

Abstract
Background. High-throughput genome-wide techniques have facilitated the identification of previously unknown host proteins involved in cellular human immunodeficiency virus (HIV) infection. Recently, 3 independent studies have used small interfering RNA technology to silence each gene in the human genome to determine the importance of each in HIV infection. Genes conferring a significant effect were termed HIV-dependency factors (HDFs).
Methods. We assembled high-density panels of 6380 single-nucleotide polymorphisms (SNPs) in 278 HDF genes and tested for genotype associations with HIV infection and AIDS progression in 1633 individuals from clinical AIDS cohorts.
Results. After statistical correction for multiple tests, significant associations with HIV acquisition were found for SNPs in 2 genes, NCOR2 and IDH1. Weaker associations with AIDS progression were revealed for SNPs within the TM9SF2 and EGFR genes.
Conclusions. This study independently verifies the influence of NCOR2 and IDH1 on HIV transmission, and its findings suggest that variation in these genes affects susceptibility to HIV infection in exposed individuals.
More than 25 years after the discovery of the human immunodeficiency virus (HIV) [1], the HIV/AIDS epidemic continues to present an urgent global challenge. The majority of antiretroviral therapies combat HIV by inhibiting viral proteins. To date, the Food and Drug Administration has approved only 1 anti-AIDS drug that targets a host cellular pathway: maraviroc, which binds to the human chemokine (C-C motif) receptor 5 (CCR5), preventing viral entry into CD4+ macrophages [2]. The development of maraviroc was facilitated by the discovery that homozygosity for the common CCR5-D32 allele effectively blocks infection in HIV-exposed persons [3, 4]. As HIV-1 strains continue to acquire resistance to antiretroviral drugs, the identification of novel pharmacological targets is imperative [4]. These targets will probably include human proteins required for HIV replication.
Recent genome-wide screens to discover HIV-dependency factors (HDFs) used small interfering RNAs (siRNAs) to inhibit host gene expression, enabling the identification of genes necessary for HIV infection and replication [5–7]. The first siRNA screen identified 281 HDFs [5], and 2 subsequent studies reported 295 and 232 HDFs [6, 7]. Interestingly, only 3 genes were implicated as HDFs in all 3 studies, with 27 genes overlapping in 2 of 3 studies (Table 1, part A). The limited degree of overlap suggests that differences in study methods (including the siRNA libraries used) exerted a sizable influence on experimental outcome. The likelihood of nonoverlapping false-positive and false-negative findings in the studies underscores the need for alternative approaches to verify HDFs discovered by siRNA screens; these may include genetic association studies in AIDS cohorts. We performed a high-density single-nucleotide polymorphism (SNP) screen in 278 HDF genes identified by Brass et al [5] and tested whether genetic variation in HDFs influences HIV infection or AIDS progression in clinical AIDS cohorts previously employed to identify CCR5Δ32, HLA, KIR, and other AIDS restriction genes [8, 9].
Table 1.

Association Analyses of HIV-Dependency Factors Identified by Multiple Functional Genomics Studies
Materials and Methods
Study population. Genotyping was performed in 1633 white individuals from 6 cohorts (Table 2). Informed consent was obtained from all patients. Subjects were included in 1 or more of the following clinical categories: (1) high-risk HIV-exposed seronegative; (2) HIV seroconverter; or (3) HIV seroprevalent with ⩾8 years of AIDS-free follow-up.
Table 2.

Demographics of Study Populations Used in Human Immunodeficiency Virus (HIV)-Dependency Factor Gene Association Analyses
SNP selection. Of the 281 HDF genes identified by Brass et al [5], 3 were excluded owing to missing or unverified genomic coordinates in the National Center for Biotechnology Information (NCBI) database Build 35 (C8orf14, FLJ46066, and CXorf50). HDF regions were defined as the gene and flanking upstream (5 kb) and downstream (1 kb) sequences. A total of 1064 SNPs in these regions were predicted to affect gene expression or function in dbSNP (NCBI Build 35), PupaSuite (http://pupasuite.bioinfo.cipf.es), or SNPeffect (http://snpeffect.vib.be). An additional 7406 tagging SNPs were selected using Tagger (http://www.broadinstitute.org/mpg/tagger), for a total of 8470 SNPs.
