

Abstract
Introduction. Acute hepatitis C virus (HCV) infection is rarely studied, but virus sequence evolution and hostvirus dynamics during this early stage may influence the outcome of infection. Hypervariable region 1 (HVR1) is genetically diverse and under selective pressure from the host immune response. We analyzed HVR1 evolution by frequent sampling of an acutely infected HCV cohort.
Methods. Three or more pretreatment samples were obtained from each of 10 acutely infected subjects. Polymerase chain reaction amplification was performed with multiple primer combinations to identify the full range of sequences present. Positive samples were cloned and sequenced. Phylogenetic analyses were used to assess viral diversity.
Results. Eight of the 10 subjects were coinfected with at least 2 HCV subtypes. Multiple subtypes were detected in individual samples, and their relative proportions changed through acute infection. The subjects with the most complex subtype structure also had a dynamic viral load; however, changes in viral load were not directly linked to changes in subtype.
Conclusions. This well-sampled cohort with acute HCV infection was characterized by dynamic coinfection with multiple viral subtypes, representing a highly complex virologic landscape extremely early in infection.
Hepatitis C virus (HCV) creates a major burden of disease, with an estimated 170 million infections worldwide [1]. In developed countries, the main risk factor is injection drug use (IDU), although there have been differences in the contribution of risk factors over time, including nosocomial exposure to unsafe blood transfusions before the isolation and identification of HCV in 1989 [2]. Recently, genotype 3 incidence has overtaken genotype 1 in IDU patients in the United Kingdom, resulting in cocirculation of the 2 strains [3].HCV prevalence in current IDUs is up to 70% [4, 5], although seroprevalence can approach 100%, indicating that most IDUs have been exposed to the virus.
Cohorts of patients with acute hepatitis C are studied relatively rarely because of the mostly asymptomatic nature of early infection; an estimated 17% of all incident cases present with symptomatic acute hepatitis per year in the United States [6]. There are only 9 acutely infected cohorts from North America, collectively identifying 674 patients [7, 8]. Acutely infected cohort for which sampling has been repeated at short time intervals are particularly valuable, because this early stage provides a critical window of opportunity in which to study viral sequence evolution and hostvirus dynamics, which may define the outcome of infection [9].
The acute period of HCV infection may be followed by rapid clearance of virus from the blood or, more commonly, by long-term persistence of viral RNA. In some cases, which may result in either outcome, an intermediate state is observed that is characterized by unstable viremia and partial or transient control. The mechanisms that determine these different outcomes are not fully understood. There is evidence that robust T cell responses and neutralizing antibody responses contribute to successful control, whereas viral variation in envelope and T cell epitopes, together with down-regulation of T cell responses, may contribute to viral persistence (reviewed in [10]).
Hypervariable region 1 (HVR1) is the most genetically diverse part of the HCV genome. Located at the N-terminus of envelope protein 2 (E2), it is a neutralizing antibody target and is therefore under intense immune pressure for sequence variation. There are conflicting results regarding its role during early infection. Farci et al [11] demonstrated that antibodies raised to an HVR1 peptide are capable of preventing HCV infection in chimpanzees, and an early humoral response to HVR1 in humans has been associated with virus clearance [12]. More recently it has been suggested that divergent quasispecies of HVR1 may provide a mechanism for persistence, with major HCV clones generating considerably divergent minor “decoy” clones, which are preferentially neutralized [13]. HVR1 also plays an important biological role in cell entry; it is thought to be involved in target cell recognition and virus attachment [14]. It is commonly used to examine differences between closely related strains.
In the present study, we analyzed HVR1 evolution in an acutely infected cohort, with intensive sampling to include very closely spaced time points. In particular, this cohort included subjects in whom highly dynamic viral loads (termed yo-yo) had been observed. We therefore examined how dynamic changes in viral load were related to mutations in HVR1. We observed rapid changes in the HVR1 sequence in the majority of subjects in the cohort and demonstrated that this was due to the presence of multiple coexisting strains, reflecting an intensely dynamic virologic landscape at this early stage of infection.
Methods
Samples. Ten subjects from Vienna, Austria, who had recently tested positive for HCV RNA by reverse-transcription polymerase chain reaction (RT-PCR) were identified. Eight (subjects 1, 3–6, and 8–10) were selected from a cohort of 20 who were referred for study between 2003 and 2005; 2 (subjects 2 and 7) were selected from 7 referrals between 2005 and 2006. Most patients were identified at the emergency outpatient unit; usually 1 patient with acute HCV infection was seen at this unit every month, with no single outbreak of multiple HCV cases. The remainder were referred from their physicians for antiviral therapy. Asymptomatic acute HCV infection was identified during examinations due to drug abuse (n = 1) or follow-up after medical procedures (n = 2). Informed consent was obtained from all subjects. Inclusion criteria were as follows: (1) subjects had to have available at least 3 frozen plasma or serum samples from acute infection, defined as 180 days after infection or after the onset of symptoms in cases where the date of infection was unknown; (2) subjects had to be treatment naive for at least 3 consecutive acute-stage samples; and (3) subjects could not have had a known previous infection. The samples had been included in an immunological study of acute HCV infection [15]. The 53 samples analyzed were obtained from 0 to 473 days after the onset of symptoms, at a range of 4–225 days apart, and spanned the acute infection phase. Samples were also obtained from the chronic phase for those who progressed to chronicity.
