

Abstract
The all-ferrous, carbene-capped Fe4S4 cluster, synthesized by Deng and Holm (DH complex), has been studied with density functional theory (DFT). The geometry of the complex was optimized for several electronic configurations. The lowest energy was obtained for the broken-symmetry (BS) configuration derived from the ferromagnetic state by reversing the spin projection of one of the high spin (Si = 2) irons. The optimized geometry of the latter configuration contains one unique and three equivalent iron sites, which are both structurally and electronically clearly distinguishable. For example, a distinctive feature of the unique iron site is the diagonal Fe⋯S distance, which is 0.3 Å longer than for the equivalent irons. The calculated 57Fe hyperfine parameters show the same 1:3 pattern as observed in the Mössbauer spectra and are in good agreement with experiment. Broken-symmetry analysis of the exchange interactions in the optimized geometry for the 1:3, MS = 4, BS configuration confirms the prediction of an earlier study that the unique site is coupled to the three equivalent ones by strong antiferromagnetic exchange (J > 0 in JΣj<4Ŝ4 · Ŝj) and that the latter are mutually coupled by ferromagnetic exchange (J′ < 0 in J′Σi<j<4Ŝi · Ŝj). In combination, these exchange couplings stabilize an S = 4 ground state in which the composite spin of the three equivalent sites (S123 = 6) is antiparallel to the spin (S4 = 2) of the unique site. Thus, DFT analysis supports the idea that the unprecedented high value of the spin of the DH complex and, by analogy, of the all-ferrous cluster of the Fe-protein of nitrogenase, results from a remarkably strong dependence of the exchange interactions on cluster core geometry. The structure dependence of the exchange-coupling constants in the FeII-(μ3-S)2-FeII moieties of the all-ferrous clusters is compared with the magneto–structural correlations observed in the data for dinuclear copper complexes. Finally, we discuss two all-ferric clusters in the light of the results for the all-ferrous cluster.
1. Introduction
Presently the all-ferrous state of a Fe4S4 cluster with tetrahedral coordination sites has been established in four molecules. These systems include the Fe-protein of nitrogenase in Azotobacter vinelandii,1 the activator protein of the dehydrogenase from Acidaminococcus fermentans,2 and the synthetic complexes [Fe4S4(CN)4]4- 3 and [Fe4S4(Pri2NHCMe2)4]0 (Pri2NHCMe2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene);4 the latter complex being hereafter referred to as the Deng–Holm (DH) complex. The observation of a [Fe4S4]0 state in the Fe-protein raised the possibility that an Fe4S4 cluster, which normally delivers one electron at a time by alternating between [Fe4S4]1+/2+, may also donate two electrons in a concerted manner, using the redox couple [Fe4S4]0/2+, thereby doubling the efficiency of the nitrogen reduction process in terms of the number of Mg-ATP molecules hydrolyzed.5 A recent comparison of the structural and spectroscopic data for the all-ferrous Fe-protein and the DH complex has revealed that their [Fe4S4]0 clusters are remarkably similar.6 Crystal structure and Mössbauer spectra show that the irons in the all-ferrous state appear as two subsets with a 1:3 ratio. A similar pattern has been observed in all-ferrous Fe8S8 clusters, suggesting a modular formulation of these super clusters in terms of their cubane constituents.7 The system spins of the DH complex and the protein bound clusters are 4. The S = 4 ground-state spin results from anti-parallel alignment of the spin of the unique iron to the three parallel aligned spins of the equivalent irons: S = 3 Sloc – Sloc = 2 Sloc = 4. The same expression for the system spin in terms of the local spins was found to hold for an all-cobaltous (Sloc = 3/2) [Co4S4]0 cluster with spin S = 3.8 Analysis of the spin-state energies with the Heisenberg–Dirac–van Vleck (HDvV) Hamiltonian, , shows that the condition for having the 1:3, S = 4 ground state is a combination of three strong antiferromagnetic exchange-coupling constants (J = J4,i > 0 for i = 1, 2, 3) and three weaker antiferromagnetic (or even ferromagnetic) couplings between the equivalent sites (0 < J′ = Jij ≪ J or J′ < 0 for i < j < 4).6 To investigate if this strong condition, which stipulates highly dissimilar coupling constants for rather similar superexchange units, can be fulfilled in a computational setting, we have performed broken-symmetry density functional calculations. The quality of the broken-symmetry DFT approximation for the S = 4 state has been monitored by comparing the predictions for the cluster structure and the 57Fe hyperfine parameters with the experimental data. The results will be discussed in the context of magneto–structural data available in the literature.
