

Abstract
Prospective studies of the persistence of human papilloma-virus (HPV) variants are rare and typically small. We sequenced HPV-16 variants in longitudinal pairs of specimens from 86 women enrolled in the ASCUS-LSIL Triage Study. A change of variants was identified in 4 women (4.7% [95% confidence interval, 1.3%-11.5%]). Among women with intervening HPV results (n = 60), a variant switch occurred in 2 of 11 who had evidence of intervening negativity for HPV-16, compared with 1 of 49 who consistently tested positive (P = .11). These results suggest the possibility that rare misclassification of transient infections as persistent infections occurs in natural history studies of type-specific HPV infections.
Studies of the natural history of human papillomavirus (HPV) infection define viral persistence at a type-specific level—that is, as the same genotype of HPV DNA detected during a certain time period. Intervening negativity is usually assumed to be the result of fluctuation in viral DNA load to below a detectable threshold [1]. In practice, it is difficult to distinguish type-specific persistence from recurrent infection.
In light of the intratypic diversity of the HPV genome [2], the persistence of type-specific HPV infection may be better defined by analyses of nucleotide alterations in viral isolates [3]. For any given type of HPV, isolates that differ by <2% in the DNA sequence of the L1 (conserved major capsid) gene are designated as variants. Because HPV evolution is slow (genetic drift occurs at approximately the same rate as in the human genome) [4], spontaneous mutations in HPV variants during the life span of infected hosts occur extremely rarely. Accordingly, different variants in consecutive samples from an individual most likely represent separate infections.
In the present study, we compared sequence variations in HPV-16 variants in pairs of specimens collected prospectively from a subset of women who were enrolled in a nested case-control study investigating the clinical relevance of HPV-16 variants.
Methods. Cervical samples examined were from the case-control study of women enrolled in the ASCUS-LSIL Triage Study (ALTS), a randomized, multicenter, clinical trial designed to evaluate strategies for the management of equivocal and mildly abnormal cervical cytology. These women were followed semiannually over 2 years for HPV typing and detection of cervical lesions. A detailed description of the ALTS design and study population has been presented elsewhere [5].
Case patients were women who had (1) cervical intraepithelial neoplasia grade 2 or 3 (CIN2–3) initially diagnosed during follow-up and (2) HPV-16 DNA detected by polymerase chain reaction (PCR)-based reverse line blot [6] at follow-up visits concurrent with or before the diagnosis of CIN2–3. Control patients were selected from those who did not have a diagnosis of CIN2–3 during the entire study period and had HPV-16 DNA detected at 1 or more follow-up visits. The study protocol was approved by the institutional human subject review board of the University of Washington.
We characterized HPV-16 variants for 1 follow-up specimen per woman and for the enrollment sample from those who had a baseline HPV-16 infection. Of 255 ALTS participants enrolled in the nested case-control study, 86 (45 case and 41 control patients) were positive for HPV-16 variants at enrollment and at least 1 follow-up visit and were therefore eligible for the present study. For case patients, the follow-up sample was selected from visits at which CIN2–3 was initially diagnosed. If the sample from that visit was HPV-16 negative, the one from immediately before the time of CIN2–3 diagnosis was selected. Samples from control patients were frequency-matched to those from case patients on the timing of sample collection for variant analysis. All of the tested specimens derived from Dacron cervical swabs.
HPV-16-positive samples were initially assayed by PCR-based direct DNA sequencing of PCR products, as described elsewhere [7]. A viral isolate was designated as a distinct variant if, compared with the prototype and other variants identified in the study, there was 1 or more nucleotide alterations in the region from nucleotide positions 7723 to 567 (corresponding to the 3 'part of the long control region and the entire E6 region). In accordance with lineages categorized previously [8], HPV-16 variants were classified as European (E), Asian (As), Asian American (AA), African 1 (Af1), and African 2 (Af2).
Direct sequencing of PCR products detects only the predominant variants. To examine multiple-variant infections and whether the predominant variant in one sample was present as a minor variant in the other, we further assayed 40 pairs of enrollment and follow-up samples by PCR-based subcloning and sequencing. These samples were randomly selected except that we included all those that had displayed a change of variants in the assay of direct sequencing of PCR products. Briefly, PCR products were cloned into plasmids by means of a TOPO TA Cloning kit (Invitrogen), in accordance with the manufacturer's protocol. Plasmid DNAs were purified using a QIA Miniprep kit (Qiagen). Ten clones per sample were selected for sequencing. A predominant or minor variant was defined on the basis of the relative proportions of the clones in which the variant was present. A minor variant was defined if it was detected in 2 or more clones, with the exception of apparent recombinants. Consideration of these recombinants has been reported elsewhere [9].