HCV RNA was initially detected and genotyped by line probe assay (LiPA) (INNO-LiPA II; Innogenetics) on the first available serial sample. Initial qualitative HCV RNA RT-PCR was performed using the Cobas Amplicor HCV test (Roche Diagnostic Systems), which has a lower detection limit of 50 IU/mL. Viral loads were determined using the Cobas Amplicor HCV Monitor test (version 2.0; Roche), which has a detection limit of 600 IU/mL. Samples from 1 patient with HCV genotype 1 infection who cleared HCV from serum (subject 6) were also HCV RNA negative by an ultrasensitive Taq-Man assay (Roche; detection limit, 10 IU/mL).
The 225-base pair (bp) sequence analyzed spans the C-terminal end of envelope glycoprotein E1 and the N-terminal end of E2 and includes HVR1 (81 bp). This corresponds to positions 1395–1619 inclusive on the H77 reference sequence (GenBank accession no. AF009606 [16, 17]).
RNA extraction, complementary DNA synthesis, and amplification. Plasma (500 µmL) was concentrated by highspeed centrifugation (23,600 g for 1 h) at 4°C. Viral RNA was extracted using a QIAmp Viral RNA MiniKit (Qiagen) and reverse-transcribed into complementary DNA in 20 µL using the Superscript II system (Invitrogen), in accordance with the manufacturer's instructions, with 2 pmol of gene-specific primer RC21_rev (5′-GCTTGCGAGTGCCCCGGGAG-3′) [18].
PCR amplification was performed with High Fidelity Taq DNA polymerase (Roche) in nested reactions to amplify the 225-bp region, in accordance with to the manufacturer's instructions. The primary and secondary reaction primers are detailed in Table 1. Each initial reaction contained 1 µg of DNA; 2.5 µL of the first-round PCR product was used in the second round. All samples were tested with each primer combination to ensure maximum coverage of genotypes. A negative PCR result was assumed to mean that the genotype was not present. PCR conditions were as follows: 94°C for 2 min; 10 cycles of 30 s at 94°C, 30 s at the primer-specific annealing temperature, and 30 s at 72°C; followed by 20 cycles of 30 s at 94°C, 30 s at the primer-specific annealing temperature, and 30 s increasing by 5 s every cycle at 72°C; and a final extension of 72°C for 7 min. PCR products were analyzed on 1.2% agarose gel and purified using the QIAquick Gel Extraction kit (Qiagen). All purified samples were cloned; this was repeated for samples with a positive result for >1 primer pair (subject 6 on days 53, 81, and 93; subject 7 on days 17, 199, and 204), and the resulting sequences were grouped together. Other samples may have had >1 genotype amplified by a single primer pair.
Table 1.

Methodological Details: Primer Information and List of Reference Sequences Used to Generate Phylogenetic Trees
Cloning, amplification, and sequencing of viral populations. Amplicons were cloned into One Shot Chemically Competent E. coli by means of the pCR4-TOPO vector (Invitrogen). Colonies were grown overnight, and plasmid DNA was purified using the Montage Plasmid Miniprep96 kit (Millipore). Successful transformation was confirmed by EcoRI digestion (New England Biolabs), and positive clones were sequenced bidirectionally using the ABI Prism BigDye Terminator system (version 3.0; Applied Biosystems) on an ABI 3100 DNA automated sequencer with the primers M13For and M13Rev (Invitrogen). Sequences were assembled and edited using the SeqMan program in the DNAstar suite (version 3.2; Lasergene) and were deposited in GenBank under accession numbers GU364182- GU365816.
Phylogenetic and statistical analyzes. Sequences were aligned using the online MAFFT tool hosted on the EMBL-EBI web site (http://www.ebi.ac.uk/Tools/mafft/index.html) [19]. All clones were regenotyped using an online HCV subtyping tool (http://www.bioafrica.net/virus-genotype/html/subtypinghcv.html) [20]. The Kimura 2-parameter model was implemented using MEGA software (version 4.0) [21] to create neighbor-joining phylogenetic trees (see Table 1 for a full list of reference sequences). Bootstrap analyzes were performed with 1000 replicates. Statistical analysis was performed in R software (version 2.10.1) [22].
