

Abstract
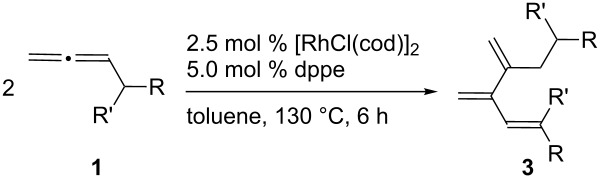
A rhodium(I)/dppe catalyst promoted dimerization of monosubstituted allenes in a stereoselective manner to give cross-conjugated trienes, which are different from those obtained by a palladium catalyst.
Keywords: allene, cross-conjugated triene, dimerization, rhodium, stereoselective
Introduction
Cross-conjugated trienes, known as [3]dendralenes [1], are attractive synthetic precursors used for consecutive double [4 + 2] cycloaddition reactions [2–4] to provide a rapid access to polycyclic carbon frameworks. Thus, a number of methods for the preparation of the parent 3-methylenepenta-1,4-diene [5] and its substituted derivatives [6–17] has been developed. Among these, transition-metal-catalyzed dimerization of allenes presents a unique entry to substituted cross-conjugated trienes. For example, a nickel(0)/triphenylphosphine complex catalyzes a dimerization reaction of 3-methylbuta-1,2-diene to afford 2,5-dimethyl-3,4-bismethylenehex-1-ene [18–19]. The nickel-catalyzed reaction, however, leads to a complex mixture of products when monosubstituted allenes such as penta-1,2-diene and 1-phenylpropa-1,2-diene are employed [20]. On the other hand, a palladium-catalyzed dimerization reaction of monosubstituted allenes produces substituted cross-conjugated trienes 2 in high yield (Scheme 1) [21]. We report here that dimerization of monosubstituted allenes is also catalyzed by a rhodium(I)/dppe complex to form cross-conjugated trienes 3, which are different from those obtained with the palladium catalyst.
Scheme 1.

A new entry to substituted cross-conjugated trienes.
Results and Discussion
We initiated our study using undeca-1,2-diene (1a) as the model substrate and a rhodium(I) complex as the catalyst (Table 1). When 1a was treated with a catalytic amount of [RhCl(cod)]2 (2.5 mol %, cod = cycloocta-1,5-diene) in toluene at 130 °C for 12 h, 2a was formed in 40% NMR yield along with another minor dimerized product (13% NMR yield) and unidentified compounds (Table 1, entry 1). The structure of the minor dimerized product was determined to be (E)-10,11-dimethyleneicos-8-ene (3a) by 1H and 13C NMR spectroscopy. Thus, the two isomeric dimers, one identical to the isomer obtained by the palladium-catalyzed reaction and the other a different isomer, were produced by the rhodium-catalyzed reaction. Next, several phosphine ligands were examined (Table 1, entries 2–5). To our delight, the use of the dppe ligand suppressed the formation of 2a and the unidentified compounds, and increased the NMR yield of 3a to 96% (86% isolated yield, Table 1, entry 4). A complex mixture of products was obtained when the reaction temperature was lowered from 130 °C to 90 °C (Table 1, entry 6). Moreover, the use of [Rh(OH)(cod)]2 and Rh(acac)(cod) as the precatalyst resulted in a decrease of the reaction rate (Table 1, entries 7 and 8).
Table 1.
Optimization of reaction conditionsa.
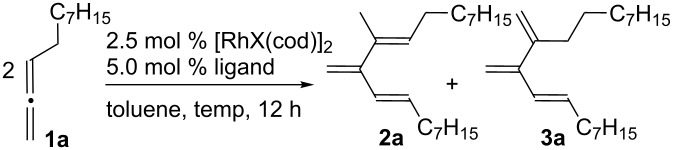 | |||||
| Entry | X | Ligandb | T (°C) | Yield of 2a (%)c |
Yield of 3a (%)c |
| 1 | Cl | none | 130 | 40 | 13 |
| 2 | Cl | PPh3d | 130 | 24 | 18 |
| 3 | Cl | dppm | 130 | 24 | 37 |
| 4 | Cl | dppe | 130 | <5 | 96 (86) |
| 5 | Cl | dppp | 130 | 17 | 50 |
| 6 | Cl | dppe | 90 | 38 | 24 |
| 7 | OH | dppe | 130 | 40 | 10 |
| 8 | acace | dppe | 130 | 44 | <5 |
aReactions conducted on a 0.4 mmol scale.
bdppm = 1,1-bis(diphenylphosphino)methane, dppe = 1,2-bis(diphenylphosphino)ethane, dppp = 1,3-bis(diphenylphosphino)propane.
cNMR yield using mesitylene as an internal standard. Isolated yield given in parenthesis.
dUsing 10 mol % of PPh3.
eUsing 5.0 mol % of Rh(acac)(cod).