2. Methods
The DFT calculations were performed with the Amsterdam Density Functional package (ADF 2008 version 01).9 The geometries for three electronic configurations, labeled MS = 8, MS = 4, and MS = 0 according to their total magnetic quantum number, were optimized using the functional/basis set VWNBP/TZP. The MS = 8 configuration represents the magnetic substate |S = 8, MS = 8> of the ferromagnetic state and the other two are broken-symmetry configurations.10 The total magnetic quantum number, MS, of these configurations is the sum of the local magnetic quantum numbers (MS1, MS2, MS3, MS4), which are given by (2, 2, 2, 2) for MS = 8, (2, 2, 2, −2) for MS = 4, and (2, 2, −2, −2) for MS = 0. The calculations for the BS configurations were initiated using the keyword SPINFLIP. The lowest energy was obtained for the MS = 4 configuration with the optimized MS = 8 and MS = 0 configurations being, respectively, 10,447 cm-1 and 819 cm-1 higher in energy. To test for trapping in local minima, we initiated the geometry optimization for the lowest MS = 4 configuration with two geometries, a structure obtained from the X-ray structure by truncating the side chains of the carbene ligands (see Figure 1) and the same truncated structure in which the core was symmetrized by averaging iron–iron distances to 2.70 Å. The two optimizations yielded the same optimized geometry. The MS = 4 configuration has preponderant S = 4 character (∼70%, see Supporting Information), suggesting that the S = 4 state is the ground state of the cluster and that both the geometry and the properties calculated for the MS = 4 configuration are good approximations for those for the ground state. In order to verify whether the S = 4 state is the ground state we have evaluated the exchange-coupling constants appearing in the HDvV Hamiltonian (see Introduction). Since the HDvV Hamiltonian gives a valid description of the exchange splittings only when the spin state energies have been determined for the same geometry (cf. refs. 6 and 11), we have evaluated the SCF energies for the MS = 8, MS = 4, and MS = 0 configurations in single point calculations for the same structure, for which we have adopted the optimized geometry of the MS = 4 configuration. These three energies allowed us to evaluate the exchange-coupling constants J and J′ in the MS = 4 optimized structure as described in section 3.3.
Figure 1.
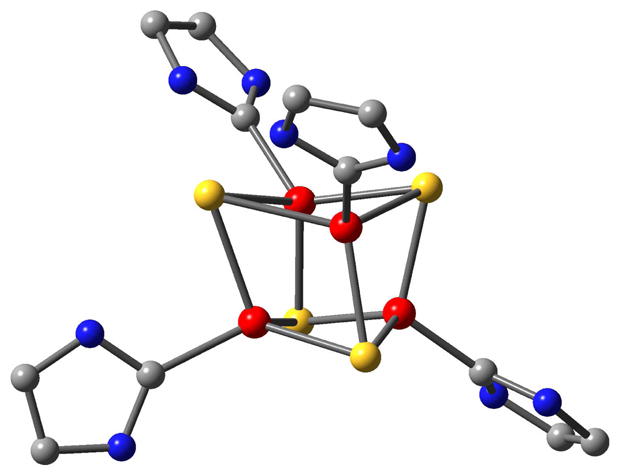
Optimized structure of [Fe4S4(imidazol-2-ylidene)4]0, the computational model used for the DH complex. The unique iron site, labeled Fe4 in the text, is located at the bottom on the right. Color code: carbon (grey), nitrogen (blue), iron (red), and sulfur (yellow). Hydrogen atoms are not shown for the sake of clarity.
The exchange-coupling constants were also calculated with the OPBE functional, which has been recommended for evaluating relative spin state energies,12 to estimate the functional dependency of the results. In these calculations J and J′ were evaluated from the OPBE/TZP SCF energies for the MS = 8, MS = 4, and MS = 0 configurations obtained from single point calculations for the same VWNBP/TZP-optimized MS = 4 structure used above.
