, in PBSCM-AGR. Binding was allowed to proceed for 1 h on ice, terminated by six washes with ice-cold PBSCM, and prepared for scintillation counting as described above.
Statistical analysis.
Chronoamperometric data were analyzed statistically with two-factor (treatment, time), repeated-measure ANOVA with time as the repeated measure. For cell-based assays, the two-tailed paired or unpaired Student's t test was used for determining statistical significance.
Results
In vivo clearance of 5-HT and DA by SERT in rat hippocampal CA3 region
To assess whether SERTs expressed by intact brain serotonergic neurons are capable of transporting DA, we compared the in vivo clearance times of exogenously applied substrates in the CA3 region of the rat hippocampus and in the rat dorsal striatum (Fig. 1). The CA3 region has abundant 5-HT projections and SERT activity but negligible NET activity (Daws et al., 1998) and little DA innervation or DAT expression (Verney et al., 1985), whereas the opposite is true for the dorsal striatum. We used high-speed chronoamperometry to monitor clearance of 5-HT and DA after pressure ejection of a constant volume of neurotransmitter repeated at 5 min intervals from a micropipette positioned ∼300 μm from the carbon fiber microelectrode. This resulted in maximal signal amplitudes equivalent to ∼1 μm concentrations at the electrode. We compared 5-HT/DA clearance times (T80 values; see Materials and Methods) because we have found them to be the most sensitive and stable parameter reflecting neurotransmitter clearance. Baseline clearance times of both transmitters in CA3 were comparable (5-HT, 73.4 ± 8.3 s; DA, 79.5 ± 4.1 s; mean ± SEM; both n = 10), suggesting similar rates of clearance. To verify that SERT was responsible for clearance of both 5-HT and DA, we examined the effect of citalopram, a potent and highly SERT-selective blocker (Owens et al., 1997; Tatsumi et al., 1997). Peripheral injection of 10 mg/kg citalopram (intraperitoneally) caused a rapid and significant prolongation in clearance times for both 5-HT and DA, relative to controls (Fig. 1A,B) (5-HT, F(1,8) = 8.79, p = 0.018; DA, F(1,8) = 10.45; p = 0.012). In contrast, the DA clearance time in the DAT-rich dorsal striatum was unaffected by citalopram (Fig. 1C) (F(1,4) = 2.63, p = 0.18), underscoring the idea that DA clearance from the CA3 region is mediated by SERT, rather than by uptake by a catecholamine transporter or by degradation.
Figure 1.
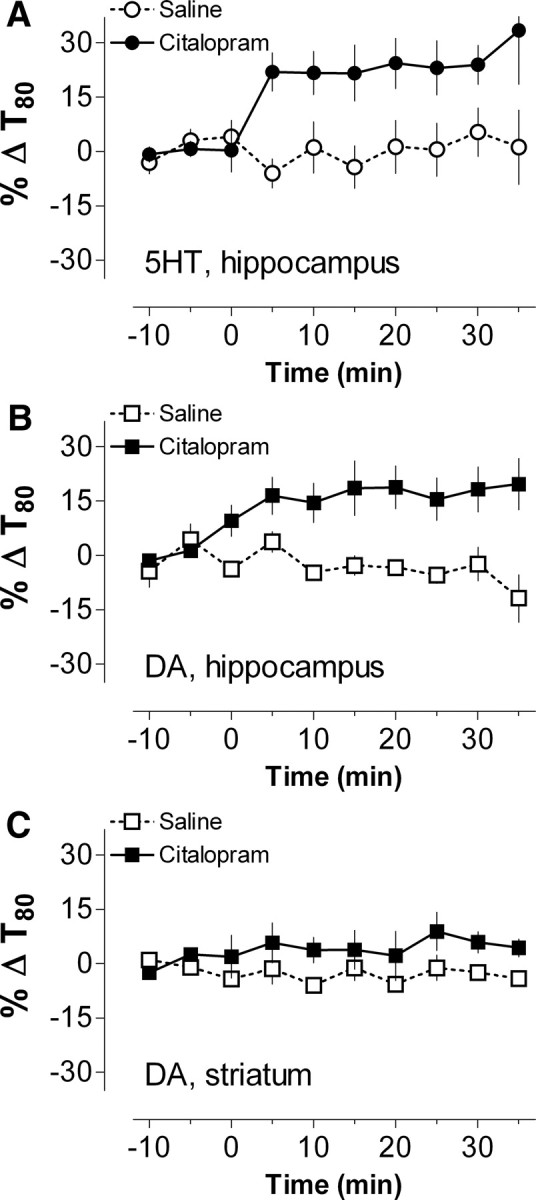
Blockade of SERT increases in vivo clearance time for exogenous 5-HT and DA in hippocampus but not in striatum of anesthetized rats. A–C, The in vivo clearance times (T80 values) for exogenously applied 5-HT (A) and DA (B) in the rat hippocampal CA3 region and DA in the dorsal striatum (C) were measured electrochemically. Systemic injection of the selective SERT inhibitor citalopram (10 mg/kg, i.p., at time 0; filled symbols) significantly prolongs clearance times relative to saline-injected controls (open symbols) for both 5-HT and DA in the CA3 region. In contrast, citalopram had no significant effect on DA clearance time in dorsal striatum where DA is cleared primarily by DAT (C). Data are presented as percentage change of the respective baseline values (−10 to −5 min) and represent mean ± SEM (n = 3–6 rats per group).
Uptake of DA by cloned hSERT
Because DA is a relatively weak inhibitor of 5-HT transport, most studies have overlooked the possibility that DA might serve as a substrate for SERT. Instead of looking at inhibition of 5-HT uptake, we decided to confirm that DA could be transported by hSERT and to characterize the properties of hSERT-mediated DA uptake. We used transiently transfected COS-7 cells as a model system to facilitate high expression of hSERT. In COS-7 cells, we observed that hSERT transports DA with an apparent affinity (Km) of 78 μm compared with 0.76 μm (Table 1) (t(14) = 10.79, p < 0.0001) for 5-HT transport. The Km for DA was approximately fourfold higher than its potency to inhibit 5-HT transport (360 μm for inhibition of 5-HT transport versus 84 μm for transport of DA) (Table 2) (t(10) = 16.45, p < 0.0001). In contrast, 5-HT showed a Km for transport similar to its IC50 for inhibition of DA transport (t(10) = 0.3089, p = 0.76). Although the Km for DA transport by hSERT was substantially higher than that for 5-HT transport, we noted that hSERT has a Vmax for DA transport that is four to five times higher than that for 5-HT transport (Fig. 2, Table 1) (t(7) = 4.058, p < 0.01). Consequently, hSERT transports DA faster than 5-HT at extracellular substrate concentrations above 25 μm. These results confirm that the cloned hSERT readily transports DA in COS-7 cells, a property we also observed using the Xenopus oocyte expression system (M. Sonders, unpublished observations).
Table 1.