Results
Patient characteristics. HCV strains from 10 acutely infected high-risk patients were analyzed at the E1/E2 HVR1 locus, using 53 samples collected at a median interval of 17.5 days (range, 4–225 days; data not shown). Table 2 summarizes the virologic features and clinical outcome for each subject. IDU was the main risk factor for 60% subjects; the remainder were a mixture of nosocomial, sexual, and unknown transmission. There was a mixture of subtypes, with most subjects presenting with genotype 1 (6/10 [60%]), as assessed by LiPA genotyping. HCV genotype did not correlate with peak RNA level or outcome of infection. Seven subjects were symptomatic (subjects 1–3, 5– 7, and 9), and 3 were asymptomatic (subjects 4, 8, and 10). Four subjects cleared the virus, 2 spontaneously (subjects 1 and 2) and 2 through successful antiviral treatment (subjects 7 and 8). A fifth (subject 6) had an end-of-treatment response and may have gone on to clear. Two subjects did not respond to treatment (subjects 9 and 10), and 3 progressed to chronic infection without being treated (subjects 3–5). Subjects 4, 5, 9, and 10 were followed up as they progressed to chronic infection; hence, data for subjects 9 and 10 includes samples obtained during treatment as well as the requisite 3 pretreatment acutestage samples.
Table 2.

Summary of Clinical and Virologic Features of Patients with Acute Hepatitis C Virus (HCV) Infection
Phylogenetic analysis of HVR1 sequences during acute infection. We analyzed the longitudinal evolution of HCV by sequencing HVR1 and the surrounding E2 region over time. Online subtyping of clonal sequences identified multiple variants of HCV subtypes 1a, 1b, 2k, and 3a in the cohort (Figure 1). Analysis of individual subjects revealed that the majority of the cohort (80%) were infected with >1 HCV subtype (Figure 2). Strikingly, multiple infections of 3 subtypes were observed in 6 members of the cohort (60%). HVR1 subtyping did not always correlate fully with LiPA genotyping, with the latter failing to detect any mixed-genotype infections (Table 3). However, in 8 of 10 cases the subtype identified by LiPA was detected during the course of infection by HVR1 analysis.
Figure 1.
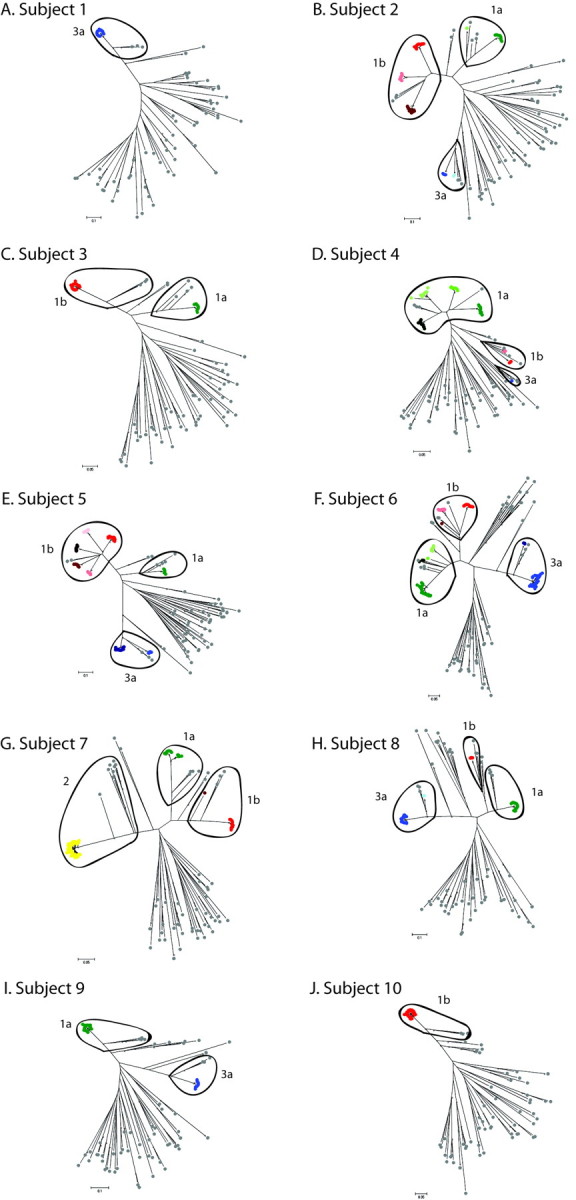
Neighbor-joining trees based on the 225-base pair E2 fragment, which includes hypervariable region 1, showing all sequences from all subjects throughout acute hepatitis C virus infection. Colored and gray spots represent patient samples and reference sequences from all verified genotypes, respectively. Different colors are used to distinguish distinct viral clades. Sample sequences are colored according to subtype: shades of green indicate 1a; red, 1b; yellow, 2; and blue, 3a. Marked ovals contain all sequences (sample and reference) assigned to the annotated subtype. The same shade in different graphs does not imply the same clone. Asterisks indicate nodes with >75% bootstrap support.
Figure 2.

