

Abstract
The Gs and Gi pathways interact to control the levels of intracellular cAMP. Although coincident signaling through Gs and Gi-coupled receptors can attenuate Gs-stimulated cAMP levels, it is not known if prior activation of the Gi pathway can affect signaling by Gs-coupled receptors. We have found that activated Gαo/i interact with RGS20, a GTPase activating protein for members of the Gαo/i family. Interaction between Gαo/i and RGS20 results in decreased cellular levels of RGS20. This decrease was induced by activated Gαo and Gαi2 but not by Gαq, Gαi1 or Gαi3. The Gαo/i-induced decrease in RGS20 can be blocked by proteasomal inhibitors lactacystin or MG132. Activated Gαo stimulates the ubiquitination of RGS20. The serotonin-1A receptor that couples to Go/i reduces the levels of RGS20 and this effect is blocked by lactacystin, suggesting that Go/i promotes the degradation of RGS20. Expression of RGS20 attenuates the inhibition of β-adrenergic receptor-induced cAMP levels mediated by the serotonin-1A receptor. Prior activation of the serotonin-1A receptor results in loss of the RGS20-mediated attenuation, and the loss of attenuation is blocked when lactacystin is included during the prior treatment. These observations suggest that Go/i-coupled receptors, by stimulating the degradation of RGS20, can regulate how subsequent activation of the Gs and Gi pathways controls cellular cAMP levels, thus allowing for signal integration.
Keywords: RGS20, Go/i, cAMP
1. Introduction
One of the earliest examples of networking between signaling pathways is the interaction between the Gs and Gi pathways in regulating cellular cAMP levels [1]. It has long been known that different receptors couple with Gs or Gi to either stimulate or inhibit adenylyl cyclases [2–4]. These effects are dependent on the isoforms of adenylyl cyclase [5]. When stimulated simultaneously, the Gi-coupled receptors can lower the increase in levels of cellular cAMP achieved by stimulation of Gs-coupled receptors. Tang and Gilman [6] demonstrated that, through Gβγ subunits, Gi-coupled receptors could stimulate cAMP levels when adenylyl cyclase 2 or related isoforms were present. Thus it appears there are extensive interactions between the Gs and Gi pathways in a co-incident manner.
Cellular cAMP levels not only are important regulators of final downstream effectors of direct targets of cAMP, such as ion channels, exchange factors or protein kinase A, but also regulate signal flow through many other signaling pathways through gating [7]. Given this versatile capability of both acting as a direct linear signaling pathway as well as modulating other pathways, it would be very useful for cells to integrate signals that regulate cAMP levels across time scales. Such integration has been a long standing interest of our laboratory. In 1984, using MDCK cells, our laboratory had shown that stimulation of the glucagon receptor/Gs pathway resulted in heterologous desensitization which was accompanied by increases in the levels of Gαi [8]. More recently, we have sought to discover regulatory mechanisms whereby prior stimulation of the Gi pathway affected stimulation of the Gs pathway. We had found initial clues for such mechanism when we discovered that activated Gαo/i interacted with RGS20 in a yeast two-hybrid screen designed to identify direct interactors of Gαo [9,10]. RGS20, a member of the large family of GTPase activating proteins for the Gi, Gq and G12/13 members of Gα subunits, was initially identified as a selective GAP for Gαz [11,12], although subsequently it has been shown that it has GAP activity towards other members of the Gi family [13]. In this study, we characterize the interactions between Gαo/i and RGS20. Like the other Gαo/i interacting protein Rap1GAPII [10], we find that Gαo/i subunits stimulate the ubiquitination and proteasomal degradation of RGS20. This change in the level of RGS20 allows for differential interactions between the Gs and Gi pathways such that prior stimulation of the Gi pathway leads to subsequent enhancement of inhibition of Gs-stimulated cellular cAMP levels by concurrent stimulation of the Gi pathway.
2. Experimental procedures
2.1. Materials
All cell culture materials were from Invitrogen (Invitrogen Corporation, Carlsbard, CA). Isoproterenol, forskolin, UK 14,304, mouse monoclonal M2-FLAG (5 different lots over a 3 year period) and anti-actin (A4700) antibodies were from Sigma (Sigma-Aldrich, St. Louis, MO). 8-OH-DPAT and NAN-190 were from Tocris. Mouse monoclonal antibody anti-Gαo (A2) and rabbit polyclonal p27 (c19) antibody were from Santa Cruz. Mouse monoclonal anti-β catenin antibody was from BD transduction Labs. Protein molecular weight standards were from SM Biotech or Amersham. The inhibitors E64, MG132 and lactacystin were from Calbiochem. Restriction enzymes, DNA T4 ligase and calf intestinal phosphatase (CIP) were from New England Biolabs. Oligonucleotides were synthesized by MWG. 100 bp and 1 kb DNA ladder, 100 mM dNTP, DNA polymerase Platinum Pfx and competent bacterial cells (One Shot TOP10) were from Invitrogen. Acrylamide/bisacrylamide 37.5:1 solution and Protein-G-Agarose were from Roche.
2.2. Cell culture
Cells were obtained from ATCC, (Rockville, Maryland) and were cultured at 37 °C in a 5% CO2 humidified atmosphere in DMEM supplemented with 10% FBS (Gibco-Invitrogen), 0.2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (+1 mM sodium pyruvate for Neuro2a cells). When required cells were serum starved for 16 h by incubating them in the same media supplemented with 0.1% BSA (COS-7) or 0.5% BSA (Neuro2a).