The magnetic hyperfine coupling (A) and electric field gradient (EFG) tensors of the 57Fe sites in the optimized geometry for the MS = 4 configuration were evaluated with the keywords ESR and QTENS. Relativistic effects were accounted for in scalar ZORA (thus, excluding small contributions from spin–orbit coupling induced orbital momentum to the A values).
All calculations were spin unrestricted and used for the SCF convergence and integration accuracy parameters the settings 10-5 and 4.0, respectively.
3. Results and Discussion
In the following sections we present the DFT results for the structure, the 57Fe hyperfine parameters, and the exchange-coupling constants for the optimized MS = 4 configuration of the (truncated) DH complex and compare them with X-ray and spectroscopic literature data for this system.
3.1 Structure
The DFT optimized geometry for the MS = 4 configuration (II in Figure 2) has an approximately C3v symmetric core with the three-fold axis running through one of the irons (Fe4); the remaining irons (Fe1-3) are nearly equivalent by symmetry. Averages and standard deviations for selected metric parameters for the two types of site are listed in Table 1. The DFT distances between equivalent sites are shorter than the distances between the unique site and the equivalent sites by 0.09 Å. Concomitantly, the FeiSFej (i < j < 4) angles are smaller than the Fe4SFej angles by 6.0°. The corresponding differences for the X-ray structure of the DH complex, also listed in Table 1, are 0.10 Å and 3.6°. The changes in the Fe⋯Fe distances as obtained by DFT are about equal to the changes observed in the X-ray structure but the angular changes predicted by DFT are considerably larger. At constant Fe-S bond length, small changes in Fei⋯Fej distance, Δ(FeiFej), are related to changes in the FeiSFej angle, Δ(FeiSFej), by the expression
Figure 2.
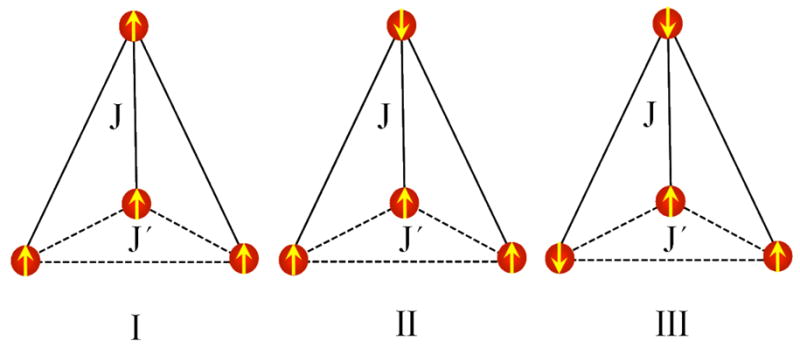
Spin configurations with magnetic quantum numbers MS = 8 (I), 4 (II), and 0 (III) for which the energies (EI, EII, and EIII) were calculated with DFT in the optimized geometry for II (see section 2) to obtain the exchange-coupling constants J and J′ for the 1:3 structure (section 3.3). The unique site (Fe4) is located at the top.
Table 1. Selected Structural Parameters for S = 4 Ground State of DH Complex.
| Fei (Fej) site(s) |
Fei⋯Fej (Å) | FeiSFejb (°) | Fei⋯Sic (Å) | Si⋯Sjc (Å) | Fei-S f (Å) | Fei-Ci (Å) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-ray a | DFT | X-ray a | DFT | X-ray a | DFT | X-ray | DFT | X-ray | DFT | X-ray | DFT | |
| i = 4, j < 4 d | 2.732 e | 2.631 | 72.09 | 70.53 | 4.06 | 4.15 | 3.804 | 3.951 | 2.312 | 2.242 | 2.07 | 1.93 |
| i < j < 4 d | 2.633 | 2.552 | 68.411 | 64.522 | 3.894 | 3.822 | 3.683 | 3.631 | 2.341 | 2.367 | 2.121 | 2.051 |
From ref. 4.
Average over two bridges.
Sulfide Si is diagonal to Fei.
Average over three equivalent irons.
Subscript gives standard deviation in the least significant decimal(s).