Kinetic parameters for 5-HT and DA uptake kinetics by hSERT WT and hSERT Y95F mutant
| Construct | 5-HT |
DA |
||
|---|---|---|---|---|
| Km (μm) | Vmax (pmol · well−1 · min−1) | Km (μm) | Vmax (pmol · well−1 · min−1) | |
| hSERT WT | 0.76 ± 0.13 | 0.66 ± 0.15 | 78 ± 5.7*** | 2.7 ± 0.65 |
| hSERT Y95F | 0.57 ± 0.12 | 0.56 ± 0.10 | 12 ± 1.0 | 2.4 ± 0.52 |
Uptake was performed in transiently transfected COS-7 cells plated in 96-well plates and allowed to proceed for 10 min at room temperature. Values are expressed as mean ± SEM; n = 8. Significance levels are indicated for hSERT WT versus hSERT Y95F by the two-tailed t test (***p < 0.001).
Table 2.
IC50 values for inhibition of [3H]5-HT or [3H]DA uptake by hSERT WT and hSERT Y95F
| hSERT WT |
hSERT Y95F |
|||
|---|---|---|---|---|
| [3H]5-HT | [3H]DA | [3H]5-HT | [3H]DA | |
| 5-HT | 1300 ± 220 | 1400 ± 50 | 1100 ± 170 | 850 ± 40 |
| DA | 360,000 ± 21,000 | 84,000 ± 7000*** | 59,000 ± 3000 | 11,000 ± 1300*** |
| Cocaine | 8000 ± 2000 | 270 ± 20* | 1700 ± 300 | 77 ± 6.0** |
| MDMA | 4400 ± 840 | 2700 ± 330* | 3100 ± 430 | 940 ± 110*** |
| Paroxetine | 140 ± 42 | 25 ± 12* | 450 ± 120 | 85 ± 40* |
| Fluoxetine | 710 ± 200 | 71 ± 19** | 1200 ± 310 | 140 ± 37** |
| Citalopram | 89 ± 17 | 16 ± 5.6** | 1200 ± 240 | 92 ± 18*** |
| GBR 12935 | 16,000 ± 3000 | 17,000 ± 2700 | 9800 ± 1500 | 8300 ± 1200 |
All values are given as nm and represent mean value ± SEM; n = 3–9. Significance levels are indicated for [3H]5-HT versus [3H]DA by the two-tailed t test (*p < 0.05; **p < 0.01; ***p < 0.001).
Figure 2.

hSERT WT transports DA with a higher maximal velocity than 5-HT. COS-7 cells transfected in 48-well plates with hSERT WT were incubated with 200 nm [3H]5-HT or 200 nm [3H]DA and increasing concentrations of unlabeled 5-HT or DA for 10 min at 37°C, followed by washing and scintillation counting. Mean and SEM from a representative experiment run in triplicate are shown.
hSERT Y95F mutation has enhanced transport affinity for DA
In the course of characterizing a number of hSERT mutations near the putative substrate and sodium binding site, we observed that one mutant, Y95F, exhibited transport kinetics for 5-HT very similar to wild type but much more robust DA uptake activity. We decided to explore whether this mutant might have properties in common with the DA-transporting conformation of hSERT WT. As shown previously by others (Barker et al., 1998), hSERT Y95F displayed kinetics of uptake for 5-HT similar to hSERT WT (Table 1). However, a comparison of the kinetic parameters of DA uptake by hSERT WT with those of Y95F showed a clear difference in apparent substrate Km values: hSERT Y95F had a Km value of 12 μm for DA uptake, compared with 78 μm for DA uptake by hSERT WT (Table 1) (t(14) = 11.28, p < 0.0001). Nonetheless, like the hSERT WT, the Km for transporting DA for the Y95F mutant was significantly lower than its IC50 for DA inhibition of 5-HT uptake (11 μm vs 59 μm) (Table 2) (t(10) = 16.76, p < 0.0001). The apparent transport affinity of 5-HT was similar to the potency of 5-HT for inhibition of DA transport, as was the case for hSERT WT (t(10) = 0.8887, p = 0.40). The Vmax for DA transport in the hSERT Y95F mutant was similar to the WT and approximately four to five times higher than its Vmax for 5-HT transport (Table 1) (t(14) = 3.332, p < 0.01). Thus, at substrate concentrations above 3 μm, hSERT Y95F transports DA at a higher rate than 5-HT. Together, these data suggest that the Y95F mutant retains normal 5-HT uptake kinetics but displays a selective enhancement in its affinity for DA transport.
DA transport by hSERT WT and Y95F requires high Na+ and Cl−
hSERT belongs to the neurotransmitter sodium symporter (NSS) family, the members of which use the Na+ gradient across the cell membrane to drive substrate transport. Preliminary experiments indicated that DA transport by hSERT required higher Na+ concentrations for maximal activity than 5-HT transport. To examine the difference in cation dependency between 5-HT and DA transport in greater detail, we compared DA and 5-HT transport by hSERT under conditions in which Na+ concentrations were reduced in the uptake buffer and when Na+ was completely substituted by Li+. When Na+ was eliminated in the uptake buffer by substitution with 140 mm Li+, hSERT was still able to catalyze 20% of 5-HT uptake observed in the presence of Na+, whereas no DA transport was detected under these conditions (data not shown). Because it became evident that choline displayed a direct inhibitory action on DA uptake, we used N-methyl-D-glucamine (NMDG) as a monovalent cation substitute for Na+. In solutions using NMDG to substitute for varying concentrations of Na+, DA transport requires significantly higher Na+ concentrations to achieve a maximum velocity (EC50 ∼ 80–100 mm) than does 5-HT transport (EC50 ∼ 10–15 mm) (Fig. 3A,C) (t(12) = 3.638, p < 0.01). These results indicate that the DA transport process displays an ∼10-fold lower Na+ affinity when compared with 5-HT transport.
Figure 3.

Transport of DA requires higher concentrations of sodium and chloride than 5-HT transport. A–D, COS-7 cells transiently expressing hSERT WT or hSERT Y95F were incubated with increasing concentrations of sodium (A, C) or chloride (B, D) and 100–200 nm [3H]5-HT or 100–200 nm [3H]DA for 10 min at room temperature. NMDG chloride was substituted for NaCl in A and C to maintain the chloride concentration at 140 mm, and sodium gluconate was substituted for NaCl in B and D to maintain the sodium concentration at 140 mm and to keep overall osmolarity constant. Nonspecific uptake was determined by parallel uptake in mock-transfected cells and subtracted. Mean and SEM from representative experiments run in duplicate are shown.
Although Y95 resides in close spatial proximity to the putative Na+ binding site in the hSERT (Forrest et al., 2007), we observed only modest differences between hSERT WT and Y95F in the Na+ dependence of 5-HT uptake (Fig. 3A). Both transporters displayed apparent Na+ affinities in the 10–15 mm range, with hSERT Y95F consistently giving slightly higher values than WT. Y95F also displays a slightly higher EC50 for the Na+ dependence of DA transport than hSERT WT.