Number of hepatitis C virus (HCV) strains detected in each subject during acute infection. Sequences are amalgamated across all samples for each subject; subtypes were identified using an online HCV subtyping tool. Within-subtype clades were identified by comparison with reference sequences from GenBank (Table 1).
Table 3.

Comparison of Line Probe Assay (LiPA)- Derived Genotype with Hypervariable Region 1 (HVR1)-Derived Genotype
Phylogenetic analyzes showed that highly divergent clades were present even within subtypes when compared with a standard bank of reference sequences from GenBank (Figure 1). These distinct clades are defined as monophyletic groups of sampled sequences that cannot expand to include other sample sequences without also encompassing 1 or more reference sequences. The lowest level of HVR1 sequence diversity observed was single infection with 1 viral clade throughout the acute period (subjects 1 and 10) (Figure 1A and 1J and Figure 3A and 3J). At the intermediate level, we observed multiple HCV subtypes in subjects, each consisting of 1 dominant viral clade (subjects 3 and 7–9) (Figure 1C and 1G–1I and Figure 3C and 3G–3I), and subjects with the highest levels of sequence diversity were coinfected with multiple subtypes, each with multiple distinct intrasubtypic clades (subjects 2 and 4–6) (Figure 1B and 1D–1F and Figure 3B and 3D–3F). The number of distinct viral clades observed in each subject ranged from 1 to 9, with 40% of subjects showing 7 or more clades (Figure 2). Clades of different subtypes were characterized by widely differing amino acid motifs in both the surrounding E1/E2 region and within HVR1. The differences between same-subtype clades were largely accounted for by sequence variation in HVR1; however, there also were a small number of conserved differences in the surrounding E1/E2 region. Within clades, the substitutions distinguishing different quasispecies were evenly distributed both inside and outside HVR1. Intersubtype variation was supported by bootstrap values of at least 95% (data not shown), whereas support for intrasubtype variation was much lower because of the short sequence length. Intrasubject diversity was not associated with clinical outcome, peak viral load, or peak ALT level.
Figure 3.
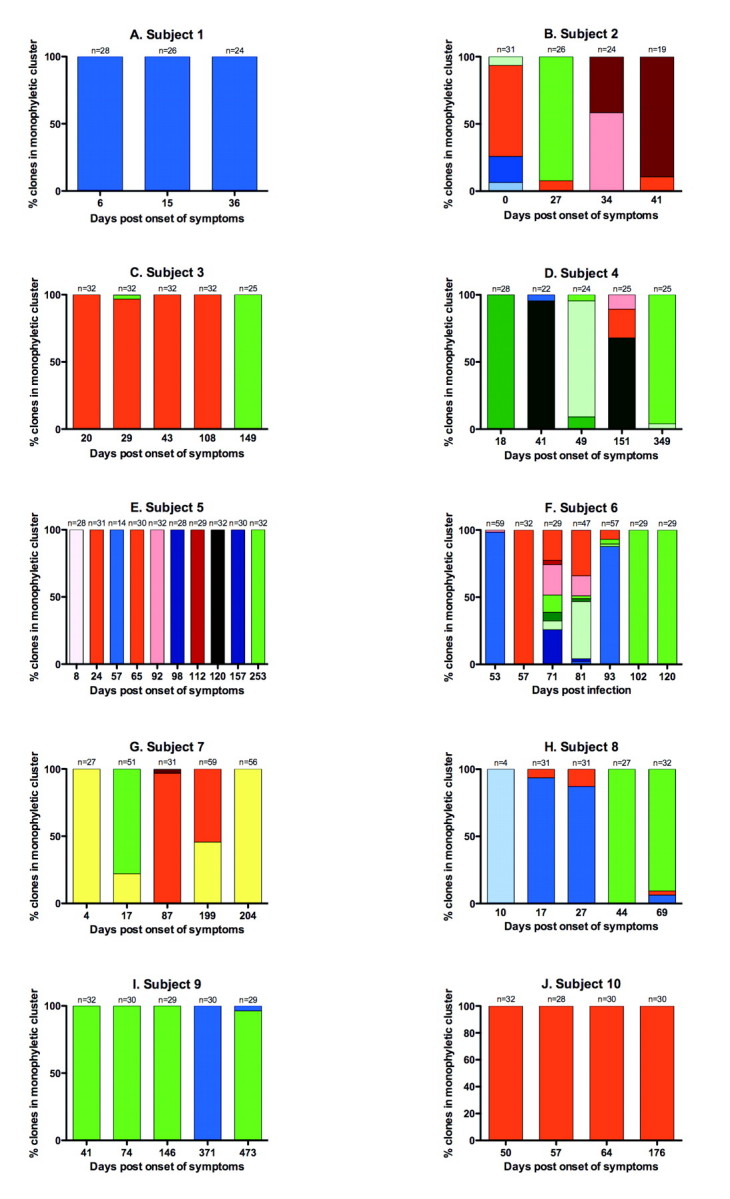
Hepatitis C virus (HCV) quasispecies clades in subjects through acute infection. Different colors represent within-patient clades, expressed as a percentage of the total clones retrieved; the colors used for each viral clade correspond to those in Figure 1. Clades are colored according to subtype: shades of green indicate 1a; red, 1b; yellow, 2; and blue, 3a. The same shade in different graphs does not imply the same clone. The number of clones analyzed is shown above each time-point bar. Note that samples from subject 6 on days 53, 81, and 93 and from subject 7 on days 17, 199, and 204 were cloned twice with different primer combinations and the results amalgamated; hence, where multiple distinct clones are present the percentages do not accurately reflect clone proportions.