Average over bound sulfurs.
| (1) |
Using this expression we obtain for the DFT / X-ray angular increments Δ(FeiSFej) = 6.0° / 3.6° the distance changes Δ(FeiFej) = 0.20 Å / 0.12 Å. Thus, the increments in the FeiSFej bond angle and the associated Fei⋯Fej distance occur in the X-ray structure at approximately fixed Fe-S distances but are in the DFT structure accompanied by changes in the Fe-S bond length (see Table 1). This discrepancy between the DFT and X-ray structures is also reflected in the diagonal Fei⋯Si distances (Table 1), which for the unique site (i = 4) are longer than for the equivalent sites (i = 1-3) by 0.33 Å and 0.17 Å in the DFT and X-ray structures, respectively. The larger distortion in the DFT structure may be related to an overestimation of the exchange-coupling constants by this method (see section 3.3). The average DFT / X-ray values for the Fe⋯Fe and S⋯S distances are 2.59 Å / 2.68 Å and 3.79 Å / 3.74 Å, respectively, showing that the tetrahedron defined by the four irons is smaller than the one spanned by the four sulfurs as in all known Fe4S4 clusters.13 The edges of the 4-Fe tetrahedron in the DFT structure are shorter than in the X-ray structure by 0.09 Å, while those for the 4-S tetrahedron are 0.05 Å longer. Thus, the DFT structure displays a more pronounced size difference between the 4-Fe and 4-S tetrahedra than the X-ray structure. This feature is reflected in the average FeSFe angles, 67.5° / 70.2° (DFT / X-ray), which are more acute in the DFT structure than in the X-ray structure. Despite these differences, the agreement of the DFT structure with the X-ray structure is quite satisfactory.
3.2 Hyperfine parameters
Table 2 lists the values for the 57Fe quadrupole splitting (ΔEQ), the asymmetry parameter of the EFG (η), and the isotropic contribution to the magnetic hyperfine coupling constants for the unique and equivalent iron sites in the DH complex as obtained from Mössbauer (Mb) spectroscopy and DFT calculations for the MS = 4 configuration (II in Figure 2). The overall agreement between the DFT and Mössbauer results for these parameters is excellent, which inspires confidence in the quality of the DFT solution. Both methods give a large, positive ΔEQ for the unique site and a negative ΔEQ of about half the magnitude for the equivalent sites. The isotropic contribution to the local magnetic hyperfine coupling constants for the unique site is about half the value for the equivalent sites, both in experiment and theory. The discrepancy between the Mb and DFT results for aiso is actually larger than apparent from Table 2 due to the presence of a positive orbital contribution that is not included in the DFT number.14 The Fermi contact term obtained from DFT is often smaller than observed as this method has the propensity to overestimate the covalent reduction of the spin population in the iron 3d shell. The results of Table 2 reveal that the 1:3 differentiation of the iron sites in the structure of the DH complex must have a marked impact on their orbital structures. The parameter values in Table 2 can be compared with the Mössbauer parameters for the two high-spin FeII sites in the [Fe4S4]1+ cluster in the ferredoxin from reduced Bacillus stearothermophilus:15 ΔEQ = 1.89 mm/s, η = 0.32, aiso = −88 kG,16 and isomer shift δ = 0.58 mm/s. The quadrupole splitting in the ferredoxin is positive, like for the unique site in the DH complex but smaller in size (Table 2). The aiso value for the ferrous sites in the [Fe4S4]1+ cluster is close to the value listed for the unique site in Table 2, suggesting that the unique site in the DH complex has a greater resemblance with the ferrous sites in common ferredoxins than the equivalent sites. The 57Fe isomer shift for the ferrous sites in [Fe4S4]1+ (0.58 mm/s) is bracketed by the values δ4 = 0.54 mm/s and δ1-3 = 0.61 mm/s for the DH complex. The difference in the latter values correlates with a small difference in the Fe–carbene distances (Table 1). In absence of an isomer-shift calibration for the computational procedure described in section 2, we give here no computational estimate for this quantity.
Table 2. 57Fe Quadrupole splitting, Asymmetry Parameter, and Isotropic Contribution to Magnetic Hyperfine Coupling of Iron Sites in DH Complex.
| Fei site |
ΔEQ (mm/s) | ηd | aisoie (kG) | |||
|---|---|---|---|---|---|---|
| Mb c | DFT b | Mb c | DFT | Mb c | DFT f | |
| i = 4 | 2.96 | 2.51 | 0.59 | 0.57 | −78 | −66 |
| i = 1-3 a | −1.53 | −1.26 | 0.07 | 0.22 | −156 | −113 |
Average over equivalent sites.