Genotyping and quality control. A total of 2929 SNPs were genotyped with the Illumina BeadChip kit (Illumina) and 5541 with the Affymetrix SNP Array 6.0 (Affymetrix) in the study population described above. Genotyping was performed in the Genotyping Facility at the Laboratory of Genomic Diversity, Center for Cancer Research, National Cancer Institute (NCI-Frederick, Frederick, MD). Scatterplots were visually inspected for all BeadChip SNPs and for Affymetrix 6.0 SNPs that were not in Hardy-Weinberg equilibrium, using Illumina BeadStudio or an in-house program to visualize Affymetrix intensity output. SNPs were excluded owing to assay failure (<95%call rate), inconclusive clustering, or minor allele frequency of <1%. All subjects had a call rate of ⩾92%, with an average of 98.1%.
Evaluation of tag SNPs. The web-based Tagger program [10] was used to assess haplotype coverage in the HDF regions identified by Brass et al [5]. Coverage was determined using the HapMap CEU panel for SNPs with a minimum allele frequency of 2%. Pairwise and multimarker predictors were calculated with an R2 threshold of 1.0.
Statistical analysis. Thirty-nine nonindependent statistical tests were used to examine associations between genotypes and HIV-related outcomes (3 HIV infection tests, 36 AIDS progression tests) (Table 3). Because there is an a priori evidence-based hypothesis that genes identified as HDFs by Brass et al [5] are involved in clinical HIV/AIDS outcomes, significance level thresholds were adjusted for the number of genes (n = 278) or SNPs (n = 6380) examined in this study, with the latter correction being more conservative (perhaps ultraconservative, owing to SNPs in linkage disequilibrium). Thresholds were also adjusted for the number of genetic hypotheses tested (HIV infection, n = 3; AIDS progression, n = 36). We report P values adjusted for both the gene test (no. of genes times no. of tests [ngenes × ntests]) and the SNP test (no. of SNPs times no. genes tests of tests [nSNPs × ntests]).
Table 3.


Statistical Hypotheses Used in Human Immunodeficiency Virus (HIV)-Dependency Factor Gene Association Analyses
SNP effect prediction. To predict SNP effects on gene expression or function, we entered highly significant SNPs into the SNPinfo web server (http://snpinfo.niehs.nih.gov) [11], which considers the potential impact of a SNP on transcriptional regulation, splicing, microRNA binding, and protein sequence and structure. The GENEVAR database (http://www.sanger.ac.uk/humgen/genevar) was also used to examine genotype-expression correlations in lymphoblastoid cell lines from the HapMap population.
Gene ontology and network analysis. The DAVID database was used to classify genes by their gene ontology terms [12, 13]. To visualize biomolecular interaction networks containing gene products of interest, interactions were gathered from the IntAct database [14] and the HIV-1, Human Protein Interaction Database [15–17] and imported into Cytoscape (version 2.6.2) [18, 19]. Tissue expression was determined using the BioGPS database (version 1.0.7.4885; http://biogps.gnf.org). The HIV-1, Human Protein Interaction Database was used to identify which genes are known to interact with HIV-1; a χ2 test was used to test for differences between the numbers of gene products in the database for different sets of genes.
Results
Based on the in vitro evidence generated by functional genomics studies, we hypothesized that genetic variants in HDFs would influence clinical HIV/AIDS outcomes. To evaluate this hypothesis, we performed a high-density screen of the genetic variation in HDF genes [5] comprising a panel of 8470 SNPs (Table 4). Of these, 6380 SNPs passed quality control metrics (Table 4) and were used for association testing. These SNPs had an average density of 1 per 3.6 kb of nucleotide sequence (range, 1 SNP per 0.1–22.8 kb). The median autosomal haplotype coverage in white subjects was 90.9% (Table 4), compared with 70.8% for the Affymetrix SNP Panel 6.0 and 80.0% for the Illumina Human Hap550 SNP array (data not shown).
Table 4.