Like several of the other NSS family members, substrate transport by hSERT is chloride dependent, and thus we next examined the Cl− dependence of 5-HT and DA transport to determine whether the requirement for chloride differs between the two substrates. The apparent affinity for Cl− in 5-HT transport was 0.5–1 mm for both hSERT WT and Y95F (Fig. 3B), demonstrating that only a low concentration of Cl− is needed for 5-HT transport to proceed. When we performed the same assay for DA uptake, we saw a dramatic difference in Cl− affinity (Fig. 3D). The apparent affinities for Cl− measured for DA uptake were ∼100 mm, indicating an ∼100-fold lower apparent affinity for Cl− during DA transport, when compared with 5-HT transport (t(5) = 22.99, p < 0.0001). DA transport by the Y95F mutant displayed a similar 100-fold lower apparent affinity for Cl− (t(4) = 13.82, p < 0.001). Thus, DA transport requires significantly higher Na+ and Cl− concentrations to support transport than does 5-HT transport by hSERT. These results suggest that the ion binding sites in the DA-transporting hSERT conformation may have an altered geometry or accessibility when compared with 5-HT-transporting hSERT conformation. Two papers describing the Cl− binding site (Forrest et al., 2007; Zomot et al., 2007) placed the putative binding site in close spatial proximity to Y95. However, because both hSERT WT and Y95F have similar chloride dependence, these results indicate that an altered Cl− affinity does not explain the increase in apparent DA affinity in the hSERT Y95F mutant.
DA uptake by hSERT is more sensitive to inhibitors than 5-HT uptake by hSERT
An initial survey of SERT inhibitors indicated that they displayed differential potencies when tested against 5-HT versus DA uptake by hSERT. We decided to study this phenomenon more thoroughly by testing several drugs for their potency to inhibit 5-HT and DA uptake in parallel. The drugs we chose to test were the nonselective biogenic amine transporter inhibitor cocaine; the SSRIs paroxetine, fluoxetine, and citalopram; the DAT inhibitor GBR 12935; and the biogenic amine transporter substrate 3,4- methylenedioxymethamphetamine (MDMA). GBR 12935 was unique among the tested inhibitors in that this weak inhibitor showed no difference in potency between 5-HT and DA uptake by either hSERT WT (t(6) = 0.1769, p = 0.87) or Y95F (t(6) = 1.460, p = 0.19). The remaining inhibitors all displayed a significantly higher potency for inhibiting DA versus 5-HT uptake activity of hSERT. Cocaine, in particular, displayed much higher potency for inhibiting hSERT-mediated DA uptake than 5-HT uptake and was 20- to 30-fold more potent at inhibiting DA uptake (Fig. 4, Table 2) (WT: t(4) = 3.849, p < 0.05; Y95F: t(4) = 5.310, p < 0.01). The SSRIs showed a 3- to 13-fold higher potency for inhibiting DA uptake by hSERT, with citalopram showing the greatest and paroxetine showing the least difference in potencies. The substrate MDMA showed a twofold to threefold lower IC50 for inhibiting DA over 5-HT uptake (WT: t(10) = 1.840, p < 0.05; Y95F: t(10) = 4.901, p < 0.001). As demonstrated previously (Barker et al., 1998), hSERT Y95F displayed higher affinity than WT for cocaine and lower affinity than WT for citalopram. It is noteworthy that although hSERT WT and Y95F showed differences in affinities for these compounds, all compounds were still more potent at inhibiting DA transport when compared with 5-HT transport in the Y95F mutant.
Figure 4.

DA transport by hSERT is more sensitive to cocaine than 5-HT transport. Transiently transfected COS-7 cells were incubated for 10 min with increasing concentrations of cocaine, followed by the addition of [3H]5-HT or [3H]DA to give final concentrations of 100 nm [3H]5-HT or 200 nm [3H]DA, respectively. The uptake was allowed to continue for 10 min at room temperature before washing. Mean and SEM from representative experiments run in duplicate are shown.
5-HT inhibits DA uptake noncompetitively, but the two substrates display mutually exclusive binding at an external site
As shown in Table 2, DA is an efficient substrate but a poor inhibitor of 5-HT transport at hSERT (the DA IC50 was fivefold lower for blocking DA transport than for blocking 5-HT uptake), whereas the 5-HT IC50 values for inhibition of DA and 5-HT transport are not significantly different. This unexpected observation led us to investigate whether 5-HT and DA act competitively or noncompetitively to inhibit transport of the other substrate. In kinetic analysis of transport inhibition (Fig. 5A), 5-HT inhibited DA uptake by hSERT WT in a noncompetitive fashion. This was manifested in the observation that increasing concentrations of unlabeled 5-HT depressed the Vmax of DA uptake without affecting the Km value of DA. Conversely, we found that DA is a competitive inhibitor of 5-HT uptake by hSERT WT (Fig. 5B), an observation confirmed by Schild analyses (data not shown). These results contradict a simple transport model with a single substrate site where each substrate would compete for the same binding site. In such a model, one would expect to see competitive inhibition for all substrates and curves with no change in Vmax but changes in Km value after addition of increasing amounts of unlabeled heterologous substrate. One possible explanation for these results is that 5-HT interacts with a site in the hSERT that cannot be accessed by DA.
Figure 5.

5-HT is a noncompetitive inhibitor of DA uptake, but DA is a competitive inhibitor of 5-HT uptake. A, B, Eadie-Hofstee plots derived from data collected in COS-7 cells that were transiently transfected with plasmids containing hSERT WT and incubated with increasing concentrations of [3H]DA (A) or [3H]5-HT (B) and fixed concentrations of unlabeled 5-HT (A) or unlabeled DA (B) for 10 min at room temperature. Original data are shown in the inset. The parallel curves in A are typical for a noncompetitive inhibitor that displays an unchanged Km value but a decrease in the Vmax with the addition of an inhibitor, demonstrating that 5-HT inhibits DA uptake noncompetitively. In contrast, B shows that DA is a competitive inhibitor of 5-HT uptake, because it changes only the Km but not the Vmax. Nonspecific uptake was determined in parallel in mock-transfected cells and subtracted from specific uptake. C, D, Dixon plots from experiments in which stably transfected HEK-hSERT WT cells were incubated for 1 h on ice with 0.2 nm [125I]RTI-55 and increasing concentrations of unlabeled DA with or without a fixed concentration of unlabeled 5-HT (C) or with 0.5 nm [125I]RTI-55 and increasing concentrations of unlabeled 5-HT with or without a fixed concentration of unlabeled DA (D). Original data are shown in the inset. The parallel curves in C and D indicate that DA and 5-HT have mutually exclusive binding sites. Nonspecific uptake was determined by parallel [125I]RTI-55 binding in the presence of 100 μm unlabeled 5-HT and subtracted. Mean and SEM from representative experiments run in duplicate are shown.