ΔEQ = 2-1 e Q Vzz (1 + η2/3)1/2, using 57Fe (I = 3/2) quadrupole moment Q = 0.17 barn.
Taken from Mössbauer study in ref. 6; A-values in Table 1 of ref. 6 are presented in the coupled-spin Ŝ · Ai · Îi convention (S = 4) and have been converted here to local a-values, using spin projection factors: a4 = −(5/2)A4 and a1-3 = (15/7) A1-3.
Asymmetry parameter of EFG, η = (Vxx − Vyy)/Vzz where the Vqq are the eigenvalues of the EFG tensor, using convention |Vxx| ≤ |Vyy| ≤ |Vzz|.
Isotropic part of magnetic hyperfine coupling tensor in the convention with Si = 2 and Ii = 1/2 (nuclear ground state) and 3/2 (nuclear excited state) are the electronic and nuclear spins of 57Fe site i.
Values obtained from the Ai-values listed in ADF output for the BS MS = 4 configuration II (S = 4 convention), with the relations a4 = −2 A4 and a1-3 = +2 A1-3.
3.3 Exchange-coupling constants
As the [Fe4S4]0 core in the optimized structure for the BS, MS = 4 configuration (II in Figure 2) has approximately C3v symmetry, the HDvV Hamiltonian for describing the exchange interactions between the iron sites can be expressed as
| (2) |
where c is a spin-independent constant and J and J′ are exchange-coupling constants. The expectation values of the HDvV Hamiltonian for the spin configurations of Figure 2, En = 〈n|ℋ̂|n〉, n = I, II, and III, are given by the expressions EI = c + 12J + 12J′, EII = c − 12J + 12J′, and EIII = c − 4J − 4J′. These equations can be solved to obtain c and the exchange-coupling constants, yielding c = (EI + 3 EIII)/4, J = (EI − EII)/24, and J′ = (EI + 2 EII − 3 EIII)/48. The configurational energies obtained with VWNBP/TZP (section 2) are EI = −277.9169 eV, EII = − 279.8062 eV, and EIII = −278.8276 eV. Substitution of these values in the expressions for couplings gives J = 635 cm-1 (antiferromagnetic) and J′ = −176 cm-1 (ferromagnetic). With these exchange-coupling constants, the |S123 = 6, S = 4> state is free of spin frustration and is the ground state of the HDvV Hamiltonian. (In the case that J′ is a weak antiferromagnetic coupling, this state may remain the ground state but the couplings between Si and Sj (i < j < 4) will then be “frustrated”.) The first two excited states of ℋ̂ are |S123 = 5, S = 3> and |S123 = 6, S = 5> with excitation energies of 2326 cm-1 and 3175 cm-1, respectively. The OPBE results (see section 2) are J = 469 cm-1 and J′ = −229 cm-1, which yield again a |S123 = 6, S = 4> ground state and excitation energies of 2312 cm-1 and 2345 cm-1, respectively.
The large difference between the values for J and J′ implies a remarkably strong dependence of the exchange-coupling constants on the structure of the associated FeII (μ3-S)2FeII units. Assuming that the exchange-coupling constant between any pair of irons in the [Fe4S4]0 cluster is given by the same function, denoted
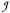 (θ), of the average bond angle, θ = (FeSFe)av, taken over the two ligand bridges connecting the irons, and adopting the linear approximation
(θ) ≈ (d
/dθ) (θ − θo), the angular slope can be expressed as d
/dθ ≈ (J − J′)/[(Fe4SFei)av − (FeiSFej)av], (i < j < 4). Substitution of the DFT values for J ≡
(70.5°) and J′ =
(64.5°) and the angular DFT averages, 70.5° and 64.5°, listed in Table 1, gives the slope d
DFT/dθ = 135 cm-1/degree and yields, after a simple evaluation, the value for the zero point θo = 65.8° where
(θ) vanishes (for OPBE: d
DFT/dθ = 116 cm-1/degree and θo = 66.5°). The d
DFT/dθ value for the DH complex is comparable to the steep slope observed for the CuII(μ2-OH)2CuII bridges reported by Hatfield, Hodgson, and coworkers17 for which d
/dθ ≈ 80 cm-1/degree, where θ is the CuOCu angle. However, the rate of change in the total exchange splitting between the top and ground levels of the spin ladder for a FeII-(μ3-S)2-FeII unit in the DH complex (which is 10 × d
/dθ vs. d
/dθ in dicopper(II) complexes), is predicted to be more than an order of magnitude larger than in the copper complexes.