HIV-Dependency Factor Polymorphisms Genotyped in This Study
We used 1633 individuals from HIV/AIDS cohorts to test for genetic associations, with clinical endpoints including HIV infection and AIDS progression outcomes (time to <200 CD4+ cells/mL, the 1987 and 1993 Centers for Disease Control and Prevention [CDC] definitions of AIDS, and AIDS-related death) (Table 3). To adjust for statistical artifacts generated by multiple comparisons (ie, 278 genes, 6380 SNPs, and 39 statistical tests) we computed odds ratios and P values using 3 methods: first, uncorrected P values for every SNP-test combination; second, a gene-test correction, with a significance threshold of P = .05/(ngenes × ntests) (eg, for the HIV infection phenotype and β = 0.05, the significance threshold is P = .05/[278 × 3] = 6.00 × 10−5); and third, a SNP-test correction, with a significance threshold of P = β/(nSNPs × ntests) (eg, for the HIV infection phenotype and β = 0.05, the significance threshold is P = .05/[6380 × 3] = 2.61 × 10−6). The gene-test correction may be somewhat less stringent owing to multiple SNPs tested for associations in each gene, whereas the SNP-test correction may be overly conservative, because local linkage disequilibrium within any HDF region may lead to haplotypes in which SNPs are correlated and nonindependent [20–22]. Higher confidence was placed on association P values that fell below the SNP-test threshold, whereas P values that fell between the gene-test and SNP-test thresholds were considered plausible but with a lower degree of statistical confidence that they reflect true genetic associations.
Six tests in 2 genes (nuclear receptor corepressor 2 and isocitrate dehydrogenase 1, abbreviated NCOR2 and IDH1, respectively) were significantly associated with HIV infection by the gene-test criterion. Two of the SNPs within the NCOR2 region were significant by the highly conservative SNP-test criterion (Table 5). The SNP association with the lowest P value was in NCOR2 (Figure 1A). Of 89 SNPs tested within the NCOR2 gene, 16 were associated with HIV infection with unadjusted P < .05, and 7 of these had unadjusted P < .01 (data not shown). NCOR2, also called SMRT, is a member of the nuclear corepressor family of histone deacetylases, which are components of the transcription silencing machinery [23]. NCOR2 is highly expressed in hematopoetic cells.
Table 5.

Human Immunodeficiency Virus (HIV)-Dependency Factor Single-Nucleotide Polymorphism (SNP) Associations with HIV Infection
Figure 1.

Genotype distribution for single-nucleotide polymorphisms that are highly associated with human immunodeficiency virus (HIV) infection. A, Seroconverters (SC) were significantly more likely to carry the variant T allele for rs1794942 in the NCOR2 gene, compared with high-risk HIV-exposed uninfected subjects (HREU) (A). The dominant model is shown. In a codominant genetic model for rs7578859 (located near the IDH1 gene), the variant C allele conferred protection against HIV infection in a dose-dependent manner, with 2 copies of the C allele providing the highest degree of protection (ie, the CC genotype was found at a higher frequency in HREU than in SC) and a single copy conferring less protection (B). OR, odds ratio.
A SNP near IDH1 was also strongly associated with HIV acquisition (Figure 1B; Table 5). Mutations in this metabolic enzyme occur frequently in malignant gliomas [24]. It has been suggested that decreased activity of IDH1 (eg, due to mutation or genetic variation) impairs the generation of a-ketoglutarate and nicotinamide adenine dinucleotide phosphate, which could stabilize downstream genes such as HIF-1β [24], which are involved in the regulation of immune cells under stress conditions [25].
To explore AIDS progression, we performed 36 association tests (Table 3) to give a gene-test correction threshold of P < 5.00 × 10−6 and a SNP-test threshold of P < 2.18 × 10−7. Although no SNPs met the experiment-wide corrected level of significance (248,820 SNP-test combinations), SNP-test associations in 2 gene regions merit comment. The 4 most significant SNP-test association P values obtained across the entire data set, generated by 2 SNPs within 5 kb of each other in the transmembrane 9 superfamily 2 (TM9SF2) gene, fell just short of the gene-test significance threshold (range, Pp.051 to P = .078) (Table 6). Furthermore, 18 of 26 SNPs within the TM9SF2 region displayed unadjusted P < .05 for ⩾1 AIDS progression test, and 6 had unadjusted P < .01 (data not shown). TM9SF2 is an evolutionarily conserved endosomal transmembrane protein [26]. Genetic studies in Drosophila melanogaster demonstrated that flies lacking 1 of the TM9SF genes developed abnormal hemocyte cytoskeletons, interfering with cell adhesion and phagocytosis to the detriment of cellular immunity [27]. Transcripts of TM9SF2 are highly expressed in peripheral blood cells.