To further explore the relationship between the binding sites for the two substrates, we tested whether 5-HT and DA share a common binding site in the hSERT. To do this, we used HEK-293 cells stably expressing hSERT WT and performed a whole-cell binding assay on ice with the high-affinity cocaine analog [125I]RTI-55 in the presence of a fixed concentration of 5-HT and increasing concentrations of DA or a fixed concentration of DA and increasing concentrations of 5-HT, respectively. We plotted the data in the form of Dixon plots (Fig. 5C,D). As evident from the plots, the lines are parallel indicating that the binding sites for 5-HT and DA are mutually exclusive and thus most likely share at least one common externally accessible binding site overlapping the RTI-55 binding site in the outward-facing hSERT (Segel, 1975).
Secondary site involvement in 5-HT and DA transport
The crystal structure of the bacterial leucine transporter LeuTAa, (Yamashita et al., 2005), which is an ortholog of the biogenic amine transporters, has provided a structural framework in which to consider the mechanism of substrate transport by SERT. Shi et al. (2008) recently proposed that LeuTAa catalyzes leucine–sodium symport by a mechanism in which substrate binding to a secondary site on the same protomer triggers intracellular release of sodium ions and substrate from a primary binding site further down the pathway. One residue in LeuTAa, I111, forms part of the extracellular secondary leucine binding site in LeuTAa, and mutation of I111 to cysteine renders the carrier incapable of binding substrate at the secondary site. When I179, the corresponding residue in hSERT, is mutated to cysteine within a parent construct devoid of reactive cysteines (C109A), 5-HT transport activity is reduced 30–50% and can be completely inactivated by the sulfhydryl-modifying reagent MTSET (Chen et al., 1997). To explore whether this putative second substrate binding site might differentiate between 5-HT and DA, we next examined whether the putative secondary site might be a site accessible only to 5-HT and not to DA. In this case, MTS modification of I179C should eliminate 5-HT transport, as shown previously, but retain DA transport. Because we found hSERT Y95F to be virtually identical to hSERT WT in all measured features except apparent DA transport affinity and inhibitor affinity, we subsequently took advantage of Y95F and used it in concert with other hSERT mutants to elevate DA uptake activity at low DA concentrations.
Introducing the I179C mutation into a Y95F/C109A background resulted in a decrease in 5-HT uptake (∼50%). Treatment of the Y95F/C109A mutant with 10 mm MTSET decreased 5-HT uptake activity (>90%) after 10 min. Although DA was still able to inhibit 5-HT uptake in inhibition assays (IC50 twofold to threefold higher than Y95F/C109A), the Y95F/C109A/I179C mutant displayed almost nondetectable transport of DA (<2%) before or after MTSET treatment. These results suggest that in the I179C mutant, DA still binds and inhibits substrate translocation at the primary binding site and that the putative secondary site plays a significant role in DA translocation.
5-HT and DA induce similar 5-HT efflux, but little or no DA efflux, in preloaded COS-7 cells
It is well established that SERT can mediate efflux of intracellular substrates during inward transport of extracellular substrates. We next considered the relationship of 5-HT and DA with respect to substrate efflux/exchange by hSERT. Using COS-7 cells transfected with hSERT WT and preloaded to equivalent concentrations with either [3H]5-HT or [3H]DA (1.00 ± 0.19 pmol of [3H]5-HT per well; 0.92 ± 0.19 pmol [3H]DA per well), we studied efflux initiated by the addition of a high concentration of 5-HT or DA to the extracellular medium (Fig. 6). We found that for cells preloaded with [3H]5-HT, there was substantial and comparable efflux using either 5-HT or DA as the substrate to initiate efflux (Fig. 6A). However, when cells were preloaded with [3H]DA, we observed no significant efflux of [3H]DA in the presence of 5-HT or DA (Fig. 6B). Experiments with hSERT Y95F gave results similar to the ones described for hSERT WT (data not shown). The hSERT substrate transport cycle is thought to be composed of multiple steps with binding and unbinding of ions and substrate as well as larger conformational changes of the hSERT protein to open and close “gates” (Rudnick, 2006). That [3H]DA is effluxed to a much lower degree with either 5-HT or DA added extracellularly suggests that either hSERT has a very low capacity to efflux DA or that the affinity for DA at the internal binding site is much lower than for 5-HT.
Figure 6.

5-HT and DA added extracellularly cause efflux of preloaded [3H]5-HT but little or no efflux of preloaded [3H]DA. COS-7 cells transfected with hSERT WT were preloaded with [3H]5-HT (A) or [3H]DA (B) for 45 min at room temperature followed by washes every 5 min for 45 min to give similar concentration of intracellular substrate. Cells were then incubated with PBS alone or with PBS containing 20 μm 5-HT or 2 mm DA. At specified time points, half of the supernatant was collected and prepared for scintillation counting. Released substrate is expressed as the radioactivity in the complete supernatant fraction as a percentage of the radioactivity present in the cells before efflux was initiated. The mean and SEM of six wells are plotted for efflux. Significance levels are indicated for control versus 5-HT or DA by the two-tailed t test (***p < 0.001).
hSERT spends more time in an outward-facing conformation when transporting DA than when transporting 5-HT
If hSERT has a lower internal affinity for DA than for 5-HT and this difference significantly impacts the reversal rate of the transporter, it would imply that hSERT engaged in DA transport would spend more time in an outward-facing conformation compared with a transporter engaged in 5-HT transport. We next sought to measure whether this is indeed the case. In studies using SERT cysteine mutants and membrane binding assays, 5-HT and cocaine have previously been shown to shift the equilibrium of SERT to inward- or outward-facing conformations, respectively (Zhang and Rudnick, 2006). Recently, Torres-Altoro et al. (2010) described an hSERT mutant, Q332C, the transport activity of which was susceptible to MTS reagents. Moreover, coincubation with 5-HT hindered MTS inactivation of transport by Q332C whereas cocaine coincubation facilitated it, suggesting that these ligands differentially alter the MTS accessibility of the residue and conformation of the protein (Torres-Altoro et al., 2010). Thus, we were interested to test whether DA changes SERT conformation in a manner different from 5-HT. To this end, we constructed the Y95F/C109A/Q332C mutant and assayed for MTSET inactivation of hSERT in the absence of any ligand or in the presence of 40 μm 5-HT, 4 mm DA, 40 μm cocaine, or 160 μm ibogaine. Ibogaine was included because it has previously been demonstrated to shift the equilibrium of SERT toward an inward-facing conformation (Jacobs et al., 2007). When expressed in cells, the C109A mutant is not inactivated by MTSET in buffer alone or with any ligand (data not shown). As shown in Figure 7, we confirm the previous observation that MTSET inactivation of hSERT Q332C is protected by 5-HT (t(12) = 6.247, p < 0.0001) and potentiated by cocaine (t(8) = 5.652, p < 0.001). We also observed protection from MTSET inactivation by ibogaine (t(8) = 5.652, p < 0.001), corroborating the idea that protection or potentiation of Q332C modification by MTSET reflects a shift in the conformational equilibrium of hSERT to more inward-facing or outward-facing conformations, respectively. DA, on the other hand, gives only a very small protection to MTSET inactivation compared with PBS (t(12) = 2.404, p < 0.05) and offers significantly less protection than 5-HT (t(12) = 5.146, p < 0.001), indicating that hSERT indeed spends less time in an inward-facing conformation when transporting DA than during 5-HT transport.