Within-host quasispecies dynamics. Of the subjects, 70% showed coexistence of >1 subtype at individual time points, as well as different clades within those subtypes (Figure 3). Only the singly infected cohort members (subjects 1 and 10) and subject 5 had 1 detectable variant in each sample.
The detectable variants also changed markedly through acute infection in the 8 subjects with coinfection. The most complex dynamics were seen in subjects with the most diverse infections (subjects 2 and 4–8); all showed multiple switches in the dominant variant detected at each time point. In addition to sequential replacements by previously undetected variants, all coinfected subjects showed a dominance of variants seen earlier during infection but not detected in intervening samples. Marked changes in quasispecies profile occurred extremely rapidly; for example, in subject 6 we observed a complete switch from a 3a-dominated to a 1b-dominated infection in just 4 days (days 53–57) (Figure 3F).
Six cohort members (subjects 2, 4–6, 8, and 9) had infections in which both genotypes 1 (either 1a or 1b) and 3 were present. In all of these cases, the genotype 1 strain was dominant in the last sample available, although the follow-up time varied between subjects. This was statistically significant (P = .041 with the Yates continuity correction).
Relationship between viral sequence dynamics and viral load dynamics. This cohort was enriched for individuals showing yo-yo viral load dynamics, defined as a minimum 2 log decline in viral load followed by a resurgence of viral RNA. Therefore, we addressed whether this viral load profile was related to viral species switching. We observed that subjects with the most detected clades also showed a complex fluctuating pattern of viremia during acute infection (subjects 2 and 4–7) (Figure 4). This was not treatment induced, unlike the sharp decline in viral load observed in subject 10. However, the timing of the viremic peak did not coincide in any case with the changes in the dominant variant. Subject 1 also showed fluctuating viremia, yet only 1 HCV variant was detected. Overall, of the 6 patients showing yo-yo viral load dynamics, 5 also had the most complex viral sequence dynamics, with 3 viral subtypes. Of the 4 subjects without yo-yo dynamics, only 1 was coinfected with 3 subtypes.
Figure 4.

Hepatitis C virus (HCV) load (solid lines with diamonds) and alanine aminotransferase (ALT) levels (hatched lines with circles) during acute infection, pegylated interferon (pgIFN) treatment (subjects 6–10; solid lines), or progression to chronicity (subjects 4, 5, 9, and 10). Time points from which samples were analyzed by hypervariable region 1 (HVR1) phylogenetics are indicated by an inverted triangle; these correspond to the time points shown in Figure 3. Line probe assay (LiPA) genotyping was performed on samples indicated by a solid circle.
Discussion
The results of the present study highlight the diversity and dynamics of acute HCV infection in HVR1 in a small, wellsampled cohort. The anticipated patterns of immune selection were completely masked by the far greater effects of viral diversity, with genotypes 1, 2, and 3 all present in the cohort. We make 2 general observations: first, an extremely high level of within-host diversity, with multiple subtypes coexisting both at single time points and throughout acute infection; and second, the unstable dynamics of acute HCV infection with the apparent disappearance and reemergence of variants over a very short time scale.
We minimized the effects of any methodological shortfalls where possible. The short length of the fragment analyzed limited the robustness of the phylogenetic trees when distinguishing within-subtype variants, but there was sufficient diversity to accurately assess differences between subtypes. This was an unavoidable limitation due to the difficulties in amplifying longer sequences or other genomic regions. PCR is not the ideal tool for assessing clonal diversity because the limited number of clones results in underestimation of the variation present. However, all samples were cross-checked with multiple genotype- specific primers for genotypes 1a, 2, and 1b/3a in order to identify the broadest range at all time points, and all positive samples were cloned. In cases with only 1 dominant viral variant, this affects the resulting clonal proportions very little (subject 6 on days 53 and 93 and subject 7 on day 204); however, where a mixture of genotypes were retrieved the overall proportions do not accurately reflect the viral variants present in the sample (subject 6 on day 81 and subject 7 on days 17 and 199). PCR is also susceptible to random effects and systematic bias. Performing limiting dilutions would annul bias but we were unable to amplify samples by this method, possibly because of poor sample quality. PCR may also overestimate diversity due to incorporation of errors during viral sequence copying. The PCR error rate for this protocol was previously estimated as 5.86×10−6 errors/bp/cycle for 35 cycles. Combined with the estimated rate of RT error (3.0×10−5– 6.4×10−5 errors/bp), we would expect 1 error every 2110 bp, or once every 9.4 sequences [23]. Finally, samples were obtained from peripheral blood and may not be a true representation of quasispecies diversity in the liver [24]. Despite potential quantitative errors inherent in the methods used, the qualitative observations of dynamic HCV subtype coinfection are robust, and if anything the true diversity may be underestimated.