Table 6.

Human Immunodeficiency Virus (HIV)-Dependency Factor Single-Nucleotide Polymorphism (SNP) Associations with AIDS Progression
A second region with a strong association signal involved the epidermal growth factor receptor (EGFR) gene, in which 1 SNP (rs4948007) produced highly significant unadjusted P values (range, P = 2.06 × 10−5 to P = 3.37 × 10−5) (Table 6). This SNP also produced unadjusted P < .01 in 25 of the 36 AIDS progression association tests (data not shown). Furthermore, 19 of 120 EGFR SNPs had unadjusted P < .01 in ⩾1 AIDS progression association test. EGFR encodes a well-studied epidermal growth factor receptor that has been implicated in the development of multiple human malignancies yet is responsive to monoclonal antibody therapeutics, such as cetuximab and panitumumab [28]. Expression of EGFR is also known to be influenced by HIV-1 Gag activity, which may indirectly contribute to cellular HIV-1 spread [29].
A list of HDF SNPs that displayed associations at lower levels of significance is presented in part A of Table 7. SNPs in 60 genes passed a test-corrected statistical threshold for infection [P = .017 = 0.05/3 tests], and 53 genes contained SNPs that were below the same significance limit for progression [P = 1.39 × 10−3 = 0.05/36 tests]. Ten of the HDF genes associated with HIV infection and 7 of the associated progression genes are known to interact with HIV (Table 7, part A). These proportions (16.7% and 13.2%, respectively) are similar to the proportion of HIV-interacting HDFs identified by Brass et al [5] (14.0%). As expected, all of these sets of HDFs are greatly enriched for proteins that interact with HIV in comparison to the genome (∼7.2%). Reported interactions between HDF gene products and HIV-1 proteins are shown in Figures 2A and 2B (infection and progression, respectively), with node size indicating genes meeting significance thresholds and color intensity indicating the degree of significance.
Table 7.


Single-Nucleotide Polymorphisms in HIV-Dependency Factors Identified by Brass [5], Konig [6], and/or Zhou [7] with Unadjusted Infection or Progression Association of P < .05
Figure 2.
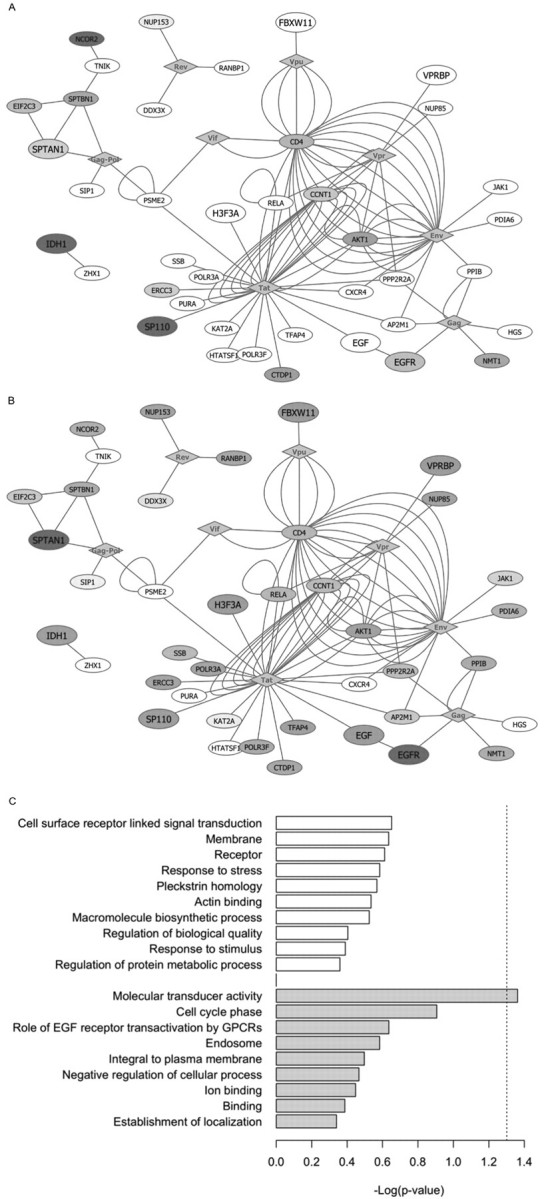
Extended network and gene ontology analyses of human immunodeficiency virus (HIV)-dependency factors (HDFs) of interest. The results of the single-nucleotide polymorphism (SNP) associations tested in this study were examined in 2 subsets of genes: HDFs with curated interactions with HIV-1 and HDFs that demonstrated low-level significant associations with HIV/AIDS outcomes in our study. A, Network visualization of HDFs identified by Brass et al [5] associated (test-adjusted P < .05) with HIV infection in this study that are known to interact with HIV-1. Data from the IntAct protein-protein interaction database [17–19] and the HIV-1, Human Protein Interaction Database [15, 16] were used to assemble networks in Cytoscape [20], with each edge representing a separate interaction. Shade intensity corresponds to the lowest P values for HIV infection in this study (Table 7, part A) for HDF genes, with dark gray indicating a smaller P value. Larger node size indicates that ⩾1 SNP in a gene was moderately associated with HIV infection. Gray diamonds represent HIV-1-encoded proteins. B, Same as panel A, but for AIDS progression. C, Functional groups (generated through clustering analysis performed using the DAVID tool) identified for genes containing SNPs with low-level infection and progression associations in this study. The negative logarithm of the P values of the functional terms associated with the clusters identified in the 60 infection HDFs are shown in white, whereas those associated with clusters found in the 53 AIDS progression genes are displayed in gray. The dotted line denotes statistical significance at −log (.05). EGF, epidermal growth factor; GPCRs, G protein-coupled receptors.