Figure 7.

Effect of substrates and ligands on reactivity of hSERT Y95F/C109A/Q332C mutant with MTSET. A, COS-7 cells transfected with hSERT Y95F/C109A/Q332C were incubated for 10 min with the indicated ligand or buffer alone followed by addition of increasing concentrations of MTSET for 10 min. Cells were then washed extensively, and uptake of [3H]5-HT was measured as described in Materials and Methods. All steps were performed at room temperature. Mean and SEM from a representative experiment run in duplicate is shown. B, Average and SEM of the rate of reactivity from at least nine individual experiments. Nonspecific uptake was determined by using mock-transfected cells and subtracted. Significance levels are indicated versus control, unless otherwise indicated, by the two-tailed paired t test (*p < 0.05; ***p < 0.001).
Subunit interactions differ between 5-HT and DA uptake
Although some of the differences (increased Vmax, different inhibitor sensitivity, mutually exclusive binding, capacity for efflux) between DA and 5-HT transport by hSERT can be explained by differences in the dynamic equilibrium conformations of the carrier in the presence of each substrate, it remains difficult to reconcile the noncompetitive interactions between substrates during transport without postulating different binding sites within a protomer. One possibility is that the noncompetitive relationship could be related to differences in the cooperative interactions between subunits during DA and 5-HT transport processes, and thus we next tried to address this question. By cotransfecting different ratios of MTS-sensitive and MTS-insensitive SERT constructs in cells and assaying for MTS-insensitive uptake, Kilic and Rudnick (2000) demonstrated that transport activity did not decline linearly as the concentration of MTS-sensitive SERT increased but instead appeared less sensitive to inactivation than predicted if the two protomer forms function independently. This observation implies that inactivation of one subunit within an hSERT multimer may facilitate function of a remaining subunit and that subunit interactions have a significant impact on 5-HT transport. To examine whether the transport of DA is similarly influenced by cooperative effects between subunits, we examined the effects of cotransfection of different ratios of hSERT and either an MTS-sensitive (Fig. 8A,B) or a nonfunctional hSERT mutant (Fig. 8C,D) on DA transport. As shown previously, substitution of a cysteine residue for I172 in TM3 in a C109A hSERT MTS-insensitive background generates a transporter that shows no 5-HT transport activity after modification with MTSEA (Chen et al., 1997). We cotransfected the Y95F/C109A/I172C mutant and MTS-insensitive Y95F/C109A hSERT mutant constructs in different ratios in COS-7 cells and assayed for MTSEA-insensitive 5-HT and DA uptake. As reported by Kilic and Rudnick (2000), we also found a nonlinear relationship between subunit inactivation and inhibition of 5-HT transport. In the case of 5-HT transport by hSERT, assuming random assembly of protomers into multimers, transport activity remained higher than that predicted by the ratio of sensitive-to-insensitive transporters (Fig. 8A, compare data points with dotted line that models independent transport by protomers). On the other hand, for DA transport, our results fit better with a model where protomers behave independently in DA transport and the reduction in DA transport is proportional to the number of MTS-sensitive protomers in a multimeric transporter (Fig. 8B, compare data points with dotted line). The Y95F/C109A/I172C mutant had a twofold lower Km for DA than the Y95F/C109A, which is the basis for the increased DA uptake in the control (Fig. 8B).
Figure 8.

Absence of cooperative interactions between subunits during DA but not 5-HT transport. A, B, COS-7 cells transiently transfected with variable ratios of MTS-insensitive hSERT Y95F/C109A and MTS-sensitive hSERT Y95F/C109A/I172C were treated with 0.25 mm MTSEA for 10 min, washed, and assayed for uptake of either 150 nm [3H]5-HT (A) or 300 nm [3H]DA (B) for 10 min. C, D, COS-7 cells transiently transfected with variable ratios of hSERT Y95F and transport-inactive hSERT Y95F/D98G were assayed for uptake of either 150 nm [3H]5-HT (C) or 300 nm [3H]DA (D) for 10 min. All steps were performed at room temperature. Shown is the average and SEM of six independent experiments, each performed in triplicate. Values from each experiment were normalized to the activity of MTSEA-treated 100% hSERT Y95F/C109A (A, B) or 100% Y95F (C, D). The dotted line shows the activity expected if the activity of a multimer is proportional to the fraction of noninactivated protomers.
As an alternative approach that does not entail MTS modification, we used the hSERT D98G mutant, previously shown to be expressed at the membrane surface at levels equivalent to the hSERT WT but inactive at 5-HT transport (Barker et al., 1999). We first demonstrated that hSERT D98G displays no DA uptake activity, and then we constructed the transport-inactive Y95F/D98G mutant and cotransfected it with hSERT Y95F in different ratios in COS-7 cells and assayed for 5-HT and DA transport. Our results from this experiment (Fig. 8C,D) are similar to the results obtained with the MTSEA-sensitive mutant and also suggest that DA is transported in hSERT protomers that function independently, whereas 5-HT transport depends on cooperative interactions between subunits.
Discussion
Studies have shown that DA is a poor inhibitor of 5-HT transport by SERT in rodent brain, as well as by the cloned SERT (Azzaro and Rutledge, 1973; Hoffman et al., 1991; Ramamoorthy et al., 1993; Corey et al., 1994; Barker et al., 1999). Nevertheless, DA accumulation by a process with the pharmacological sensitivity of SERT has been observed in native brain tissues (Berger and Glowinski, 1978; Kelly et al., 1985) and in hSERT-expressing cell lines (Saldana and Barker, 2004), although the mechanism underlying these seemingly discrepant results has never been examined.
Conventional alternating access models for ion-coupled cotransport predict that substrate and ion binding events drive changes in transporter conformation, which lead to coordinated closing and opening of external and internal gates and access of substrates to the internal medium. In some carriers, including SERT, binding of additional counter-transported ions reorients the carrier to the outward facing form (Rudnick, 2006). Although we found that 5-HT and DA have mutually exclusive extracellular binding sites on hSERT, our results do not agree with a simple cotransport model. First, uptake assays for 5-HT and DA show that for DA, the apparent affinity for uptake is significantly higher than its potency for inhibiting 5-HT uptake (Km < IC50). Second, the two substrates do not show reciprocal competitive inhibition for transport. We found that 5-HT is a noncompetitive inhibitor of DA uptake, whereas DA inhibited 5-HT uptake competitively. The observation that 5-HT inhibits DA transport noncompetitively implies that there is a distinct binding site for the noncompetitive inhibitor that is separate from the primary binding site for DA in hSERT. One explanation for the noncompetitive inhibition of DA transport by 5-HT is that 5-HT has an additional binding site, not accessed by DA, either located on the same protomer or, alternatively, with the two sites interacting allosterically on different protomers.