An exact confidence interval was calculated for the proportion of sample pairs with a change of HPV-16 variants. The difference in lengths of time intervals of sample pairs between women with and those without a change of variants was tested by the Student t test. The Fisher exact mid- P test [10] was used to compare frequencies of a change of variants between women with and those without intervening HPV-16-negative test results (OpenEpi, version 2.3; http://www.OpenEpi.com). This analysis was restricted to those who had at least 1 intervening visit with HPV test results. The 2-tailed P value was adjusted for the number of intervening visits with HPV-16 test results.
Results. Among the 86 study subjects, the mean age was 24.3 (standard deviation [SD], 5.5) years at the time of enrollment. A change of predominant variants between enrollment and follow-up samples was detected in 4 participants (4.7% [95% confidence interval, 1.3%-11.5%]) by direct sequencing of PCR products, including 2 with and 2 without a switch of variant lineages. The mean length of time between collection of 2 samples was 18.9 (SD, 4.9) and 15.6 (SD, 7.8) months for women with and those without a change of variants, respectively (P = .40). Among those without a change of variants, HPV-16 E, AA, Af1, and Af2 were detected in 57, 14, 5, and 6 pairs of samples, respectively.
As shown in Table 1, the first 2 pairs of samples with a change of variants displayed identical sequences in each of the 10 clones. The E variants in pair 1 differed by 2 nucleotides at positions 12 and 131. Pair 2 had the AA variant in the enrollment sample and the E variant in the follow-up sample. Neither the predominant nor the minor HPV-16 AA variant in the enrollment sample of pair 3 was identical to the variant in the follow-up sample. For sample pair 4, the predominant E variant (with 2 recombinants as minor variants) was detected in the enrollment sample, and the Af2 variant was detected in the follow-up sample. The results for these 4 pairs of samples were confirmed by a second run of PCR-based subcloning with another 10 clones per sample sequenced (data not shown). Among the 36 pairs without a variant change that were randomly selected for PCR-based subcloning and sequencing, an infection with >1 variant was detected in 6 samples; none of the minor variants were detected consecutively (data not shown).
Twenty-six women did not have any intervening HPV test result, including 23 without an intervening visit, 1 without an HPV test at 1 visit, and 2 without an HPV test at 2 visits (Table 2). One woman without an HPV test at 2 scheduled intervening visits had a change of variants. Of the 60 women with at least 1 intervening HPV test result, 11 (18.3%) had 1 or 2 HPV-16- negative results at intervening visits. A change of variants appeared to be more likely (although not statistically significantly so) among women with than among women without an intervening negative HPV-16 test result (2/11 vs 1/49; Padjusted = .11). The results remained the same when women without an HPV test at the intervening visit were excluded.
Table 2.
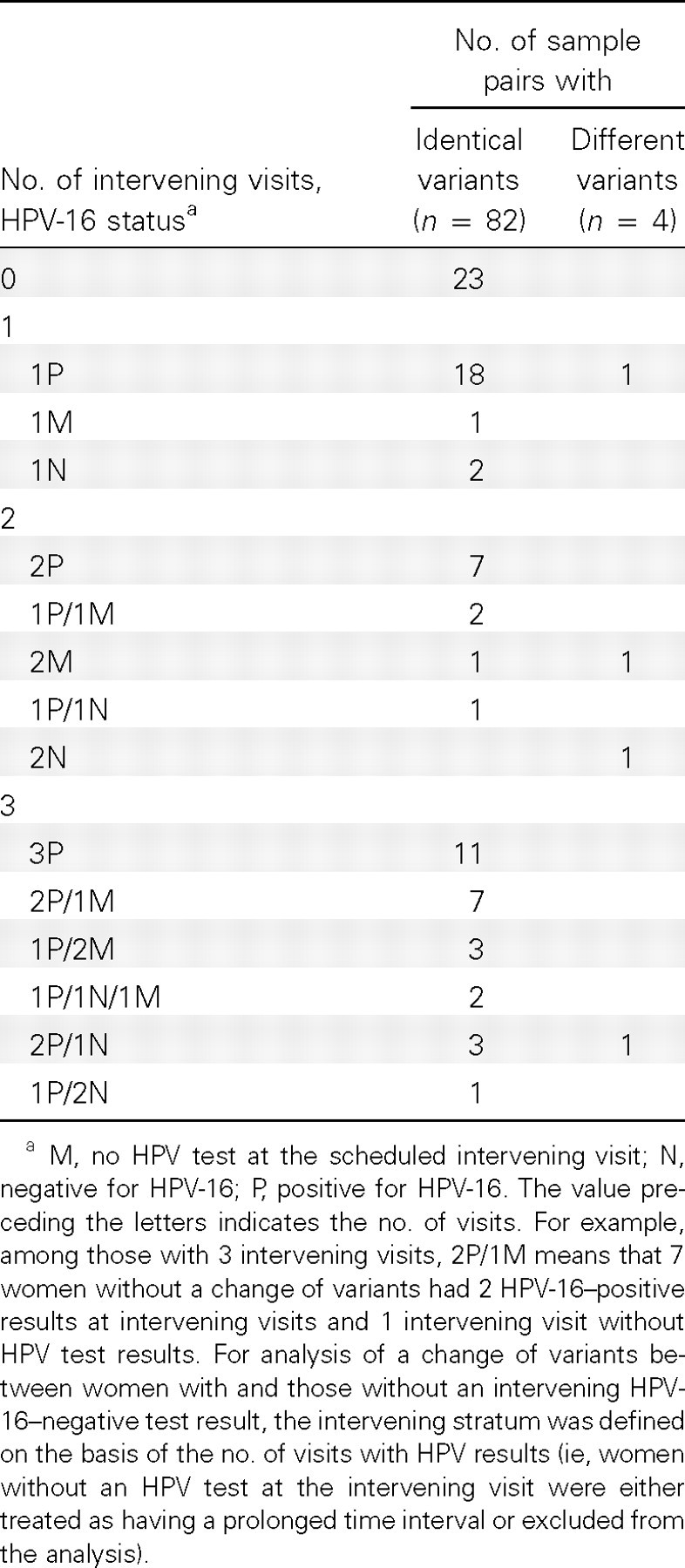
Intervening Human Papillomavirus Type 16 (HPV-16) Status between Enrollment and Follow-up Visits at Which Samples Were Assayed for HPV16 Variants
Discussion. In this study of HPV-16 isolates in pairs of enrollment and follow-up samples, we found a change of variants in only 4.7% of 86 women during ∼1.5 years of follow-up. Such changes cannot be explained by spontaneous mutations, given the slow evolutionary process of the HPV genome. In addition, they are not likely to be due to PCR errors, because the same changes were detected in 3 independent PCR runs (1 for direct sequencing of PCR products and 2 for subcloning and sequencing). Although it is possible that the predominant variant in one sample was present as a minor variant in the other, we did not see evidence of this.