We propose that the dimerization reaction proceeds through the pathway outlined in Scheme 2. Initially, two molecules of 1a coordinate to a rhodium(I) center at the terminal carbon–carbon double bonds from their sterically less-hindered sides. Oxidative cyclization occurs in a head-to-head manner to form the five-membered rhodacyclic intermediate A [22–25], which is in equilibrium with another rhodacyclic intermediate B via σ–π–σ isomerization. Then, β-hydride elimination takes place with B to form rhodium hydride C stereoselectively. Finally, reductive elimination from C yields 3a together with the catalytically active rhodium(I) complex. It is also conceivable, however, that oxidative cyclization of two molecules of 1a occurs in a tail-to-tail manner to directly furnish B. The other isomer 2a could be formed through allylic 1,3-migration of rhodium from C and subsequent reductive elimination.
Scheme 2.

A proposed reaction pathway.
Under the optimized reaction conditions using dppe as the ligand, various monosubstituted allenes 1b–j were subjected to the catalytic dimerization reaction (Table 2). In most cases, essentially one isomer 3 was formed, and the other isomer 2 was barely detectable in the 1H NMR spectrum of the crude reaction mixture (<5%). Allenes 1b–i possessing a primary alkyl group reacted well to afford the corresponding products 3b–i in yields ranging from 63% to 90% (Table 2, entries 1–8). Functional groups such as benzyloxy, siloxy, hydroxy and cyano groups were tolerated in the alkyl chain under the reaction conditions. Cyclohexylpropa-1,2-diene (1j) possessing a secondary alkyl group also participated in the dimerization reaction (Table 2, entry 9). On the other hand, 1,1-disubstituted allenes such as 3-methylbuta-1,2-diene and 3-pentylocta-1,2-diene failed to undergo the dimerization reaction, in contrast to the nickel-catalyzed reaction [18–19].
Table 2.
Synthesis of cross-conjugated trienes by the allene dimerization reactiona.
 | |||||
| Entry | 1 | R | R' | 3 | Yield (%)b |
| 1 | 1b | C5H11 | H | 3b | 85 |
| 2 | 1c | CH2Ph | H | 3c | 78c |
| 3 | 1d | Cy | H | 3d | 83d |
| 4 | 1e | CH2OBn | H | 3e | 70 |
| 5 | 1f | (CH2)3OBn | H | 3f | 78 |
| 6 | 1g | (CH2)3OSiMe2t-Bu | H | 3g | 90 |
| 7 | 1h | (CH2)3OH | H | 3h | 63 |
| 8 | 1i | (CH2)3CN | H | 3i | 75 |
| 9 | 1j | –(CH2)5– | 3j | 60d | |
aReactions conducted on a 0.4 mmol scale.
bIsolated yield unless otherwise noted.
cThe product was accompanied by a small amount of an unidentified impurity.
dNMR yield using mesitylene as an internal standard.
Next, we examined the consecutive double [4 + 2] cycloaddition reaction of the cross-conjugated trienes obtained in the present study. Triene 3a was treated with 4-phenyl-1,2,4-triazoline-3,5-dione (4, PTAD), a highly reactive dienophile, in toluene at 0 °C (Scheme 3). The conversion of 3a was complete within 1 h, and after chromatographic isolation, bisadducts 5a and 5a’ were obtained in 75% and 6% yields, respectively. The major bisadduct 5a resulted from initial addition to the more congested diene moiety of 3a (site β). When tetracyanoethylene (6, TCNE), which was a less reactive dienophile than 4, was used, [4 + 2] cycloaddition also occurred preferentially at site β, but only once on heating at 60 °C for 24 h.
Scheme 3.

[4 + 2] cycloaddition reaction of 3a with PTAD and TCNE.
Conclusion
In summary, we have developed a new dimerization reaction of monosubstituted allenes catalyzed by a rhodium(I)/dppe complex, allowing the stereoselective formation of substituted cross-conjugated trienes. It is interesting that the rhodium catalyst and the palladium catalyst gave different types of cross-conjugated trienes.
Experimental
General procedure for rhodium-catalyzed dimerization of monosubstituted allenes
To a side-arm tube equipped with a stirrer bar, was added [RhCl(cod)]2 (4.9 mg, 2.5 mol %) and dppe (7.7 mg, 5 mol %). The tube was evacuated and refilled with argon three times. Then, toluene (4 mL) and substrate 1 (0.4 mmol) were added via syringe and the tube was closed. After heating at 130 °C for 6 h, the reaction mixture was cooled to room temperature, passed through a pad of Florisil® and eluted with ethyl acetate (≈ 90–100 mL). The filtrate was concentrated under reduced pressure and the residue purified by preparative thin-layer chromatography to give product 3. Although the isolated 3 was relatively labile, it could be kept at −30 °C for days without any detectable decomposition or polymerization.
Supporting Information
Experimental details and spectroscopic data for new compounds.
Acknowledgments
This work was supported in part by MEXT (Grant-in-Aid for Challenging Exploratory Research, No. 21656033). T. T. is grateful for Research Fellowship from JSPS for Young Scientists.
This article is part of the Thematic Series "Allene chemistry".
Contributor Information
Tomoya Miura, Email: tmiura@sbchem.kyoto-u.ac.jp.