It has been reported previously that greater quasispecies diversity may be associated with progression to chronicity [25], but this relationship was not observed in this (albeit small) cohort. Instead, we observed the coexistence of numerous distinct clades leading to a range of clinical outcomes. Previous studies have noted intra- and intergenotypic superinfection in chronic HCV infection, where bulk sequencing shows the HCV strain to be replaced by an unrelated variant [26–30]. Many have produced conflicting results, reporting prevalences of mixed genotypes in chronic infection ranging from 0% to 56% ([31, 32]; reviewed in [33]). No coinfection of different HCV subtypes was observed in a study of 12 subjects with acute HCV infection after transfusion [9], yet levels of diversity similar to our results were found in 12 non-IDU subjects with chronic infection [13]; this was postulated to have arisen through the role of HVR1 as an immunological “decoy,” but coinfection was not considered. Herring et al [26] reported a 20% prevalence of HCV coinfection in an IDU cohort in San Francisco, California, and suggested that this was the result of little immunological cross-protection against alternative quasispecies by the adaptive immune response. They later detected heterogeneous quasispecies, analogous to our within-subtype clades, in one-third of a recently infected cohort that included IDUs, transfusion recipients, and plasma donors [34]. HVR1 is the most variable region of the HCV genome, so it is possible that strong within-host antibody-driven selection here is driving the apparent divergence between within-subtype variants. However, the additional differences between these variants in the more conserved E1/E2 region surrounding HVR1 support the hypothesis that they diverged before entering the host.
The apparent dominance of genotype 1 over genotype 3 infection when both infect the same host was an intriguing finding. This may result from differences in adaptive immune responses or in the response to innate cytokines or from viral interference between genotypes. This is consistent with the higher treatment response rates for genotype 3 than genotype 1, with sustained virologic response rates of 75%–80% versus 40%–45%, respectively, for combination pegylated interferon and ribavirin treatment [35, 36]. However, studies of patients who experience spontaneous resolution of acute HCV infection have shown no consistent link to the infecting genotype [37–39], and the variance in follow-up times in these 6 subjects (range, 41–473 days; standard deviation, 184 days) limits the validity of the comparison.
We observe a general pattern where subjects infected with the highest number of variants also have a fluctuating course of viral replication (subjects 2 and 4–7). In particular, subjects 4 and 5, who were followed up for long periods without treatment, showed highly complex viral diversity and dynamics. A similar pattern of viremia has been described previously in other subjects with acute infection, in health care workers exposed to needlestick accidents [40, 41] and other IDU cohorts [42], and in the original cohort from which the present subgroup was obtained [15]. The combination of subtype coinfection, changing quasispecies profile, and fluctuating viral load points to an unstable system, with changes driven by both virus and host factors. The most intuitive explanation of the multiple subtypes in these subjects is their epidemiology; they are mostly IDUs and therefore are likely to be highly exposed to HCV through contact with contaminated needles and other injection equipment. It is possible that repeated exposure could have occurred before diagnosis or that coinfection of multiple virions within 1 infectious dose led to the existence of multiple strains in the subject at the time the first sample was obtained. Ongoing exposure is unlikely, because the variants that dominate later during acute infection are not newly acquired; they are quasispecies from clusters detected in previous samples, sometimes at low levels, but in many cases forming a dominant earlier clade. The location of this cohort may have facilitated the detection of multiple genotypes, given that a range of genotypes are known to be circulating in central Europe [43, 44]. In other IDU cohorts in areas with a limited range of genotypes, this phenomenon may be less apparent. Furthermore, these results may not be representative of HCV infections in general; the cohort is enriched for subjects with fluctuating viremia, in whom it may be easier to detect mixed infections compared with the stable high-level replication observed during chronic infection, where the dominant variant may mask others present at low frequency.
In conclusion, this cohort provides a rich within-host data set and highlights the complex diversity and dynamics of acute HCV infection. Most subjects were coinfected with multiple HCV subtypes, which is associated with high levels of exposure. This raises important questions for vaccine design, with a monovalent HCV vaccine unlikely to be effective in western European populations.
Footnotes
Potential conflicts of interest: none reported.
Presented in part: Epidemics Conference, Monterey, California, 1–3 December 2008 (oral presentation).
Financial support: Wellcome Trust; Medical Research Council; James Martin 21st Century School, Oxford; US National Institutes of Health (grant U19 AI082630); UK National Institute for Health Research Biomedical Research Centre Programme, Oxford.