The structure of LeuTAa shows substrate and two sodium ions bound in the middle of the transporter molecule near helices proposed to shift and allow alternating access (Yamashita et al., 2005). Recent work described a secondary binding site in LeuTAa that, when occupied, triggers the intracellular release of substrate and Na+ bound at the primary site (Shi et al., 2008). We created a mutant (Y95F/C109A/I179C) disrupting the putative secondary site and found that it transported 5-HT but not DA, even though DA could still inhibit 5-HT transport. These observations suggest that the putative secondary site has a critical function in DA transport. The fact that DA still inhibits 5-HT transport with similar affinity to that observed with WT hSERT, even though DA is not transported in theY95F/C109A/I179C mutant, suggests that DA inhibits 5-HT transport at the primary binding site. It also suggests that the secondary substrate site observed in LeuTAa cannot account for the noncompetitive inhibition of DA transport by 5-HT. The disruption of DA transport by a mutation in this secondary site could result from either a direct effect of destroying the secondary binding site or an indirect effect that prevents conformational changes in hSERT required for DA transport.
Tyrosine 95 is part of the predicted primary substrate binding site (Celik et al., 2008). Our findings that the Y95F mutant has an increased apparent affinity for DA and an increased potency for inhibition of 5-HT transport by DA supports the notion that DA inhibits 5-HT transport at the primary binding site. The requirement for higher Na+ and Cl− concentrations to drive DA transport suggests that a local change in the ion binding sites takes place during the DA transport cycle that does not occur during 5-HT transport. It has been shown that SERT, as well as NET and DAT, is able to bind substrate in the absence of Na+ and Cl− (Humphreys et al., 1994; Sonders et al., 1997; Schwartz et al., 2003). Moreover, there is no well established order for ion and substrate binding to SERT, and it is possible that DA binds to SERT before Na+ and Cl−. Because the ion binding sites are in close proximity to the substrate binding site, the initial binding of DA could perturb the ion binding sites leading to lower ion affinities.
The observation that all tested inhibitors, except GBR12935, exhibit higher potency for inhibiting DA transport than 5-HT transport implies that there may be conformationally distinct sites for antagonists on a single SERT protein or within a functional oligomeric complex, and these may differentially inhibit DA and 5-HT uptake. It is also possible that antagonists could stabilize several conformations of SERT that have distinct capacity to transport DA and 5-HT. If SERT forms an oligomeric complex in which substrates and antagonists can bind to different protomers, this too could afford an explanation for the different antagonist potencies. Regardless of the exact mechanism, it seems likely that the effects we observe for DA transport with respect to ion and inhibitor sensitivities reflects a distinct conformation required for DA transport.
We also explored the idea that the binding sites for 5-HT in separate protomers interact in a cooperative manner and tested whether this interaction was preserved during DA transport. Interestingly, as shown by others (Kilic and Rudnick, 2000), 5-HT transport in hSERT multimers is less sensitive to biochemical inactivation of individual protomers than would be anticipated if all protomers operate independently. One interpretation of this result is that transport activity in one protomer inhibits simultaneous 5-HT transport in other protomers and that when one protomer is inactivated, the remaining protomers are more active. In contrast, we found that inactivation of protomers directly correlated with attenuation of DA transport. These differences between 5-HT and DA transport could contribute to the differences observed in maximal transport rates of 5-HT and DA. If SERT protomers transport DA independently, but 5-HT is only transported by a multimer, our Vmax results suggest that the 5-HT-carrying protomer may inhibit transport by the other protomers in a multimeric complex. We also hypothesize that the kinetic differences in substrate inhibition reflect allosteric interactions between subunits that influence steps in the transport cycle other than initial extracellular substrate binding events.
Figure 9 depicts models of DA and 5-HT transport by hSERT. We propose that the transformation of the inward-facing ion- and substrate-bound transporter to the empty outward-facing transporter is faster after hSERT has transported DA than 5-HT. As demonstrated by MTSET inactivation of the Q322C mutant transporter, this leads to a greater proportion of hSERT facing outward during DA transport than during 5-HT transport. Additionally, in our model, hSERT in an inward-facing conformation has a low affinity for DA and ions. This feature of the hSERT fits well with the observation that cells preloaded with [3H]DA display no appreciable efflux after extracellular application of either 5-HT or DA. Our kinetic studies also clearly show that in parallel assays, the Vmax (and thus the turnover number) of hSERT is fourfold higher for DA than for 5-HT. If the rate-limiting step of transport for hSERT is carrier reorientation, as proposed for many transporters [e.g., DAT (Erreger et al., 2008)], these data would be consistent with a faster reorientation after DA import. We hypothesize that hSERT may reorient with different velocities after 5-HT and DA import because it does so from distinct conformations, perhaps related to differences in the mechanism of substrate and ion dissociation or the potassium dependence of reorientation. This idea is also consistent with results from the transporter complementation experiments that impute a role for hSERT multimers only in 5-HT transport. The higher potency of individual pharmacological inhibitors in blocking DA versus 5-HT transport by hSERT is also best explained if different conformations of hSERT carry the two substrates.
Figure 9.

Models of 5-HT and DA transport by the hSERT. A simplified drawing of state diagrams summarizing the basic features and favored equilibria of 5-HT (left) and DA (right) transport by SERT. Only chloride ion, sodium ion, and substrate is shown. The star shape denotes the accessibility of the reactive cysteine (Q332C) to MTSET modification. Below each diagram is a drawing of the transporter itself illustrating subunit–subunit interactions.
We also confirmed that SERT can take up DA in vivo in the CA3 region of the hippocampus of anesthetized rats, but how significant a role SERT plays in DA clearance will vary between brain regions and physiological states. Regions with low DAT expression that receive dual innervation with DA- and 5-HT-containing fibers in close apposition may be most relevant, particularly when SERTs are close to DA release sites where DA concentrations are higher. A number of brain areas that express SERT (Sur et al., 1996) and receive DA projections are known to express comparatively little DAT, such as the prelimbic medial prefrontal cortex (Ciliax et al., 1995; Sesack et al., 1998), the nucleus accumbens shell (Van Bockstaele and Pickel, 1993; Nirenberg et al., 1997), and the basolateral amygdala (Revay et al., 1996), all regions implicated in a diverse range of motivated behaviors (Kalivas et al., 1999). In these locations, the relative absence of DATs may facilitate DA diffusion to distant receptors and supports a role for noncognate transporters such as SERT and NET in regulating paracrine or “volume” neurotransmission (Sesack et al., 1998; Zoli et al., 1999). Therapy with antidepressants that inhibit SERT may increase DA locally in these areas. Our findings suggest that SERT could play an enhanced role in clearing DA in DAT knock-out mice and provide mechanistic insights as to why DAT/SERT double knock-outs were needed to abolish cocaine reward (Sora et al., 2001).
Footnotes
This work was supported by Howard Hughes Medical Institute and National Institutes of Health Grants P01 DA12408 and R37 DA07595 (S.G.A.) and R01 DA004216 and K05 DA015050 (N.R.Z.). Citalopram was generously provided by J. Hyttel (H. Lundbeck A/S), and MDMA was obtained from the NIDA Drug Supply System (K. Gormley). We are very grateful to Dr. Peter Pörzgen for cloning the hSERT cDNA from human placenta and for his helpful comments. We also thank Dr. Geoffrey Murdoch, Dr. Fusun Kilic, and our colleagues in the Amara group for their thoughtful input and comments.