Masahiro Murakami, Email: murakami@sbchem.kyoto-u.ac.jp.
References
- 1.Hopf H. Nature. 2009;460:183–184. doi: 10.1038/460183a. [DOI] [PubMed] [Google Scholar]
- 2.Tsuge O, Wada E, Kanemasa S. Chem Lett. 1983;12:239–242. doi: 10.1246/cl.1983.239. [DOI] [Google Scholar]
- 3.Kwon O, Park S B, Schreiber S L. J Am Chem Soc. 2002;124:13402–13404. doi: 10.1021/ja028086e. [DOI] [PubMed] [Google Scholar]
- 4.Brummond K M, You L. Tetrahedron. 2005;61:6180–6185. doi: 10.1016/j.tet.2005.03.141. [DOI] [Google Scholar]
- 5.Bradford T A, Payne A D, Willis A C, Paddon-Row M N, Sherburn M S. J Org Chem. 2010;75:491–494. doi: 10.1021/jo9024557. [DOI] [PubMed] [Google Scholar]
- 6.Woo S, Squires N, Fallis A G. Org Lett. 1999;1:573–576. doi: 10.1021/ol990695c. [DOI] [Google Scholar]
- 7.Chang H-M, Cheng C-H. J Org Chem. 2000;65:1767–1773. doi: 10.1021/jo991651k. [DOI] [PubMed] [Google Scholar]
- 8.Brummond K M, Chen H, Sill P, You L. J Am Chem Soc. 2002;124:15186–15187. doi: 10.1021/ja027588p. [DOI] [PubMed] [Google Scholar]
- 9.Chin C S, Lee H, Park H, Kim M. Organometallics. 2002;21:3889–3896. doi: 10.1021/om020410r. [DOI] [Google Scholar]
- 10.Kang B, Kim D-H, Do Y, Chang S. Org Lett. 2003;5:3041–3043. doi: 10.1021/ol035014z. [DOI] [PubMed] [Google Scholar]
- 11.Shi M, Shao L-X. Synlett. 2004:807–810. doi: 10.1055/s-2004-820017. [DOI] [Google Scholar]
- 12.Bräse S, Wertal H, Frank D, Vidović D, de Meijere A. Eur J Org Chem. 2005:4167–4178. doi: 10.1002/ejoc.200500330. [DOI] [Google Scholar]
- 13.Bradford T A, Payne A D, Willis A C, Paddon-Row M N, Sherburn M S. Org Lett. 2007;9:4861–4864. doi: 10.1021/ol7021998. [DOI] [PubMed] [Google Scholar]
- 14.Miller N A, Willis A C, Paddon-Row M N, Sherburn M S. Angew Chem, Int Ed. 2007;46:937–940. doi: 10.1002/anie.200603335. [DOI] [PubMed] [Google Scholar]
- 15.Shimp H L, Hare A, McLaughlin M, Micalizio G C. Tetrahedron. 2008;64:3437–3445. doi: 10.1016/j.tet.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S, Seomoon D, Lee P H. Chem Commun. 2009:1873–1875. doi: 10.1039/b823037d. [DOI] [PubMed] [Google Scholar]
- 17.Beydoun K, Zhang H-J, Sundararaju B, Demerseman B, Achard M, Xi Z, Bruneau C. Chem Commun. 2009:6580–6582. doi: 10.1039/b913595b. [DOI] [PubMed] [Google Scholar]
- 18.Englert M, Jolly P W, Wilke G. Angew Chem, Int Ed Engl. 1972;11:136–137. doi: 10.1002/anie.197201361. [DOI] [Google Scholar]
- 19.Pasto D J, Huang N-Z. Organometallics. 1985;4:1386–1395. doi: 10.1021/om00127a014. [DOI] [Google Scholar]
- 20.Pasto D J, Huang N-Z, Eigenbrot C W. J Am Chem Soc. 1985;107:3160–3172. doi: 10.1021/ja00297a022. [DOI] [Google Scholar]
- 21.Arisawa M, Sugihara T, Yamaguchi M. Chem Commun. 1998:2615–2616. doi: 10.1039/a807527a. [DOI] [Google Scholar]
- 22.Osakada K, Choi J-C, Sarai S, Koizumi T, Yamamoto T. Chem Commun. 1997:1313–1314. doi: 10.1039/a701859b. [DOI] [Google Scholar]
- 23.Lu P, Ma S. Org Lett. 2007;9:2095–2097. doi: 10.1021/ol070632r. [DOI] [PubMed] [Google Scholar]
- 24.Park J H, Kim E, Kim H-M, Choi S Y, Chung Y K. Chem Commun. 2008:2388–2390. doi: 10.1039/b718107h. [DOI] [PubMed] [Google Scholar]
- 25.Kawamura T, Inagaki F, Narita S, Takahashi Y, Hirata S, Kitagaki S, Mukai C. Chem–Eur J. 2010;16:5173–5183. doi: 10.1002/chem.200903568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and spectroscopic data for new compounds.