

Abstract
The change of phenotypic traits in domestic animals and crops as a response to selective breeding mimics the much slower evolutionary change in natural populations. Here, we describe that the dominant white phenotype in domestic pigs is caused by two mutations in the KIT gene encoding the mast/stem cell growth factor receptor (MGF), one gene duplication associated with a partially dominant phenotype and a splice mutation in one of the copies leading to the fully dominant allele. The splice mutation is a G to A substitution in the first nucleotide of intron 17 and leads to skipping of exon 17. The duplication is most likely a regulatory mutation affecting KIT expression, whereas the splice mutation is expected to cause a receptor with impaired or absent tyrosine kinase activity. Immunocytochemistry showed that this variant form is expressed in 17- to 19-day-old pig embryos. Hundreds of millions of white pigs around the world are assumed to be heterozygous or homozygous for the two mutations.
[The EMBL accession numbers for porcine KIT1*0101, KIT1*0202, KIT2*0202, and KIT2*0101 are AJ223228–AJ223231, respectively.]
There is a strong preference for white coat color in commercial pig breeding, and the selective breeding for white color can be traced back to medieval time in Europe (Wiseman 1986). The inheritance of coat color in pigs was investigated in a cross between the European Wild Boar and Large White domestic pigs (Johansson et al. 1992; Johansson Moller et al. 1996; Mariani et al. 1996). Three alleles were documented at the dominant white locus, the recessive i allele for normal color, the semidominant Ip allele for the patch phenotype, and the fully dominant I allele for the dominant white phenotype (Johansson et al. 1992); the patch phenotype has white patches and fully colored patches separated by sharp borders. We have shown previously that both the I and the Ip alleles are associated with a duplication of the KIT gene (Johansson Moller et al. 1996). Preliminary BAC cloning and fiber–FISH experiments indicate that the size of the duplication exceeds 200 kb and include the complete coding sequence (J. Kijas, A. Törnsten, P. Chardon, and L. Andersson, unpubl.).
KIT mutations cause pigmentation disorders in mice (dominant white spotting, W; Chabot et al. 1988; Geissler et al. 1988) and in humans (piebald trait; Fleischmann et al. 1991; Giebel et al. 1991). Normal migration and survival of neural crest-derived melanocyte precursors is dependent on KIT expression and the availability of its ligand, the mast/stem cell growth factor (MGF; Wehrle-Haller and Weston 1997). KIT mutations in mice are often lethal or sublethal in the homozygous condition and exhibit pleiotropic effects on the development of melanocytes, hematopoietic cells, primordial germ cells, interstitial cells in the small intestine, and may affect hearing. In contrast, the porcine dominant white allele has a more drastic effect on pigmentation than any known mouse KIT mutant, but is fully viable and fertile in the homozygous condition.
The objective of the present study was to characterize the coding sequence of KIT in white and colored animals in an attempt to reveal further mutations that could explain the fully dominant white phenotype.
RESULTS
The Segregation of Coat Color Phenotypes in a Wild Boar/Large White Intercross
The coat color phenotype in this intercross was determined by two major loci, the dominant white locus (I) encoding KIT and the extension (E) locus encoding the melanocortin receptor 1 (MC1R) (Mariani et al. 1996; J. Kijas, R. Wales, A. Törnsten, P. Chardon, M. Moller, and L. Andersson, in prep.). The two Wild Boar founders were homozygous i/i, E+/E+ and exhibited the wild-type phenotype. The eight Large White founders were homozygous I/I, Ep/Ep except one, which was classified as I/Ip, Ep/Ep because ∼50% of her progeny showed the patch phenotype (Ip/i). Color slides are available on most 200 F2 animals and the observed coat color phenotypes were analyzed according to genotypes at the I/KIT and E/MC1R loci. The former genotype was deduced by use of the dense linkage map around KIT, whereas a direct DNA test was used to distinguish the E+ and Ep alleles (J. Kijas, R. Wales, A. Törnsten, P. Chardon, M. Moller, and L. Andersson, in prep.).