We and others [11, 12] have previously demonstrated that the HPV-16 variant that is the most abundant initially remains abundant at subsequent visits. Although the presence of >1 HPV-16 variants can be detected, the minor variants are usually transient. Consistent with these findings, the present study detected minor variants in either enrollment or follow-up samples, but not both. For women with a change of variants, none of the predominant variants detected in one sample were detected in the second sample. This suggests that the change of variants was not the result of replacement of the predominant variants with the minor ones. One may argue that testing for 20 clones per sample may not be sufficient to exclude the possibility of a minor variant. However, as reported previously [9], our experience with sample pair 4 was that after sequencing a total of 105 clones we did not detect in the enrollment sample the Af2 variant that was present in the follow-up sample.
Studies of HPV variants in consecutive samples are rare, and inconsistent results have been reported. To the best of our knowledge, only 3 research groups have ever reported a change of variants over time. A case report by Franco et al [3] showed a change of HPV-16 variants in a pair of samples 7 months apart. Mayrand et al [13] demonstrated different DNA polymorphisms in 4 of 50 women by analysis of single-stranded conformation polymorphisms of HPV-16 variants in consecutive specimens. An even higher disconcordance rate was detected by pyrosequencing of HPV-16 variants in a recent study of human immunodeficiency virus-infected adults [14]. Other studies have reported identical variants in consecutive samples positive for HPV-16 [11, 12] or other high-risk types [15]. Although the inconsistent results can in part be explained by differences in study populations, interval durations, and approaches for variant characterization, most of these studies were limited by small sample size. In the present study, we additionally showed that a change of variants tended to occur among women with an intervening negative result for HPV-16, suggesting that it more likely represents a separate infection by the second variant.
It should be pointed out that the frequency of a change of variants detected in this study may not necessarily be generalizable to all women infected with HPV-16, because all ALTS participants had a referral of minor cytologic abnormalities. Additionally, although the present study included more study subjects than any previous studies, statistical power for identifying factors predictive of a change of variants was extremely limited. Finally, the present study demonstrated only a possibility, not a frequency, of recurrent type-specific infection. This is because the variant analysis was unable to distinguish persistent infection from recurrent infection for those with the same variant detected in consecutive samples. Also, it is unclear whether the second variant resulted from new acquisition or reactivation of the previously latent infection. Nevertheless, the findings of this study at least indicate a small level of misclassification of transient infection as persistent infection in studies that define persistence as detection of the same HPV type at consecutive visits, particularly for analyses that treat intervening negative test results as false-negatives.