To discern the biological significance of low-level infection and progression signals, we evaluated their gene ontology annotations to determine whether any gene ontology terms were enriched in comparison with the 278 HDFs. The primary cluster identified among the 60 HDFs with infection signals contained genes associated with cell-surface receptor-linked signal transduction, including CD4 (Figure 2C; Table 7, part B). Other clusters were related to stress response, the pleckstrin homology domain (found in proteins involved in cell signaling or in cytoskeletal rearrangements [30]), and actin binding. A significant functional cluster was identified among the 53 HDFs, with AIDS progression association signals that included genes involved in molecular transduction, such as EGFR and SP110. Secondary clusters consisted of genes with a role in regulation of the cell cycle and genes related to endosomes. The HIV infection gene clusters correlate with events that occur during early viral infection, when the virus binds to cell-surface receptors and fuses with the host cell membrane, whereas the AIDS progression gene clusters correspond to mechanisms involved in the subsequent assembly and release of viral particles [31].
To explore the potential functional effects of genetic variation in the loci highly associated with HIV acquisition (NCOR2 and IDH1) and AIDS progression (TM9SF2 and EGFR), we investigated the SNPs that met the test-adjusted threshold of significance, as well as SNPs in high linkage disequilibrium with these variants (R2 ⩾ 0.8 in HapMap CEU), using the SNPinfo web server. We also investigated correlations between these SNPs and gene expression using the GENEVAR database. None of the SNPs examined in these 4 genes was predicted to affect transcript splicing, microRNA binding, or protein sequence or structure, but 3 SNPs near IDH1 were in transcription factor binding sites and may influence transcriptional regulation (Table 7, part C). In addition, SNPs in EGFR and TM9SF2 were significantly associated with altered lymphoblastoid gene expression in the HapMap CEU population (Table 7, part C).
Since the initial HDF report [5], 2 similar studies have been published [6, 7]. The overlap between these 3 studies is surprisingly limited: 3 genes (MED6, MED7, and RELA) were identified as HDFs in all 3 studies, 24 genes were identified as HDFs by Brass et al [5] and in 1 additional study, and 3 genes overlapped between the studies by Konig et al [6] and Zhou et al [7] (Table 1, part A). We specifically explored the genetic association signals in the 27 overlapping HDFs identified by Brass et al and 1 or both of the other studies. Of the 3 HDFs found in all 3 studies, MED7 demonstrated a moderate level of association with AIDS progression (lowest unadjusted P = 1.39 × 10−3) but no associations with HIV acquisition (Table 1, part B). Of the 570 SNP-test associations within the 24 HDF genes identified by Brass et al and either Konig et al or Zhou et al that produced significant unadjusted P values (P < .05), only SNPs in IDH1 demonstrated a statistically significant association with HIV infection after a gene-test correction (Table 1, part B).