The results are summarized in Table 1. I/I and I/Ip animals were white and no apparent pigment spots were detected. I/i heterozygotes were white, but ∼50% of the animals showed clear pigment spots in the skin but with white hair or white hair intermingled with colored hair (roaning), denoted W/S in Table 1. Moreover, there was a clear indication of an effect of the E genotype on the expression of these pigment spots because 18 of 35 I/i, E+/− animals showed spots, but only 2 of 12 I/i, Ep/Ep (P = 0.036, Fisher’s exact test). It should be noted that the presence of pigment spots was scored by use of color slides, and it is possible that the difference between the two groups primarily concerns the size and visibility of the spots. The patch phenotype occurred in animals classified as Ip/i, E+/−. Patch animals were white but with fully wild-colored patches, that is, both skin and all hairs were colored; the proportion of colored coat was variable and covered between 10% and 90% of the body. The interpretation that patch is caused by a distinct allele (Ip) and does not reflect incomplete penetrance of I was proven by the fact that F2 animals with the patch phenotype only occurred in familes in which one of the F1 parents was patch. The wild-type phenotype (including juvenile stripes) was expressed by animals being i/i, E+/−. Finally, the double recessive homozygotes (i/i, Ep/Ep) exhibited black spots (colored hair and skin) on a red or white background (Mariani et al. 1996). No Ip/Ip homozygote was present in this material because only one of the eight Large White founder animals carried this allele.
Table 1.
Coat Color Phenotypes in the F2 Generation of a Wild Boar/Large White Intercross According to the Genotypes at the Dominant White (I/KIT) and Extension (E/MC1R) Loci
| I/KIT | E/MC1R | ||
|---|---|---|---|
| E+/E+ | E+/Ep | Ep/Ep | |
| I/I | white | white | white |
| I/Ip | white | white | white |
| I/i | W/S (7/15)a | W/S (11/20)a | W/S (2/12)a |
| Ip/i | patch | patch | —b |
| i/i | wild type | wild type | black spotsc |
(W/S) White but the proportion indicated showed pigmented skin spots with white hair or intermingled black and colored hair (roaning).
This phenotype could not be judged, as no good quality slides were available for the few animals with this genotype.
These pigs are white with black spots or red with black spots (see Mariani et al. 1996).
The Dominant White Phenotype Is Associated with a Splice Mutation in KIT Intron 17
Almost the entire coding sequence of KIT (2892 of 2919 bp) was determined from a colored (i/i) Hampshire pig and a white (I/I) crossbred Large White/Landrace sow by cloning and sequencing RT–PCR products. A single major transcript of expected size was identified from the colored pig, whereas two forms, one normal and one lacking exon 17 (123 bp), were obtained from the white pig. RT–PCR analysis across exon 16–19 of eight white pigs and six colored pigs revealed a complete association between the presence of a transcript lacking exon 17 and the dominant white phenotype (Fig. 1). Sequence analysis of the exon/intron borders flanking exon 17 from genomic DNA representing the I, Ip, and i alleles revealed no sequence polymorphism in intron 16, but a G to A substitution in the first nucleotide of intron 17 disrupting the highly conserved GT pair at the 5′ splice site (Fig. 2). The mutation was only found in one of the two KIT genes (arbitrarily denoted KIT2) associated with I and not in KIT sequences associated with Ip or i; the mutation obviously causes the splice defect and skipping of exon 17. Exon 17 is composed of 123 bp (41 codons) and the splice mutation is thus an in-frame mutation deleting 41 amino acids in the mature protein.
Figure 1.

Identification of a KIT transcript lacking exon 17 in pigs with the dominant white allele. Agarose gel electrophoresis of RT–PCR products of KIT exon 16–19. Samples 1–3 and 4–6 are Swedish Large White (homozygous or heterozygous for the I allele) and Swedish Hampshire (i/i) pigs, respectively. The size difference between the 424- and 301-bp fragments is due to the lack of exon 17 in the latter fragment. The two upper bands of samples 1–3 were shown to be heteroduplexes (HDs) formed between the normal and splice mutant form. The tendency for different relative amounts of the 424- and 301-bp fragments may be a PCR artifact but may also have a biological cause, e.g., whether an animal is heterozygous or homozygous for the I allele.
Figure 2.

DNA sequence comprizing 48 bp of the exon 17/intron 17 border in KIT1 and KIT2 associated with the I, Ip, and i alleles. The position of the exon/intron border is marked with a vertical line, the splice mutation (nt1G → A) with a vertical arrow, and sequence identity to the master sequence with a dash. KIT1 and KIT2 sequences from the I allele were obtained by sequencing PCR products from genomic λ clones representing the two forms while direct sequencing of PCR products from genomic DNA was carried out to determine KIT sequences associated with Ip and i.
The splice mutation creates a NlaIII restriction site allowing a simple diagnostic PCR–RFLP test. The test revealed a complete association between the presence of the splice mutation and the dominant white phenotype across breeds and within a Wild Boar/Large White intercross (Table 2). A quantitative PCR–RFLP test was consistent with the expected ratio of 2:2, 2:1, and 3:1 between the normal and splice form in I/I, I/i, and I/Ip animals, respectively (Fig. 3).