References
- 1.European Association for the Study of the Liver. EASL International Consensus Conference on Hepatitis C. J Hepatol. 1999;31:3–8. [Google Scholar]
- 2.Choo QL, Weiner AJ, Overby LR, Kuo G, Houghton M, Bradley DW. Hepatitis C virus: the major causative agent of viral non-A, non-B hepatitis. Br Med Bull. 1990;46:423–441. doi: 10.1093/oxfordjournals.bmb.a072408. [DOI] [PubMed] [Google Scholar]
- 3.Harris KA, Gilham C, Mortimer PP, Teo CG. The most prevalent hepatitis C virus genotypes in England and Wales are 3a and 1a. J Med Virol. 1999;58:127–131. doi: 10.1002/(sici)1096-9071(199906)58:2<127::aid-jmv5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 4.Micalessi MI, Gerard C, Ameye L, Plasschaert S, Brochier B, Vranckx R. Distribution of hepatitis C virus genotypes among injecting drug users in contact with treatment centers in Belgium, 2004-2005. J Med Virol. 2008;80:640–645. doi: 10.1002/jmv.21145. [DOI] [PubMed] [Google Scholar]
- 5.Aceijas C, Rhodes T. Global estimates of prevalence of HCV infection among injecting drug users. Int J Drug Policy. 2007;18:352–358. doi: 10.1016/j.drugpo.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Williams I. Epidemiology of hepatitis C in the United States. Am J Med. 1999;107:2–9. doi: 10.1016/s0002-9343(99)00373-3. [DOI] [PubMed] [Google Scholar]
- 7.Cox AL, Page K, Bruneau J, et al. Rare birds in North America: acute hepatitis C cohorts. Gastroenterology. 2009;136:26–31. doi: 10.1053/j.gastro.2008.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heller T, Hoofnagle J, Liang TJ, Rehermann B. Acute hepatitis C. Gastroenterology. 2009;136:2411. doi: 10.1053/j.gastro.2009.04.042. [DOI] [PubMed] [Google Scholar]
- 9.Farci P, Shimoda A, Coiana A, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 10.Thimme R, Neumann-Haefelin C, Boettler T, Blum HE. Adaptive immune responses to hepatitis C virus: from viral immunobiology to a vaccine. Biol Chem. 2008;389:457–467. doi: 10.1515/bc.2008.061. [DOI] [PubMed] [Google Scholar]
- 11.Farci P, Shimoda A, Wong D, et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zibert A, Meisel H, Kraas W, Schulz A, Jung G, Roggendorf M. Early antibody response against hypervariable region 1 is associated with acute self-limiting infections of hepatitis C virus. Hepatology. 1997;25:1245–1249. doi: 10.1002/hep.510250530. [DOI] [PubMed] [Google Scholar]
- 13.Korenaga M, Hino K, Katoh Y, et al. A possible role of hypervariable region 1 quasispecies in escape of hepatitis C virus particles from neutralization. J Viral Hepat. 2001;8:331–340. doi: 10.1046/j.1365-2893.2001.00305.x. [DOI] [PubMed] [Google Scholar]
- 14.Penin F, Combet C, Germanidis G, Frainais P-O, Deleage G, Pawlotsky J-M. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J Virol. 2001;75:5703–5710. doi: 10.1128/JVI.75.12.5703-5710.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aberle JH, Formann E, Steindl-Munda P, et al. Prospective study of viral clearance and CD4+ T-cell response in acute hepatitis C primary infection and reinfection. J Clin Virol. 2006;36:24–31. doi: 10.1016/j.jcv.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 16.Kuiken C, Combet C, Bukh J, et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology. 2006;44:1355–1361. doi: 10.1002/hep.21377. [DOI] [PubMed] [Google Scholar]
- 17.Kuiken C, Simmonds P. Nomenclature and numbering of the hepatitis C virus. Methods Mol Biol. 2009;510:33–53. doi: 10.1007/978-1-59745-394-3_4. [DOI] [PubMed] [Google Scholar]
- 18.Komurian-Pradel F, Perret M, Deiman B, et al. Strand specific quantitative real-time PCR to study replication of hepatitis C virus genome. J Virol Methods. 2004;116:103–106. doi: 10.1016/j.jviromet.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Oliveira T, Deforche K, Cassol S, et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics. 2005;21:3797–3800. doi: 10.1093/bioinformatics/bti607. [DOI] [PubMed] [Google Scholar]
- 21.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 22.R Core Development Team. R Foundation for Statistical Computing. 2008. R: a language and environment for statistical computing. [Google Scholar]
- 23.Sheridan I. Nuffield Department of Clinical Medicine. Oxford, United Kingdom: University of Oxford; 2006. Analysis of immune escape and viral persistence in hepatitis C virus infection; p. 353. [doctoral thesis] [Google Scholar]