(θ), of the average bond angle, θ = (FeSFe)av, taken over the two ligand bridges connecting the irons, and adopting the linear approximation
(θ) ≈ (d
/dθ) (θ − θo), the angular slope can be expressed as d
/dθ ≈ (J − J′)/[(Fe4SFei)av − (FeiSFej)av], (i < j < 4). Substitution of the DFT values for J ≡
(70.5°) and J′ =
(64.5°) and the angular DFT averages, 70.5° and 64.5°, listed in Table 1, gives the slope d
DFT/dθ = 135 cm-1/degree and yields, after a simple evaluation, the value for the zero point θo = 65.8° where
(θ) vanishes (for OPBE: d
DFT/dθ = 116 cm-1/degree and θo = 66.5°). The d
DFT/dθ value for the DH complex is comparable to the steep slope observed for the CuII(μ2-OH)2CuII bridges reported by Hatfield, Hodgson, and coworkers17 for which d
/dθ ≈ 80 cm-1/degree, where θ is the CuOCu angle. However, the rate of change in the total exchange splitting between the top and ground levels of the spin ladder for a FeII-(μ3-S)2-FeII unit in the DH complex (which is 10 × d
/dθ vs. d
/dθ in dicopper(II) complexes), is predicted to be more than an order of magnitude larger than in the copper complexes.
The X-ray value ΔθX-ray ≡ [(Fe4SFei)av − (FeiSFej)av] = 3.6° (i < j < 4) is smaller than the DFT value ΔθDFT = 6.0°, by a factor of 0.6 (Table 1). This factor may arise from the propensity of DFT calculations to overestimate the exchange-coupling constants in iron–sulfur clusters.18 If we assume that the DFT value for the exchange-coupling constant for any given geometry of the FeII(μ3-S)2FeII unit is off by a constant factor q, then the angular dependence of the true J must be given by
(θ) = q ×
DFT(θ), leading to the expression d
/dθ = q × d
DFT/dθ for the slope. Given their strong dependence on molecular geometry it seems plausible that the exchange energies have an effect on the structure of the cluster. In a previous study6 we showed that the angular dependence of the exchange-coupling constants gives rise to a spontaneous lowering of the symmetry of the [Fe4S4]0 core in the S = 4 state to C3v. The associated angular difference Δθ = (Fe4SFei)av − (FeiSFej)av (i < j < 4) was directly proportional to the force d
/dθ driving this distortion. Since the X-ray value for the angular difference, ΔθX-ray = C (d
/dθ), is smaller than the DFT value, ΔθDFT = C (d
DFT/dθ), by a factor of 0.6,19 we arrive at the relation d
/dθ = 0.6 × d
DFT/dθ, allowing us to identify the factor q in
(θ) = q ×
DFT(θ) with the factor 0.6 for the distortions: q = 0.6. Noticing that J − J′ ≈ (d
/dθ) ΔθX-ray ≈ q2 (d
DFT/dθ) ΔθDFT ≈ q2 (J − J′)DFT, it follows that (J − J′)DFT has to be reduced by the factor q2 to obtain the true value for the difference J − J′ in the physical system. Applying this factor to the individual coupling constants, we obtain J = 0.62 × 635 cm-1 = 229 cm-1, J′ = 0.62 × (−176) cm-1 = −63 cm-1, and d
/dθ = 0.6 × 135 cm-1/degree = 81 cm-1/degree (for OPBE: J = 169 cm-1, J′ = −82 cm-1, and d
/dθ = 70 cm-1/degree). Thus, the DFT values for J and J′ have a large margin of uncertainty. Somewhat accidentally, perhaps, the latter d
/dθ values are close to the slope for the aforementioned dicopper(II) complexes. In contrast, the zero point of the function
(θ) for the DH complex (θo ≈ 66°) differs significantly from the one for the bis-μ-hydroxo-copper(II) dimers (θo ≈ 97°).17 The DFT estimates given here are approximate since, (1) the geometry optimizations not only affect the FeiSFej bond angles and associated Fei⋯Fej distances, but also alter the Fe-S bond distances (see section 3.1), and (2) the large magnitude of the J changes raises concerns about the validity of the assumed linearity of the magneto–structural correlations considered here, although this is not unprecedented as we have seen in the copper dimers. As experimental J values for the FeII-(μ3-S)2-FeII bridges in Fe4S4 clusters are lacking, the DFT calculations of J are not well calibrated in these systems. The only J value for a Fe4S4 cluster currently available, J = 280 cm-1, is for the coupling between two high-spin FeIII sites and has been obtained from magnetic susceptibility measurements on the FeIII2(FeII(CN)3)2S4 cluster, in which the FeII sites are low-spin.18 This value is larger than the above estimate of 229 cm-1 for J in the DH complex, which seems reasonable given the higher oxidation state of the metal.