Table 2.
Distribution of the KIT Splice Site Mutation According to Breed and Coat Color
| Breed | Coat color | Genotype | Animals tested | Splice mutationa |
|---|---|---|---|---|
| Unrelated animals | ||||
| Large White | white | I/−b | 33 | 33 |
| Landrace | white | I/−b | 56 | 56 |
| Hampshire | colored | i/i | 5 | 0 |
| Duroc | colored | i/i | 5 | 0 |
| Pietrain | colored | i/i | 8 | 0 |
| Meishan | colored | i/i | 5 | 0 |
| Wild Boar | colored | i/i | 2 | 0 |
| Animals from a Large White/Wild Boar intercross | ||||
| white | I/I | 13 | 13 | |
| white | I/Ip | 12 | 12 | |
| white | I/i | 14 | 14 | |
| patch | Ip/i | 7 | 0 | |
| colored | i/i | 12 | 0 | |
Presence of the splice site mutation was determined by an NlaIII PCR RFLP test.
A white animal may be I/I, I/Ip, or I/i.
Figure 3.

Quantitative PCR–RFLP analysis estimating the ratio of normal to splice mutant KIT in genomic DNA from F2 animals in a Wild Boar/Large White intercross with different genotypes. (I/I) n = 13; (I/i) n = 14; (I/Ip) n = 12. The genotypes were deduced by family segregation analysis using the dense linkage map around the I locus.
The sequence analysis revealed a single sequence (KIT1*0101) from the colored pig (Table 3). Two different presumed allelic full-length transcripts (KIT1*0201 and KIT1*0202) and a single transcript lacking exon 17 (KIT2*0101) were obtained from the white crossbred pig (Table 3; the complete sequences have been deposited in the EMBL database with accession numbers AJ223228–AJ223231). The high sequence identity between the KIT1 and KIT2 sequences (99.8%–100.0%) is consistent with the KIT duplication being a recent event, which may have occurred after domestication. A total of eight nucleotide substitutions were found and two caused amino acid substitutions (Table 3). All substitutions were confirmed by sequencing several independent clones showing the variant form. In addition, an SSCP test was used to confirm the presence of three forms of exon 6 sequences within genomic DNA. Similarly, an AciI PCR–RFLP test and genomic DNA was used to confirm the polymorphism at position 2678 in exon 19. Screening >50 pigs representing different breeds revealed that the C nucleotide at this position occurred at a low frequency in both white and colored breeds; the Hampshire pig used for the cDNA cloning happened to be homozygous C/C at this position. Thus, none of the single nucleotide polymorphisms reported in Table 3 showed a complete association with the dominant white phenotype.
Table 3.
Nucleotide and Amino Acid Differences among Four KIT cDNA Sequences Identified in Colored (i/i) and Dominant White (I/I) Pigs
| Coat color | Sequence variant | Exon | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 17 | 5 (821) | 5 (828) | 6 (978) | 6 (984) | 6 (1008) | 9 (1464) | 18 (2502) | 19 (2678) | ||
| Colored | KIT1*0101 | present | AGG | ACA | AAC | GGA | GAG | ACG | CCT | GCG |
| Arg | Thr | Asn | Gly | Glu | Thr | Pro | Ala | |||
| White | KIT1*0201 | present | – – – | – – – | – – T | – – C | – – A | – – A | – – C | – T – |
| — | — | — | — | — | — | — | Val | |||
| White | KIT1*0202 | present | – A – | – – G | – – – | – – – | – – A | – – A | – – C | – T – |
| Lys | — | — | — | — | — | — | Val | |||
| White | KIT2*0201 | absent | – – – | – – – | – – T | – – C | – – A | – – A | – – C | – T – |
| — | — | — | — | — | — | — | Val | |||
The complete coding sequence of 2919 bp was determined, except for the last 27 bp of exon 21. Exon number (and nucleotide position) for each polymorphism are indicated. A dash indicates identity to the master sequence.
The quantitative PCR–RFLP test was used to screen for the presence of Ip among unrelated animals from commercial lines of white pigs. About 10% of both Swedish Landrace (7/56) and Swedish Large White pigs (3/33) were classified as I/Ip heterozygotes on the basis of an ∼3:1 ratio of normal-to-splice mutant form (cf. Fig. 3).