The first AIDS-related genome-wide association study (GWAS) was published shortly before the initial HDF study [32]. One gene, zinc ribbon domain-containing 1 (ZNRD1), was associated with disease progression in the GWAS and identified as an HDF by Brass et al [5, 32]. ZNRD1 is located in the major histocompatibility class region and encodes an RNA polymerase subunit; it was again associated with disease progression in a follow-up GWAS [33] and a candidate gene study [34], with the latter also describing its role in viral replication in vitro. In our study, after P value adjustment for the number of statistical tests, 2 SNPs in the ZNRD1 region were associated with HIV acquisition, and a third was associated with AIDS progression (Table 7, part A). None of these SNPs was in high linkage disequilibrium with the 3 SNPs associated with longtermdisease nonprogression in the candidate gene analysis [34], although the haplotype coverage of these SNPs (as well as the SNPs identified by the initial GWAS) was limited in the current study.
Discussion
We describe the use of high-throughput genotyping to interrogate the known genetic variation in 278 HDFs with a high degree of haplotype coverage (Table 4) that allowed us to investigate genotype associations with 3 HIV-1 infection endpoints and 36 AIDS progression outcomes in several European American AIDS cohorts. After P values were adjusted for the number of HDFs and statistical tests (gene-test correction), only the HIV infection phenotype produced statistically significant associations: 2 SNPs in NCOR2 and 1 SNP near IDH1 (Table 5; Figure 1). It is not entirely unexpected that the association signals for HIV infection are stronger than those for AIDS progression, because the underlying hypothesis of the siRNA studies is that the HDFs are required for infection, which may not translate into clinically measurable effects on AIDS progression [5–7].
Because the NCOR2 gene product has a role in transcriptional regulation, genetic variants that alter its expression could conceivably have downstream effects on the expression of HIV gene products, influencing viral proliferation. IDH1 is involved in cellular antioxidant regeneration [35], and a SNP causing a reduction in gene expression (and therefore enzymatic activity) might result in an increase in cellular vulnerability to HIV infection. Functional effect prediction algorithms placed the IDH1 SNPs in a transcription factor binding site, which could affect expression of this gene (Table 7, part C). While neither of these genes has been previously associated with HIV/AIDS outcomes in genetic association studies (only in siRNA screens), it is plausible that the highly significant associations between SNPs in these 2 genes and HIV infection observed in this study have a biological basis, given their cellular functionalities.
To explore the underlying biological mechanisms behind the HDFs, we used a subset of HDF genes containing SNPs that were associated with either infection (60 genes) or progression (53 genes) after P value adjustment for the number of statistical tests used (Table 7, part A). When these gene signals were mapped onto known protein-protein interactions with HIV, differences between the groups of HDF genes associated with HIV infection versus AIDS progression were apparent. Many of the genes associated with both infection and progression displayed involvement in endosomes and protein transport (eg, IDH1 and EGFR) (Figures 2A and 2B). Gene ontology analysis indicated that genes producing infection signals were enriched in annotation terms involved in cell surface signal transduction, and genes with progression signals were enriched in terms related to molecular transduction and cell cycle regulation (Figure 2C; Table 7, part B). These clusters reflect our current knowledge about the cellular pathways involved at different points in the HIV replication cycle. Approximately 15% of the genes with low-level infection or progression association signals are known to interact with HIV proteins, which is double the percentage of HIV-interacting genes in the human genome.
Three functional genomics studies have each reported >200 HDFs involved in viral replication [5–7]. Despite the high level of control that can be exerted in an in vitro environment, only 3 genes signaled in all 3 studies: MED6, MED7, and RELA (Table 1, part A). MED6 and MED7 are mediator subunits that regulate RNA polymerase II activity [36]. RELA is a nuclear factor κB subunit and a key participant in immune and inflammatory responses, which have been shown to control Tat-mediated transcription [37]. The small degree of overlap between the 3 functional genomics studies has been noted in a recent meta-analysis [38]; suggested sources of variability include experimental noise, methodological differences, and disparities in statistical techniques. In our analyses, these 3 genes were relatively unremarkable, demonstrating weak associations with AIDS progression, with unadjusted P values in the range of 10−3 to 10−2 (Table 1, part B). The lack of strong association signals may be due to limited haplotype coverage or to the absence of function- and expression-altering polymorphisms in these genes.