The strong magneto–structural correlation between J and the metal–ligand–metal bond angle in FeII-(μ3-S)2-FeII units stands in stark contrast to the reported lack of a demonstrable angular correlation for supported FeIII-(μ2-O-X)-FeIII bridges (O-X = oxo, hydroxo, alkoxo, …),20 possibly because of geometric constraints. Thus, if the sign change of J in a FeIII-(μ2-O)2-FeIII bridge were to occur at a bond angle of ∼70° as in the FeII-(μ3-S)2-FeII units of the all-ferrous clusters, this event would elude observation in any of the reported FeIII-(μ2-O-X)2-FeIII complexes, simply because their bond angles (98° − 160°)20 are far from the zero point in the angular dependence of J.
Simultaneously with the reports of the synthetic all-ferrous clusters,3,4 two all-ferric cubane clusters appeared in the literature, including an [Fe4O4]4+ cluster that is the core of an extended 8FeIII structure21 and a [Fe4S4]4+ cluster that is stabilized by bis(trimethylsilyl)amide terminal ligands.22,23 The FeIII(μ4-O)2FeIII coupling constants in the [Fe4O4]4+ moiety are weak (J ≈ 2 cm-1),21 probably due to the long Fe-O distances in this unit rather than to the nearness of the FeOFe angle (≈ 98°) to a zero point in bond-angle dependence of J. (We are not aware of any FeIII-O(2)-FeIII bridge with ferromagnetic coupling.) The J value for [Fe4O4]4+ is consistent, at least qualitatively, with the empirical relationship of ref. 20 between J and average Fe-O bridging bond length, which yields J ≈ 9 cm-1 for the distances (Fecore-O ≈ 2.05 Å) in this cluster, if one considers that the difference between 2 and 9 cm-1 is small on the scale of observed J[FeIII-O-FeIII] values, i. e., 15 cm-1 − 260 cm-1.20 The empirical J–(Fe-O) correlation predicts a value of 18 cm-1 for the coupling constants between the terminal and core irons of the 8Fe cluster (the value obtained by substituting the average, 2.00 Å, of the Fe-O distances in the Feterminal-O-Fecore bridges into the empirical correlation), which is clearly smaller than the value of ≈ 50 cm-1 deduced from magnetic susceptibility analysis,21 leaving room for some degree of bond-angle dependence of J. The [Fe4S4]4+ cluster has an S = 0 ground state with a D2d structure, in agreement with one of the two possible ground states predicted by the theoretical model presented in ref. 6 as applied to this system. However, to obtain two long and four short Fe⋯Fe distances, as observed, the model requires a negative slope, d
/dθ < 0, at the bond angle in the all-ferric system, which is about 8° larger than in the all-ferrous cluster. Establishing the interesting and consequential magneto–structural correlations in these and other metal clusters is the subject of a future investigation.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant EB0001475 and NSF Grant CHE070073 through TeraGrid resources provided by the NCSA.
Contributor Information
Eckard Münck, Email: emunck@cmu.edu.
Emile L. Bominaar, Email: eb7g@andrew.cmu.edu.