The KIT Splice Mutant Form Is Expressed in 17- to 19-Day-Old Pig Embryos
A critical issue in regard to the functional significance of the splice mutation is whether the corresponding gene product is expressed at the cell surface in pig embryos. To address this question, we developed a peptide antiserum, denoted Ex16/18, that should be specific for the splice form. Ex16/18 was used together with an antiserum (Ex21), recognizing the carboxy-terminal part of KIT, for immunolabeling of day 17–19 embryos from white (I/I) and colored parents (i/i). The results provided clear evidence that the splice form is expressed in a variety of cells in white embryos but not in pigmented embryos (Fig. 4). Immunolabeling for Ex21 was very defined in both white and colored embryos, being distributed along the various tissues but stronger in the neural tube outer walls, the cephalic mesenchyma, the notochorda, and the dermomyotoma as well as in the vascular (including blood cells) bed, both intra and extraembryonic. Ex16/18 labeled only tissues in white embryos, being quite as evenly distributed as Ex21. Further studies using embryos at different developmental stages and including an antiserum that is specific for the normal form of KIT are needed to reveal whether there are any significant differences in expression of KIT1 and KIT2 in white animals.
Figure 4.
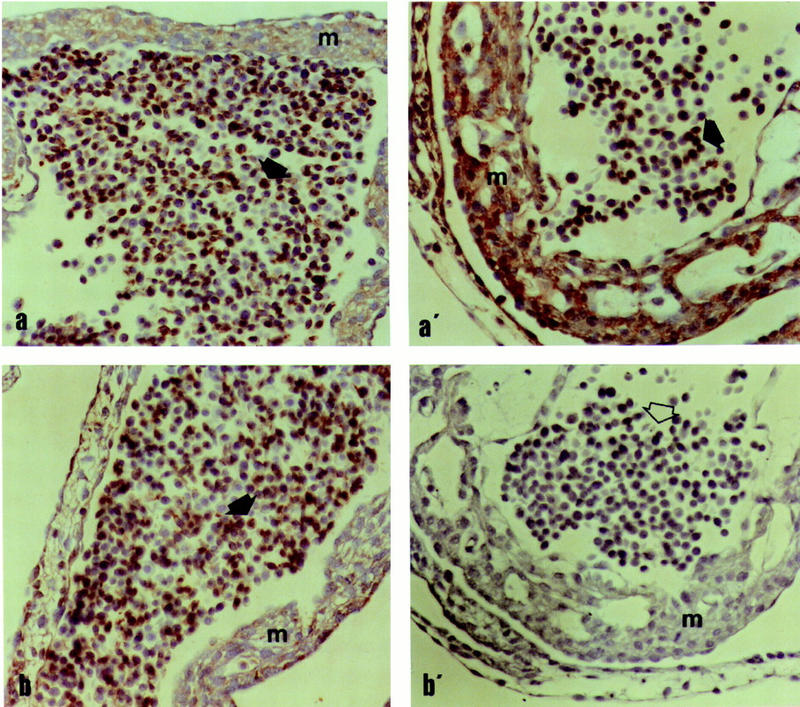
Light microphotographs of the cardiac wall (m = endomyocardium) and blood cells (arrows) in dominant white I/I (a,b) and colored embryos i/i (a′,b′) after incubation with the Ex21 (a,a′) and Ex16/18 (b,b′) antibodies. Note the negative immunolabeling of Ex16/18 in colored embryos (b′, open arrow). ABC-ELITE, magnification 200×.
Reduced Number of White Blood Cells in Pigs Homozygous for the KIT Splice Mutation
The number of peripheral blood leukocytes had been previously recorded for all 200 F2 animals in our Wild Boar intercross as part of a study with the aim to detect quantitative trait loci for immune competence traits (Edfors-Lilja et al. 1998). This provided an opportunity to test whether the splice mutation may have any mild pleiotropic effects on hematopoiesis. The numbers of cells were consistently reduced in I/I homozygotes (Table 4). Because the data are from the F2 animals in an intercross between two divergent lines, we cannot exclude the possibility that the effect is the result of a linked gene.
Table 4.
Number of White Cells According to Genotype at the Dominant White Locus
| Genotype | No. | WBC (×106/ml) | PMNL (×106/ml) | Lymphocytes (×106/ml) |
|---|---|---|---|---|
| I/I | 20 | 20.0 ± 1.5a | 9.6 ± 1.2 | 8.8 ± 0.8a |
| I/i | 82 | 23.5 ± 0.7b | 11.7 ± 0.6 | 10.2 ± 0.4a |
| i/i | 53 | 24.2 ± 1.0b | 11.0 ± 0.8 | 11.4 ± 0.5b |
No. of white cells/ml peripheral blood (×106). (WBC) white blood cells; (PMNL) polymorphonuclear leukocytes. Least square means ± s.e. are given.
a,bFigures with different letters within columns are significantly different at least at the 5% level.