- 24.Maggi F, Fornai C, Vatteroni ML, et al. Differences in hepatitis C virus quasispecies composition between liver, peripheral blood mononuclear cells and plasma. J Gen Virol. 1997;78:1521–1525. doi: 10.1099/0022-1317-78-7-1521. [DOI] [PubMed] [Google Scholar]
- 25.Ray SC, Wang Y-M, Laeyendecker O, Ticehurst JR, Villano SA, Thomas DL. Acute hepatitis C virus structural gene sequences as predictors of persistent viremia: hypervariable region 1 as a decoy. J Virol. 1999;73:2938–2946. doi: 10.1128/jvi.73.4.2938-2946.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herring BL, Page-Shafer K, Tobler LH, Delwart EL. Frequent hepatitis C virus superinfection in injection drug users. J Infect Dis. 2004;190:1396–1403. doi: 10.1086/424491. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Thomassen LV, Majid A, et al. Investigation of putative multisubtype hepatitis C virus infections in vivo by heteroduplex mobility analysis of core/envelope subgenomes. J Virol. 2008;82:7524–7532. doi: 10.1128/JVI.02220-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroter M, Feucht H-H, Zollner B, Schafer P, Laufs R. Multiple infections with different HCV genotypes: prevalence and clinical impact. J Clin Virol. 2003;27:200–204. doi: 10.1016/s1386-6532(02)00264-0. [DOI] [PubMed] [Google Scholar]
- 29.Aitken C, McCaw R, Jardine D, et al. Change in hepatitis C virus genotype in injecting drug users. J Med Virol. 2004;74:543–545. doi: 10.1002/jmv.20212. [DOI] [PubMed] [Google Scholar]
- 30.Bowden S, McCaw R, White PA, Crofts N, Aitken CK. Detection of multiple hepatitis C virus genotypes in a cohort of injecting drug users. J Viral Hepat. 2005;12:322–324. doi: 10.1111/j.1365-2893.2005.00592.x. [DOI] [PubMed] [Google Scholar]
- 31.Viazov S, Widell A, Nordenfelt E. Mixed infection with two types of hepatitis C virus is probably a rare event. Infection. 2000;28:21–25. doi: 10.1007/s150100050005. [DOI] [PubMed] [Google Scholar]
- 32.Chen YD, Liu MY, Yu WL, et al. Mix-infections with different genotypes of HCV and with HCV plus other hepatitis viruses in patients with hepatitis C in China. World J Gastroenterol. 2003;9:984–992. doi: 10.3748/wjg.v9.i5.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blackard JT, Sherman KE. Hepatitis C virus coinfection and superinfection. J Infect Dis. 2007;195:519–524. doi: 10.1086/510858. [DOI] [PubMed] [Google Scholar]
- 34.Herring BL, Tsui R, Peddada L, Busch M, Delwart EL. Wide range of quasispecies diversity during primary hepatitis C virus infection. J Virol. 2005;79:4340–4346. doi: 10.1128/JVI.79.7.4340-4346.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 36.Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann M, Meyer MF, Monazahian M, Tillmann HL, Manns MP, Wedemeyer H. High rate of spontaneous clearance of acute hepatitis C virus genotype 3 infection. J Med Virol. 2004;73:387–391. doi: 10.1002/jmv.20103. [DOI] [PubMed] [Google Scholar]
- 38.Wietzke-Braun P, Mänhardt LR, Rosenberger A, Uy A, Ramadori G, Mihm S. Spontaneous elimination of hepatitis C virus infection: a retrospective study on demographic, clinical, and serological correlates. World J Gastroenterol. 2007;13:4224–4229. doi: 10.3748/wjg.v13.i31.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris HE, Eldridge KP, Harbour S, Alexander G, Teo C-G, Ramsay ME. Does the clinical outcome of hepatitis C infection vary with the infecting hepatitis C virus type? J Viral Hepat. 2007;14:213–220. doi: 10.1111/j.1365-2893.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- 40.Racanelli V, Rehermann B. Hepatitis C virus infection: when silence is deception. Trends Immunol. 2003;24:456–464. doi: 10.1016/s1471-4906(03)00178-9. [DOI] [PubMed] [Google Scholar]
- 41.Thimme R, Oldach D, Chang K-M, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox AL, Mosbruger T, Mao Q, et al. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med. 2005;201:1741–1752. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haushofer AC, Kopty C, Hauer R, Brunner H, Halbmayer WM. HCV genotypes and age distribution in patients of Vienna and surrounding areas. J Clin Virol. 2001;20:41–47. doi: 10.1016/s1386-6532(00)00154-2. [DOI] [PubMed] [Google Scholar]
- 44.Maieron A, Metz-Gercek S, Hackl F, et al. Chronic hepatitis C in Austria, 1992-2006: genotype distribution and demographic factors. Euro Surveillance. 2010;15:19492. doi: 10.2807/ese.15.08.19492-en. [DOI] [PubMed] [Google Scholar]