References
- 1.(a) Yoo SJ, Angove HC, Burgess BK, Hendrich MP, Münck E. J Am Chem Soc. 1999;121:2534–2545. [Google Scholar]; (b) Angove HC, Yoo SJ, Burgess BK, Münck E. J Am Chem Soc. 1997;119:8730–8731. [Google Scholar]; (c) Angove HC, Yoo SJ, Münck E, Burgess BK. J Biol Chem. 1998;273:26330–26337. doi: 10.1074/jbc.273.41.26330. [DOI] [PubMed] [Google Scholar]
- 2.Hans M, Buckel W, Bill E. J Biol Inorg Chem. 2008;13:563–579. doi: 10.1007/s00775-008-0345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott TA, Berlinguette CP, Holm RH, Zhou HC. Proc Natl Acad Sci U S A. 2005;102:9741–9744. doi: 10.1073/pnas.0504258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng L, Holm RH. J Am Chem Soc. 2008;130:9878–9886. doi: 10.1021/ja802111w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 6.Chakrabarti M, Deng L, Holm RH, Münck E, Bominaar EL. Inorg Chem. 2009;48:2735–2747. doi: 10.1021/ic802192w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakrabarti M, Deng L, Holm RH, Münck E, Bominaar EL. Inorg Chem. 2010;49:1647–1650. doi: 10.1021/ic902050k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng L, Bill E, Wieghardt K, Holm RH. J Am Chem Soc. 2009;131:11213–11221. doi: 10.1021/ja903847a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ADF. Vrije Universiteit Amsterdam; 2008. http://www.scm.com/ [Google Scholar]
- 10.Noodleman L, Case DA. In: Advances in Inorganic Chemistry. Cammack R, editor. Vol. 38. Academic Press; San Diego: 1992. pp. 423–470. [Google Scholar]
- 11.Chanda A, Tiago de Oliveira F, Collins TJ, Münck E, Bominaar EL. Inorg Chem. 2008;47:9372–9379. doi: 10.1021/ic800881p. [DOI] [PubMed] [Google Scholar]
- 12.Swart M. J Chem Theory Comput. 2008;4:2057–2066. doi: 10.1021/ct800277a. [DOI] [PubMed] [Google Scholar]
- 13.Berg JM, Holm RH. In: Iron–Sulfur Proteins. Spiro TG, editor. Chapter 1 Wiley; New York: 1982. [Google Scholar]
- 14.Mouesca JM, Noodleman L, Case DA, Lamotte B. Inorg Chem. 1995;34:4347–4359. [Google Scholar]
- 15.Middleton P, Dickson DPE, Johnson CE, Rush JD. Biochem J. 1978;88:135–141. doi: 10.1111/j.1432-1033.1978.tb12430.x. [DOI] [PubMed] [Google Scholar]
- 16.The value for aloc was obtained from the experimental value given in ref. 15 for the hyperfine coupling constant for the ferrous sites in [Fe4S4]+ in the coupled spin representation, using the spin projection factor for the FeII sites in the spin coupling scheme |{9/2, (2, 2)4} ½>.
- 17.(a) Hatfield WE. In: Interrante LV, editor. Extended Interactions between Metal Ions in Transition Metal Complexes; ACS Symposium Series 5; Washington, D. C.: American Chemical Society; 1974. pp. 108–141. [Google Scholar]; (b) Dixon Estes E, Hatfield WE, Hodgson DJ. Inorg Chem. 1974;13:1654–1657. [Google Scholar]; (c) Melnik M. Coord Chem Rev. 1982;42:259–293. [Google Scholar]
- 18.Yoo SJ, Hu Z, Goh C, Bominaar EL, Holm RH, Münck E. J Am Chem Soc. 1997;119:8732–8733. [Google Scholar]
- 19.We assume here that the force constants, which occur in the definition of coefficient C, are accurately predicted by DFT, cf. ref.6.
- 20.Gorun SM, Lippard SJ. Inorg Chem. 1991;30:1625–1630. [Google Scholar]
- 21.Baran P, Boča R, Chakraborti I, Giapintzakis J, Herchel R, Huang Q, McGrady JE, Raptis RG, Sanakis Y, Simopoulos A. Inorg Chem. 2008;47:645–655. doi: 10.1021/ic7020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharp CR, Duncan JS, Lee SC. Inorg Chem. 2010;49:6697–6705. doi: 10.1021/ic100742c. [DOI] [PubMed] [Google Scholar]
- 23.Ohki Y, Sunada Y, Tatsumi K. Chem Lett. 2005;34:172–173. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.