DISCUSSION
The present study is an advance in pig coat color genetics. It shows that the fully dominant white allele is attributable to a minimum of two mutations. We also show that both I/i and Ip/i animals may show some pigmentation and that the incidence and/or the appearance of the pigment spots are more pronounced on an E+/− background than on an Ep/Ep background (Table 1). There was a marked difference in both the incidence and appearance of pigment spots in I/i and Ip/i animals. About 50% of the I/i animals exhibited pigment spots in the form of colored skin with white hair or white hair intermingled with colored hair (roaning). All Ip/i (patch) animals had fully pigmented spots or patches (skin and hair). The presence of pigment spots in I/i heterozygotes but not in I/I homozygotes implies a more severe KIT dysfunction in the developing melanoblasts of I/I animals.
Mouse KIT/W mutations can be divided into two types, regulatory mutations altering KIT expression and structural mutations resulting in a receptor with impaired or absent tyrosine kinase activity. Examples of the former type are W57, Wsh, Wbd, and Patch (Ph), which are all associated with deletions or inversion breakpoints in the 5′ flanking region of KIT (Duttlinger et al. 1995; Klüppel et al. 1997). Interestingly, these regulatory mutations are often viable and show a more striking phenotypic similarity to the pig patch (Ip) phenotype than the phenotypes associated with structural W mutations. On the basis of these comparative data, we postulate that the KIT duplication, present in both Ip and I alleles in the pig, is a regulatory mutation leading to dysregulated expression of one or both copies of KIT. It may simply be a dosage effect, that is, increased KIT expression may affect ligand availability, which in turn disturbs the migration of melanocyte precursors. The splice mutation, present only in I, is a structural mutation that most probably results in an impaired tyrosine kinase activity. Exon 17 encodes 41 amino acids of a highly conserved region of tyrosine kinases, comprising the catalytic loop and parts of the activation loop (Hubbard et al. 1994); the activation loop contains a tyrosine residue that is conserved in almost all tyrosine kinases (Hanks et al. 1988). Dysregulated KIT expression, when combined with impaired KIT signal transduction, is expected to cause a severe defect in the migration and survival of melanocyte precursors, thereby explaining the dominant white phenotype associated with the I allele. This model is supported by the fact that mice that are composite heterozygotes W/Ph are viable and show an almost completely white coat (Deol et al. 1970). A possible explanation for the enigma that the dominant white allele is fully viable despite the drastic effect on pigmentation is that the duplication allows the expression of one normal KIT receptor per chromosome, which apparently is sufficient to avoid severe pleiotropic effects on other tissues.
Our results indicated that ∼10% of Swedish Large White and Landrace pigs carry the partially dominant Ip allele in the heterozygous condition. This is unexpectedly high considering the strong selection against pigs showing pigmented spots as expected for Ip/Ip homozygotes; pigs showing pigment spots cannot be registered in the herd book of white breeds. No Ip/Ip homozygotes were included in the present study, but we have DNA-typed some purebred Landrace pigs that were white with some fully colored pigment spots (skin and hair). They lacked the KIT splice mutation and were therefore assumed to have the genotype Ip/Ip, Ep/Ep. The fairly high frequency of the Ip allele in commercial lines of pigs suggests that it may be regenerated by back mutations or has some selective advantage. The fact that KIT1 and KIT2 are very similar implies that they may show concerted evolution as a result of unequal crossing over and/or gene conversion, a well-known phenomenon for tandem clusters of closely related genes (Walsh et al. 1985; Murti et al. 1992). This possibility could be investigated by haplotype analysis of Ip alleles or by single sperm analysis. A selective disadvantage of the fully dominant I allele carrying the splice mutation is also conceivable, because pleiotropic effects on hematopoiesis and fertility are well documented for mouse KIT mutations. A small positive effect on hematopoiesis and/or fertility would be sufficient to maintain Ip and i in white lines despite strong selection for lack of pigmentation. Our results from the F2 generation of a Wild Boar/Large White intercross suggest that homozygosity for the splice mutation may be associated with a reduced number of white blood cells. This was unexpected because KIT mutations in the mouse cause severe defects only on the number of mast cells and red cells (Lev et al. 1994); there were no data available on the number of these cells in our Wild Boar/Large White intercross animals. However, the results call for further investigations, because even mild pleiotropic effects may have significant implications for commercial pig breeding considering that a large proportion of the world breeding stock are carrying this mutation.