References
- Azzaro AJ, Rutledge CO. Selectivity of release of norepinephrine, dopamine and 5-hydroxytryptamine by amphetamine in various regions of rat brain. Biochem Pharmacol. 1973;22:2801–2813. doi: 10.1016/0006-2952(73)90147-0. [DOI] [PubMed] [Google Scholar]
- Barker EL, Perlman MA, Adkins EM, Houlihan WJ, Pristupa ZB, Niznik HB, Blakely RD. High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis. An aromatic residue in transmembrane domain I dictates species-selective recognition of citalopram and mazindol. J Biol Chem. 1998;273:19459–19468. doi: 10.1074/jbc.273.31.19459. [DOI] [PubMed] [Google Scholar]
- Barker EL, Moore KR, Rakhshan F, Blakely RD. Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J Neurosci. 1999;19:4705–4717. doi: 10.1523/JNEUROSCI.19-12-04705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B, Glowinski J. Dopamine uptake in serotoninergic terminals in vitro: a valuable tool for the histochemical differentiation of catecholaminergic and serotoninergic terminals in rat cerebral structures. Brain Res. 1978;147:29–45. doi: 10.1016/0006-8993(78)90770-9. [DOI] [PubMed] [Google Scholar]
- Bunin MA, Wightman RM. Paracrine neurotransmission in the CNS: involvement of 5-HT. Trends Neurosci. 1999;22:377–382. doi: 10.1016/s0166-2236(99)01410-1. [DOI] [PubMed] [Google Scholar]
- Callaghan PD, Irvine RJ, Daws LC. Differences in the in vivo dynamics of neurotransmitter release and serotonin uptake after acute para-methoxyamphetamine and 3,4-methylenedioxymethamphetamine revealed by chronoamperometry. Neurochem Int. 2005;47:350–361. doi: 10.1016/j.neuint.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Carboni E, Silvagni A. Dopamine reuptake by norepinephrine neurons: exception or rule? Crit Rev Neurobiol. 2004;16:121–128. doi: 10.1615/critrevneurobiol.v16.i12.130. [DOI] [PubMed] [Google Scholar]
- Cass WA, Gerhardt GA. In vivo assessment of dopamine uptake in rat medial prefrontal cortex: comparison with dorsal striatum and nucleus accumbens. J Neurochem. 1995;65:201–207. doi: 10.1046/j.1471-4159.1995.65010201.x. [DOI] [PubMed] [Google Scholar]
- Cass WA, Zahniser NR, Flach KA, Gerhardt GA. Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem. 1993;61:2269–2278. doi: 10.1111/j.1471-4159.1993.tb07469.x. [DOI] [PubMed] [Google Scholar]
- Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, Wiborg O, Schiott B. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc. 2008;130:3853–3865. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- Chen JG, Sachpatzidis A, Rudnick G. The third transmembrane domain of the serotonin transporter contains residues associated with substrate and cocaine binding. J Biol Chem. 1997;272:28321–28327. doi: 10.1074/jbc.272.45.28321. [DOI] [PubMed] [Google Scholar]
- Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HB, Levey AI. The dopamine transporter: immunochemical characterization and localization in brain. J Neurosci. 1995;15:1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey JL, Quick MW, Davidson N, Lester HA, Guastella J. A cocaine-sensitive Drosophila serotonin transporter: cloning, expression, and electrophysiological characterization. Proc Natl Acad Sci U S A. 1994;91:1188–1192. doi: 10.1073/pnas.91.3.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws LC. Unfaithful neurotransmitter transporters: focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol Ther. 2009;121:89–99. doi: 10.1016/j.pharmthera.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws LC, Toney GM, Gerhardt GA, Frazer A. In vivo chronoamperometric measures of extracellular serotonin clearance in rat dorsal hippocampus: contribution of serotonin and norepinephrine transporters. J Pharmacol Exp Ther. 1998;286:967–976. [PubMed] [Google Scholar]
- Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J Neurosci. 2008;28:976–989. doi: 10.1523/JNEUROSCI.2796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Tavoulari S, Zhang YW, Rudnick G, Honig B. Identification of a chloride ion binding site in Na+/Cl -dependent transporters. Proc Natl Acad Sci U S A. 2007;104:12761–12766. doi: 10.1073/pnas.0705600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulley J, Larson G, Zahniser N. Using high-speed chronoamperometry with local dopamine application to assess dopamine transporter function. In: Michael AC, Borland LM, editors. Electrochemical methods in neuroscience. Boca Raton, FL: CRC; 2006. pp. 83–102. [PubMed] [Google Scholar]
- Hoffman BJ, Mezey E, Brownstein MJ. Cloning of a serotonin transporter affected by antidepressants. Science. 1991;254:579–580. doi: 10.1126/science.1948036. [DOI] [PubMed] [Google Scholar]
- Hoffman BJ, Hansson SR, Mezey E, Palkovits M. Localization and dynamic regulation of biogenic amine transporters in the mammalian central nervous system. Front Neuroendocrinol. 1998;19:187–231. doi: 10.1006/frne.1998.0168. [DOI] [PubMed] [Google Scholar]
- Humphreys CJ, Wall SC, Rudnick G. Ligand binding to the serotonin transporter: equilibria, kinetics, and ion dependence. Biochemistry. 1994;33:9118–9125. doi: 10.1021/bi00197a014. [DOI] [PubMed] [Google Scholar]
- Jackson BP, Wightman RM. Dynamics of 5-hydroxytryptamine released from dopamine neurons in the caudate putamen of the rat. Brain Res. 1995;674:163–166. doi: 10.1016/0006-8993(95)00019-m. [DOI] [PubMed] [Google Scholar]
- Jacobs MT, Zhang YW, Campbell SD, Rudnick G. Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J Biol Chem. 2007;282:29441–29447. doi: 10.1074/jbc.M704456200. [DOI] [PubMed] [Google Scholar]
- Jansson A, Lippoldt A, Mazel T, Bartfai T, Ogren SO, Sykova E, Agnati LF, Fuxe K. Long distance signalling in volume transmission. Focus on clearance mechanisms. Prog Brain Res. 2000;125:399–413. doi: 10.1016/S0079-6123(00)25028-0. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Romanides A. Involvement of the pallidal-thalamocortical circuit in adaptive behavior. Ann N Y Acad Sci. 1999;877:64–70. doi: 10.1111/j.1749-6632.1999.tb09261.x. [DOI] [PubMed] [Google Scholar]
- Kannari K, Shen H, Arai A, Tomiyama M, Baba M. Reuptake of L-DOPA-derived extracellular dopamine in the striatum with dopaminergic denervation via serotonin transporters. Neurosci Lett. 2006;402:62–65. doi: 10.1016/j.neulet.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Kelly E, Jenner P, Marsden CD. Evidence that [3H]dopamine is taken up and released from nondopaminergic nerve terminals in the rat substantia nigra in vitro. J Neurochem. 1985;45:137–144. doi: 10.1111/j.1471-4159.1985.tb05485.x. [DOI] [PubMed] [Google Scholar]