Data from the present study suggest that misclassification of a transient infection as a persistent infection happens even at the genotype level. Our results are relevant to natural history studies but are not intended for clinical application. While separate genotyping to detect HPV-16 and to characterize HPV-16 persistence might be shown to provide worthwhile risk stratification, we are not suggesting that repeated variant testing would be cost-effective.
Table 1.

Sequence Variation in Human Papillomavirus Type 16 Variants from Nucleotide Position 7723 to 567 Detected in 4 Pairs of Enrollment and Follow-up Samples from Women with a Change of Predominant Variants
Acknowledgments
This study was part of the project ancillary to the ALTS clinical trial but does not represent the ALTS group. We would like to thank the ALTS group for providing the biological specimens and HPV typing results.
Footnotes
Potential conflicts of interest: L.A.K. receives research funds from Merck Research Laboratories. All other authors report no potential conflicts.
Financial support: Public Health Service (grant CA133569 to L.F.X.).
References
- 1.Wheeler CM, Greer CE, Becker TM, Hunt WC, Anderson SM, Manos MM. Short-term fluctuations in the detection of cervical human papillomavirus DNA. Obstet Gynecol. 1996;88:261–268. doi: 10.1016/0029-7844(96)00120-2. [DOI] [PubMed] [Google Scholar]
- 2.de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur ausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 3.Franco EL, Villa LL, Rahal P, Ruiz A. Molecular variant analysis as an epidemiological tool to study persistence of cervical human papillomavirus infection. J Natl Cancer Inst. 1994;86:1558–559. doi: 10.1093/jnci/86.20.1558. [DOI] [PubMed] [Google Scholar]
- 4.Bernard HU, Chan SY, Delius H. Evolution of papillomaviruses. Curr Top Microbiol Immunol. 1994;186:33–54. doi: 10.1007/978-3-642-78487-3_3. [DOI] [PubMed] [Google Scholar]
- 5.Schiffman M, Adrianza ME. ASCUS-LSIL Triage Study. Design, methods and characteristics of trial participants. Acta Cytol. 2000;44:726–742. doi: 10.1159/000328554. [DOI] [PubMed] [Google Scholar]
- 6.Gravitt PE, Peyton CL, Alessi TQ, et al. Improved amplification of genital human papillomaviruses. J Clin Microbiol. 2000;38:357–361. doi: 10.1128/jcm.38.1.357-361.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xi LF, Kiviat NB, Hildesheim A, et al. Human papillomavirus type 16 and 18 variants: race-related distribution and persistence. J Natl Cancer Inst. 2006;98:1045–1052. doi: 10.1093/jnci/djj297. [DOI] [PubMed] [Google Scholar]
- 8.Yamada T, Manos MM, Peto J, et al. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J Virol. 1997;71:2463–2472. doi: 10.1128/jvi.71.3.2463-2472.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang M, Xi LF, Edelstein ZR, et al. Identification of recombinant human papillomavirus type 16 variants. Virology. 2009;394:8–11. doi: 10.1016/j.virol.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lydersen S, Fagerland MW, Laake P. Recommended tests for association in 2 × 2 tables. Stat Med. 2009;28:1159–1175. doi: 10.1002/sim.3531. [DOI] [PubMed] [Google Scholar]
- 11.Xi LF, Demers GW, Koutsky LA, et al. Analysis of human papillomavirus type 16 variants indicates establishment of persistent infection. J Infect Dis. 1995;172:747–755. doi: 10.1093/infdis/172.3.747. [DOI] [PubMed] [Google Scholar]
- 12.van Belkum A, Juffermans L, Schrauwen L, van Doornum G, Burger M, Quint W. Genotyping human papillomavirus type 16 isolates from persistently infected promiscuous individuals and cervical neoplasia patients. J Clin Microbiol. 1995;33:2957–2962. doi: 10.1128/jcm.33.11.2957-2962.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayrand MH, Coutlee F, Hankins C, et al. Detection of human papillomavirus type 16 DNA in consecutive genital samples does not always represent persistent infection as determined by molecular variant analysis. J Clin Microbiol. 2000;38:3388–3393. doi: 10.1128/jcm.38.9.3388-3393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinau M, Swan DC, Onyekwuluje JM, et al. Differences and changes in human papillomavirus 16 variant status in human immunodeficiency virus-positive adults are not uncommon. J Gen Virol. 2010;91:2068–2072. doi: 10.1099/vir.0.018663-0. [DOI] [PubMed] [Google Scholar]
- 15.Khouadri S, Villa LL, Gagnon S, et al. Human papillomavirus type 33 polymorphisms and high-grade squamous intraepithelial lesions of the uterine cervix. J Infect Dis. 2006;194:886–894. doi: 10.1086/507431. [DOI] [PubMed] [Google Scholar]