Domestic animals and crops provide unique models for evolutionary studies because they have been subjected to phenotypic selection for many generations allowing the consecutive accumulation of novel mutations. This study presents a scenario for the molecular evolution of the dominant white phenotype in which an initial gene duplication event was followed by a second hit in the form of a splice mutation. The presence of two closely linked, nearly identical, genes harboring mutations with phenotypic effects is expected to be a dynamic state in which unequal crossing-over and/or gene conversion may generate new allelic forms with different phenotypic effects. It would not be surprising if future studies reveal further genetic diversity at the porcine KIT locus.
METHODS
Animals
Blood samples for preparation of genomic DNA and mRNA were collected from animals of the following Swedish breeds: Large White, Landrace, Hampshire, and Duroc. Genomic DNA samples from unrelated Pietrain and Meishan animals were kindly provided by the Pig Improvement Company (UK) and the PiGMaP consortium, respectively. The family material consisted of an intercross between the European Wild Boar and Large White domestic pigs (Johansson et al. 1992; Johansson Moller et al. 1996; Mariani et al. 1996).
RT–PCR Analysis
mRNA was isolated from peripheral blood leukocytes from white (Landrace/Large White) and colored (Hampshire) pigs by the Message Maker mRNA isolation system (GIBCO BRL) with one cycle of poly(A)+ selection from total RNA. Poly(A)+ mRNA (100 ng) was reverse-transcribed with random primers (First-Strand cDNA Synthesis kit, Pharmacia Biotech) and the product was used at a 1:10 dilution for RT–PCR by the proofreading Advantage KlenTaq Polymerase (Clontech) according to the manufacturer’s recommendation. The following primers were used to amplify almost the entire coding sequence and 90 bp of the 5′ UTR: KIT23F (5′-GGCTCTGGGGGCTCGGCTTTGC-3′) corresponding to 5′ UTR and KIT22R (5′-TCAGACATCTTCGTGGACAAGCAGAGG-3′) corresponding to exon 21; both primers had been designed by the consensus of human and mouse KIT sequences in the GenBank database. The RT–PCR products were gel purified and cloned by the pGEM-T vector system (Promega). Plasmid clones were sequenced with vector primers and a set of internal primers, and the ABI Prism dRhodamine Terminator Cycle Sequencing Kit (Perkin Elmer). Two subclones representing each type of KIT sequence were initially sequenced and in those cases in which a discrepancy was observed (possibly caused by PCR errors) additional clones were sequenced over those particular nucleotide sites. RT–PCR analysis of KIT exon 16–19 was carried out with the primers KIT1F and KIT7R (Johansson Moller et al. 1996).
Mutation Detection
A 175-bp region including the boundary between exon 17 and intron 17 of KIT was PCR amplified from genomic DNA or from genomic λ clones containing parts of the KIT1 and KIT2 gene associated with the I allele (J. Kijas, unpubl.). PCR was carried out with primers KIT21 (5′-GTATTCACAGAGACTTGGCGGC-3′) and KIT35 (5′-AAACCTGCAAG- GAAAATCCTTCACGG-3′) with AmpliTaq Gold (Perkin Elmer). The PCR products were gel purified and sequenced directly as described above. To test for the presence of the G to A splice site mutation, the same 175-bp fragment was digested with the restriction endonuclease NlaIII, which cuts once in the normal form and twice in the splice mutant form. Products were generally visualized by ethidium bromide staining. A method for quantitation of the two forms was developed by labeling the KIT35 primer with the ABI fluorescent dye FAM. The digested PCR fragments were separated by electrophoresis with an ABI377 Prism sequencer and the peak area of fragments representing the normal and splice mutant forms were quantitated by the GeneScan software.