ZNRD1 is the only HDF that has been correlated with disease progression in an AIDS genetic association study [32–34]. The 7 SNPs identified in the GWAS [32] were outside the gene region investigated in the present study (4 were 120 kb upstream and 3 were 16 kb downstream). Two of the SNPs genotyped in this study were located within the ZNRD1 haplotype blocks identified by Ballana et al [34]; however, neither of these SNPs demonstrated significant associations in our study. We found that 2 SNPs were weakly associated with HIV acquisition (lowest unadjusted P = 6.02 × 10−4) and 1 with disease progression (unadjusted P= 1.08 × 10−3), but these SNPs did not appear to be in linkage disequilibrium in the HapMap CEU population with the signals identified by Ballana et al [34]. The weakness of the ZNRD1 signal in our study may be due to inadequate coverage of the SNPs or differences in the phenotypes used in the various analyses (<200 CD4+ cells/µL, AIDS-related death, and the 1987 or 1993 CDC definitions of AIDS, all used in the current study; <350 CD4+ cells/µL or treatment initiation [32, 33]; long-term nonprogression [34]), but the presence of low-level SNP associations in the current study would affirm the involvement of ZNRD1 in cellular HIV infection.
The high-density SNP screening of HDF genes performed here adds another dimension to functional genomics experiments in the identification of endogenous factors that affect HIV infection or AIDS pathogenesis. Importantly, our genetic association study used clinical rather than in vitro data. Strong signals in a cohort population association study such as ours provide support for a true pathogenic influence that extends the implication of in vitro studies. Follow-up experiments are required to explore the biological basis for these signals, which may offer novel therapeutic targets as well as contribute to our understanding of the cellular mechanisms of HIV/AIDS pathogenesis.
Acknowledgment
We thank Michelle Hall, Mary McNally, Lisa Maslan, David Wells, Jamie Troxler, and the student interns at the Laboratory of Genomic Diversity for generating the genotypes.
Footnotes
Potential conflicts of interest: none reported.
Presented in part: 17th Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, February 2010 (abstract 458).
Disclaimer: The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Financial support: This project has been funded in whole or in part with federal funds from the National Cancer Institute (NCI), National Institutes of Health (NIH; contract HHSN261200800001E). AIDS Link to the Intravenous Experience was supported by NIH grants R01-DA-04334 and R01-DA-12568. Multicenter AIDS Cohort Study work was supported by NIH grants UO1-AI-35042, 5-MO1-RR-00722 (GCRC), UO1-AI-35043, UO1-AI-37984, UO1-AI-35039, UO1-AI-35040, UO1-AI-37613, UO1-AI-35041, and M01 RR00425 (National Center for Research Resources grant awarded to the GCRC at Harbor-University of California, Los Angeles, Research and Education Institute). The Multicenter Hemophilia Cohort Study is supported by NCI contract N02-CP-55504 with RTI International. The Hemophilia Growth and Development Study is funded by the NIH, National Institute of Child Health and Human Development, grant 1 R01 HD41224. NCI contracts include the following: UO1-AI-35042, 5-MO1-RR-00722 (GCRC), UO1-AI-35043, UO1-AI-37984, UO1-AI-35039, UO1-AI-35040, UO1-AI-37613, and UO1-AI-35041. This project has been funded in whole or in part with federal funds from the NCI, NIH, under contract N01-CO-12400. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Gallo RC, Montagnier L. The discovery of HIV as the cause of AIDS. N Engl J Med. 2003;349:2283–2285. doi: 10.1056/NEJMp038194. [DOI] [PubMed] [Google Scholar]
- 2.Vandekerckhove L, Verhofstede C, Vogelaers D. Maraviroc: integration of a new antiretroviral drug class into clinical practice. J Antimicrob Chemother. 2008;61:1187–1190. doi: 10.1093/jac/dkn130. [DOI] [PubMed] [Google Scholar]