- Kilic F, Rudnick G. Oligomerization of serotonin transporter and its functional consequences. Proc Natl Acad Sci U S A. 2000;97:3106–3111. doi: 10.1073/pnas.060408997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthman J, Friedemann MN, Hoffer BJ, Gerhardt GA. In vivo electrochemical measurements of serotonin clearance in rat striatum: effects of neonatal 6-hydroxydopamine-induced serotonin hyperinnervation and serotonin uptake inhibitors. J Neural Transm. 1997;104:379–397. doi: 10.1007/BF01277658. [DOI] [PubMed] [Google Scholar]
- Mengual E, Pickel VM. Ultrastructural immunocytochemical localization of the dopamine D2 receptor and tyrosine hydroxylase in the rat ventral pallidum. Synapse. 2002;43:151–162. doi: 10.1002/syn.10033. [DOI] [PubMed] [Google Scholar]
- Mossner R, Simantov R, Marx A, Lesch KP, Seif I. Aberrant accumulation of serotonin in dopaminergic neurons. Neurosci Lett. 2006;401:49–54. doi: 10.1016/j.neulet.2006.02.081. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Pohorille A, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. The dopamine transporter: comparative ultrastructure of dopaminergic axons in limbic and motor compartments of the nucleus accumbens. J Neurosci. 1997;17:6899–6907. doi: 10.1523/JNEUROSCI.17-18-06899.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrholm SD, Horton DB, Dwoskin LP. The promiscuity of the dopamine transporter: implications for the kinetic analysis of [3H]serotonin uptake in rat hippocampal and striatal synaptosomes. Neuropharmacology. 2007;53:982–989. doi: 10.1016/j.neuropharm.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther. 1997;283:1305–1322. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. London: Academic; 2007. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, Ganapathy V, Blakely RD. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci U S A. 1993;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revay R, Vaughan R, Grant S, Kuhar MJ. Dopamine transporter immunohistochemistry in median eminence, amygdala, and other areas of the rat brain. Synapse. 1996;22:93–99. doi: 10.1002/(SICI)1098-2396(199602)22:2<93::AID-SYN1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Rudnick G. Chemical modification strategies for structure-function studies in transmembrane transporters. In: Quick MW, editor. Transmembrane transporters. Hoboken, NJ: Wiley; 2003. pp. 125–141. [Google Scholar]
- Rudnick G. Serotonin transporters—structure and function. J Membr Biol. 2006;213:101–110. doi: 10.1007/s00232-006-0878-4. [DOI] [PubMed] [Google Scholar]
- Saldana SN, Barker EL. Temperature and 3,4-methylenedioxymethamphetamine alter human serotonin transporter-mediated dopamine uptake. Neurosci Lett. 2004;354:209–212. doi: 10.1016/j.neulet.2003.10.032. [DOI] [PubMed] [Google Scholar]
- Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD. Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol. 2000;420:211–232. [PubMed] [Google Scholar]
- Schwartz JW, Blakely RD, DeFelice LJ. Binding and transport in norepinephrine transporters. Real-time, spatially resolved analysis in single cells using a fluorescent substrate. J Biol Chem. 2003;278:9768–9777. doi: 10.1074/jbc.M209824200. [DOI] [PubMed] [Google Scholar]
- Segel I. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. New York: Wiley; 1975. [Google Scholar]
- Sesack SR, Hawrylak VA, Matus C, Guido MA, Levey AI. Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci. 1998;18:2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sora I, Hall FS, Andrews AM, Itokawa M, Li XF, Wei HB, Wichems C, Lesch KP, Murphy DL, Uhl GR. Molecular mechanisms of cocaine reward: combined dopamine and serotonin transporter knockouts eliminate cocaine place preference. Proc Natl Acad Sci U S A. 2001;98:5300–5305. doi: 10.1073/pnas.091039298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamford JA, Kruk ZL, Millar J. Striatal dopamine terminals release serotonin after 5-HTP pretreatment: in vivo voltammetric data. Brain Res. 1990;515:173–180. doi: 10.1016/0006-8993(90)90593-z. [DOI] [PubMed] [Google Scholar]
- Sur C, Betz H, Schloss P. Immunocytochemical detection of the serotonin transporter in rat brain. Neuroscience. 1996;73:217–231. doi: 10.1016/0306-4522(96)00030-9. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Zhou FC. Differential polarization of serotonin transporters in axons versus soma-dendrites: an immunogold electron microscopy study. Neuroscience. 1999;94:821–830. doi: 10.1016/s0306-4522(99)00373-5. [DOI] [PubMed] [Google Scholar]
- Tatsumi M, Groshan K, Blakely RD, Richelson E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol. 1997;340:249–258. doi: 10.1016/s0014-2999(97)01393-9. [DOI] [PubMed] [Google Scholar]
- Torres-Altoro MI, Kuntz CP, Nichols DE, Barker EL. Structural analysis of the extracellular entrance to the serotonin transporter permeation pathway. J Biol Chem. 2010;285:15369–15379. doi: 10.1074/jbc.M109.088138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Pickel VM. Ultrastructure of serotonin-immunoreactive terminals in the core and shell of the rat nucleus accumbens: cellular substrates for interactions with catecholamine afferents. J Comp Neurol. 1993;334:603–617. doi: 10.1002/cne.903340408. [DOI] [PubMed] [Google Scholar]
- Verney C, Baulac M, Berger B, Alvarez C, Vigny A, Helle KB. Morphological evidence for a dopaminergic terminal field in the hippocampal formation of young and adult rat. Neuroscience. 1985;14:1039–1052. doi: 10.1016/0306-4522(85)90275-1. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zahniser NR, Larson GA, Gerhardt GA. In vivo dopamine clearance rate in rat striatum: regulation by extracellular dopamine concentration and dopamine transporter inhibitors. J Pharmacol Exp Ther. 1999;289:266–277. [PubMed] [Google Scholar]
- Zhang YW, Rudnick G. The cytoplasmic substrate permeation pathway of serotonin transporter. J Biol Chem. 2006;281:36213–36220. doi: 10.1074/jbc.M605468200. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Lesch KP, Murphy DL. Serotonin uptake into dopamine neurons via dopamine transporters: a compensatory alternative. Brain Res. 2002;942:109–119. doi: 10.1016/s0006-8993(02)02709-9. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Salas R, Zhang L, De Biasi M, Dani JA. Corelease of dopamine and serotonin from striatal dopamine terminals. Neuron. 2005;46:65–74. doi: 10.1016/j.neuron.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Zoli M, Jansson A, Sykova E, Agnati LF, Fuxe K. Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol Sci. 1999;20:142–150. doi: 10.1016/s0165-6147(99)01343-7. [DOI] [PubMed] [Google Scholar]
- Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI. Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature. 2007;449:726–730. doi: 10.1038/nature06133. [DOI] [PubMed] [Google Scholar]