Single-stranded conformation polymorphism (SSCP) analysis was conducted on a 174-bp fragment of KIT exon 6 amplified with primers KIT 51 (5′-ATAAAGGATTCATTAATATCTTCCCATTG-3′) and KIT 26 (5′-TCTCAGACTTGGGATAATCCTCC-3′). Electrophoresis and silver staining were as described (Johansson Moller et al. 1996). The single nucleotide polymorphism at position 2678 in exon 19 was analyzed as an AciI PCR–RFLP test. A 158-bp fragment covering 99 bp of the 3′ end of exon 19 and 59 bp of intron 19 of the KIT gene was amplified by the forward primer LA93 (5′-GAGCAGCCCCTACCCCGGAATGCCAGTTGA-3′) and reverse primer KIT56 (5′-CTTTAAAACAGAACATAAAAGCGGAAACATCATGCGAAGG-3′). Six microliters of PCR-amplified DNA was digested with 1 unit of AciI (New England Biolabs) in a total reaction volume of 10 μl at 37°C for 120 min according to the manufacturer’s recommendation. Two microliters of loading dye was added to the reaction and the restriction digests were separated by 8% native polyacrylamide gel (Protogel, 37.5:1 acrylamide:bisacrylamide, National Diagnostics, Atlanta, GA) electrophoresis in 0.5× TBE for 3 hr at 200 V in a vertical slab unit (SE600 Hoefer Scientific Instruments). Products were visualized by ethidium bromide staining.
Immunocytochemistry
Conceptuses from white (Large White × Landrace/Large White; I/I) and colored (Hampshire × Hampshire; i/i) parents were retrieved from early pregnant (days 17–19) multiparous sows and immediately fixed by immersion in a solution mixture of 8% paraformaldehyde with 0.5% glutaraldehyde (PFG 8) in phosphate buffer. The embryos were searched for normality under a stereomicroscope and dissected free from their extraembryonic membranes and embedded in paraffin. Sections of individual embryos (5 μm thick) were serially cut with a microtome and mounted on poly(A)–lysine-coated glasses. Antiserum Ex21 (previously denoted kit-C1) was raised against a peptide corresponding to amino acids 959–976 in the carboxy-terminal end of human KIT and has been used extensively for KIT detection (Blume-Jensen 1991). Antiserum Ex16/18 was made for this study by injecting rabbits with a synthetic peptide comprising the amino acid sequence of the last four amino acids encoded by exon 16 and the first four of exon 18 of pig KIT, plus a cysteine residue in its amino terminus (CASKNARLP). The peptide was coupled to keyhole limpet hemocyanine before immunization of rabbits, by use of m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS) coupling (Harlow and Lane 1988). The antisera were affinity purified by use of CNBr-activated Sepharose CL-4B (Pharmacia) columns with immobilized peptides as described (Waltenberger et al. 1993). Specificity of the affinity-purified antibodies was confirmed by the fading of stainings after preincubation with an excess molar ratio of the corresponding peptide antigen. The immunoreactivity of the Ex16/18 and Ex21 antibodies in the embryonic tissues was studied at light microscopical level with an ABC-ELITE technique performed according to the vendor’s instruction (Vector Labs, Burlingame, CA) on deparaffinated sections (Persson et al. 1997). Selected sections were photographed with a Nikon Microphot-FXA photomicroscope equipped with Nomarski interference contrast optics. Controls were run by omission of the primary antibody and by replacement of the primary antibody with an antibody (anti-cytokeratin) that gave specific staining on the ectodermal structure. Positive and negative controls were consistent throughout the incubations.
Analysis of White Blood Cells
The total and differential number of peripheral white blood cells had been determined for the F2 animals in the Large White/Wild Boar intercross (Edfors-Lilja et al. 1998). The total numbers of polymorphonuclear leukocytes and lymphocytes per ml of blood were calculated. An analysis of covariance was done by use of the GLM procedure of SAS (Anonymous 1990). The model included the KIT genotype, litter, and sex as fixed effects and live weight as covariate.
Acknowledgments
We thank U. Engström, U. Gustafsson, and Å. Jansson for excellent technical assistance. Dr. Kumar R. Juneja, Dr. Richard Wales (Pig Improvement Company), and the PiGMaP consortium kindly provided DNA samples. D.L. was supported by the Leopoldina and the Foundation Rene Touraine, Paris. The work was supported by the Swedish Research Council for Forestry and Agriculture.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Leif.Andersson@bmc.uu.se; FAX +46-18 504461.