- 3.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV- 1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 4.Menendez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 2010;85(1):210–231. doi: 10.1016/j.antiviral.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Brass AL, Dykxhoorn DM, Benita Y, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 6.Konig R, Zhou Y, Elleder D, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H, Xu M, Huang Q, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Hutcheson HB, Lautenberger JA, Nelson GW, et al. Detecting AIDS restriction genes: from candidate genes to genome-wide association discovery. Vaccine. 2008;26:2951–2965. doi: 10.1016/j.vaccine.2007.12.054. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet. 2004;36:565–574. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- 10.de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 11.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 13.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 14.Aranda B, Achuthan P, Alam-Faruque Y, et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–D531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu W, Sanders-Beer BE, Katz KS, Maglott DR, Pruitt KD, Ptak RG. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009;37:D417–D422. doi: 10.1093/nar/gkn708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinney JW, Dickerson JE, Fu W, Sanders-Beer BE, Ptak RG, Robertson DL. HIV-host interactions: a map of viral perturbation of the host system. AIDS. 2009;23:549–554. doi: 10.1097/QAD.0b013e328325a495. [DOI] [PubMed] [Google Scholar]
- 17.Ptak RG, Fu W, Sanders-Beer BE, et al. Cataloguing the HIV type 1 human protein interaction network. AIDS Res Hum Retroviruses. 2008;24:1497–1502. doi: 10.1089/aid.2008.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cline MS, Smoot M, Cerami E, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoggart CJ, Whittaker JC, De Iorio M, Balding DJ. Simultaneous analysis of all SNPs in genome-wide and re-sequencing association studies. PLoS Genet. 2008;4:e1000130. doi: 10.1371/journal.pgen.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 22.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 23.Cunliffe VT. Eloquent silence: developmental functions of class I histone deacetylases. Curr Opin Genet Dev. 2008;18:404–410. doi: 10.1016/j.gde.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan H, Bigner DD, Velculescu V, Parsons DW. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009;69(24):9157–9159. doi: 10.1158/0008-5472.CAN-09-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 26.Schimmoller F, Diaz E, Muhlbauer B, Pfeffer SR. Characterization of a 76 kDa endosomal, multispanning membrane protein that is highly conserved throughout evolution. Gene. 1998;216:311–318. doi: 10.1016/s0378-1119(98)00349-7. [DOI] [PubMed] [Google Scholar]
- 27.Bergeret E, Perrin J, Williams M, et al. TM9SF4 is required for Drosophila cellular immunity via cell adhesion and phagocytosis. J Cell Sci. 2008;121:3325–3334. doi: 10.1242/jcs.030163. [DOI] [PubMed] [Google Scholar]
- 28.Martinelli E, De Palma R, Orditura M, De Vita F, Ciardiello F. Antiepidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin Exp Immunol. 2009;158:1–9. doi: 10.1111/j.1365-2249.2009.03992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valiathan RR, Resh MD. Expression of human immunodeficiency virus type 1 gag modulates ligand-induced downregulation of EGF receptor. J Virol. 2004;78:12386–12394. doi: 10.1128/JVI.78.22.12386-12394.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(pt 1):1–18. [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez C, Hope TJ. The ins and outs of HIV replication. Cell Microbiol. 2005;7:621–626. doi: 10.1111/j.1462-5822.2005.00516.x. [DOI] [PubMed] [Google Scholar]
- 32.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fellay J, Ge D, Shianna KV, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5(12):e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballana E, Senserrich J, Pauls E, et al. ZNRD1 (zinc ribbon domaincontaining 1) is a host cellular factor that influences HIV-1 replication and disease progression. Clin Infect Dis. 2010;50:1022–1032. doi: 10.1086/651114. [DOI] [PubMed] [Google Scholar]
- 35.Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 36.Sato S, Tomomori-Sato C, Parmely TJ, et al. A set of consensus mammalian mediator subunits identified by multidimensional protein identification technology. Mol Cell. 2004;14:685–691. doi: 10.1016/j.molcel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Stroud JC, Oltman A, Han A, Bates DL, Chen L. Structural basis of HIV-1 activation by NF-kB- a higher-order complex of p50:RelA bound to the HIV-1 LTR. J Mol Biol. 2009;393(1):98–112. doi: 10.1016/j.jmb.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bushman FD, Malani N, Fernandes J, et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]