REFERENCES
- Anonymous. SAS v. 6.10. Cary, NC: SAS Institute Inc.; 1990. [Google Scholar]
- Blume-Jensen P, Claesson-Welsh L, Siegbahn A, Zsebo KM, Westermark B, Heldin CH. Activation of the human c-kit product by ligand-induced dimerization mediates circular actin reorganization and chemotaxis. EMBO J. 1991;10:4121–4128. doi: 10.1002/j.1460-2075.1991.tb04989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot B, Stephensson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- Deol MS. The relationship between abnormalities of pigmentation and of the inner ear. Proc Roy Soc (London) A. 1970;175:201–217. doi: 10.1098/rspb.1970.0019. [DOI] [PubMed] [Google Scholar]
- Duttlinger R, Manova K, Berrozpe G, Chu TY, DeLeon V, Timokhina I, Chaganti RS, Zelenetz AD, Bachvarova RF, Besmer P. The Wsh and Ph mutations affect the c-kit expression profile: c-kit misexpression in embryogenesis impairs melanogenesis in Wsh and Ph mutant mice. Proc Natl Acad Sci. 1995;92:3754–3758. doi: 10.1073/pnas.92.9.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfors-Lilja I, Wattrang E, Marklund L, Moller M, Andersson L, Fossum C. Mapping quantitative trait loci for immune capacity in the pig. J Immunol. 1998;161:829–835. [PubMed] [Google Scholar]
- Fleischman RA, Saltman DL, Stastny V, Zneimer S. Deletion of the c-kit protooncogene in the human developmental defect piebald trait. Proc Natl Acad Sci. 1991;88:10885–10889. doi: 10.1073/pnas.88.23.10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler EN, Ryan MA, Housman DE. The dominant white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- Giebel LB, Spritz R. Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci. 1991;88:8696–8699. doi: 10.1073/pnas.88.19.8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- Johansson M, Ellegren H, Marklund L, Gustavsson U, Ringmar-Cederberg E, Andersson K, Edfors-Lilja I, Andersson L. The gene for dominant white color in the pig is closely linked to ALB and PDGFRA on chromosome 8. Genomics. 1992;14:965–969. doi: 10.1016/s0888-7543(05)80118-1. [DOI] [PubMed] [Google Scholar]
- Johansson Moller M, Chaudhary R, Hellmén E, Hoyheim B, Chowdhary B, Andersson L. Pigs with the dominant white coat color phenotype carry a duplication of the KIT gene encoding the mast/stem cell growth factor receptor. Mamm Genome. 1996;7:822–830. doi: 10.1007/s003359900244. [DOI] [PubMed] [Google Scholar]
- Klüppel M, Nagle DL, Bucan M, Bernstein A. Long-range genomic rearrangements upstream of Kit dysregulate the developmental pattern of Kit expression in W57 and Wbanded mice and interfere with distinct steps in melanocyte development. Development. 1997;124:65–77. doi: 10.1242/dev.124.1.65. [DOI] [PubMed] [Google Scholar]
- Lev S, Blechman JM, Yarden Y. Steel factor and c-kit protooncogene: Genetic lessons in signal transduction. Crit Rev Oncogenesis. 1994;5:141–168. doi: 10.1615/critrevoncog.v5.i2-3.30. [DOI] [PubMed] [Google Scholar]
- Mariani P, Moller MJ, Hoyheim B, Marklund L, Davies W, Ellegren H, Andersson L. The extension coat colour locus and the loci for blood group O and tyrosine aminotransferase are on pig chromosome 6. J Hered. 1996;87:272–276. doi: 10.1093/oxfordjournals.jhered.a023000. [DOI] [PubMed] [Google Scholar]
- Murti JR, Bumbulis M, Schimenti JC. High-frequency germ line gene conversion in transgenic mice. Mol Cell Biol. 1992;12:2545–2552. doi: 10.1128/mcb.12.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson E, Sahlin L, Masironi B, Dantzer V, Eriksson H, Rodriguez-Martinez H. Insulin-like growth factor-I in the porcine endometrium and placenta. Localization and concentration in relation to steroid influence during early pregnancy. Anim Reprod Sci. 1997;46:261–281. doi: 10.1016/s0378-4320(96)01610-7. [DOI] [PubMed] [Google Scholar]
- Walsh JB. Interaction of selection and biased gene conversion in a multigene family. Proc Natl Acad Sci. 1985;82:153–157. doi: 10.1073/pnas.82.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltenberger J, Lundin L, Oberg K, Wilander E, Miyazono K, Heldin CH, Funa K. Involvement of transforming growth factor-beta in the formation of fibrotic lesions in carcinoid heart disease. Am J Pathol. 1993;142:71–78. [PMC free article] [PubMed] [Google Scholar]
- Wehrle-Haller B, Weston JA. Receptor tyrosine kinase dependent neural crest migration in response to differentially localized growth factors. BioEssays. 1997;19:337–345. doi: 10.1002/bies.950190411. [DOI] [PubMed] [Google Scholar]
- Wiseman J. A history of the British pig. Worcester, UK: Ebenezer Baylis & Son Ltd.; 1986. [Google Scholar]