2.3. Protein extraction and immuno blot analysis
Cells were washed twice in cold PBS pH 7.4 and proteins were extracted by addition of 100–250 μl per 35-mm wells of RIPA lysis buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 6 mM MgCl2, 2 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 3 mM benzamidine, 1 mM PMSF, 10 mM β-glycerophosphate, 5 mM NaF and 1 mM Na3VO4). After incubation on ice for 5 min, the cells were scraped and the lysate was collected in Eppendorf microfuge tubes. The cell lysate was mixed by rotation for 15 min at 4 °C, and then centrifuged for 10 min at 13,000 rpm (~15,000×g). Protein concentration was determined and the lysates were adjusted to the same protein concentration. Equal volumes of extracted proteins were diluted in 6X Laemmli buffer, boiled for 5 min, and stored at −20 °C for immunoblotting analysis. Proteins from total extracts were separated by 12% or 10% SDS-PAGE and transferred (300 mA, 90 min, +4 °C) onto 0.45 μm supported nitrocellulose membranes (Hybond-C Extra, Amersham) using a Mini Trans-Blot apparatus (BioRad). Non-specific binding was blocked by incubation for 1 h at RT in blocking solution (PBS pH 7.4, 0.1% Tween-20, 5% non-fat dried milk). Blots were incubated with primary antibodies overnight at 4 °C with proper dilutions in fresh blocking solution +0.1% sodium azide. After extensive washing, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Pierce), diluted 1:4000 (anti-rabbit) or 1:5000 (anti-mouse) in blocking solution with no sodium azide, and incubated for 45 min at room temperature. After washings, the immunoreactive bands were visualized using the ECL detection system (Amersham Pharmacia), according to the manufacturer’s instructions, and exposure to films (HyBlot CL, Denville Scientific). The different lots of M2-FLAG antibodies often recognize a nonspecific band of approximately 25–26 kDa and it is labeled as “nonspecific” in the figures.
2.4. Transient transfections
Transient transfections were carried out with Lipofectamine 2000 (Invitrogen), using a 1:3 (μg/μl) DNA/liposomes ratio, according to manufacturer’s instructions. Cells were seeded the day before transfection so that after 24 h they were 80–90% confluent. For 35-mm plates, 0.5–1 μg of total DNAwere used. The constructs were: pBK-FLAG-chicken-RGS20 (acc# Number AF151967),[9] pcDNA3.1(+)-Gαo and pcDNA3.1(+)-Gαo-Q205L (Gαo-Q/L, acc# AH002708), both obtained from Guthrie cDNA Resource Center (clone ID GNA0OA000 and GNA0OA00C0). The FLAG-Traf2 plasmid was a kind gift from Dr. Ze’ev Ronai. Transfection efficiency was evaluated by monitoring transfected pEGFP. For experiments of stimulation with receptor ligands, cells were serum-starved in DMEM+0.1% BSA for 16 h and used for experiments 40–48 h after transfection.
2.5. Ubiquitination assay
1.5 × 106 COS-7 cells were seeded in 100-mm dishes, and transfected after 24 h with 8 μg of DNA (4 μg of pBK-FLAG-chicken RGS20 or 1 μg FLAG-Traf2 and 4 μg pcDNA3.1(+)-Gαo). After 24 h, cells were treated with 10 μM MG132 or DMSO for 10 h, placed on ice, washed and harvested in cold PBS containing protease and phosphatase inhibitors and 10 mM N-ethyl-maleimide (NEM) (Sigma), to inhibit de-ubiquitinating activities. Cell pellets were lysed in denaturing conditions with 100 μl of denaturing lysis buffer (100 mM Tris–HCl pH 7.5, 150 mM NaCl, 2% SDS, + inhibitors) and boiled at 95 °C for 10 min. After adding 900 μl of cold non-denaturing lysis buffer (100 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, + inhibitors), lysates were sonicated and pre-cleared with 40 μl of Protein G-Agarose beads for 1 h at +4 °C. For immunoprecipitation, 700 μg of cleared lysate proteins were incubated with 4 μg of M2-FLAG antibody overnight at 4 °C. This was followed by incubation for 4 h with 30 μl of Protein-G-Agarose beads. Beads were washed three times with 1 ml wash buffer (100 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5 M LiCl, 0.01% Triton X-100) and once with TBS (100 mM Tris–HCl pH 7.5, 150 mM NaCl). Immunoprecipitated proteins were then eluted with 30 μl of 2X Lammli buffer, boiled for 5 min and resolved by SDS-PAGE on 4–12% gradient gels (NuPAGE Novex Bis–Tris, Invitrogen) followed by immunoblotting. Ubiquitinated proteins were detected by a mouse monoclonal anti-ubiquitin antibody (P4D1) (1:2000, Santa Cruz) and a secondary anti-mouse HRP-conjugated antibody recognizing only native Ig chains (mouse IgG TrueBlot, eBioscience).
2.6. GST pull-down assay
The fusion proteins GST-RGS20, GST-Gαo and GST-Gαo-Q/L were prepared using the bacterial expression vector pGEX-4T-2 in BL-21 E. Coli strain (both the vector and bacteria were obtained from Amersham Biosciences). Restriction sites for SalI/NotI were introduced immediately upstream and downstream the start and termination codons of cDNA for RGS20, Gαo and Gαo-Q/L by PCR using as templates the same constructs used in transient transfections. The PCR products were subcloned by ligation into the vector pGEX-4T-2 digested with the corresponding restriction enzymes. Bacteria were grown in 2% LB Broth Base (Gibco) +50 μg/ml ampicillin in agitation at 30 °C, and expression of the recombinant protein was induced by adding 0.1 mM IPTG for 3 h at 30 °C. The crude bacterial lysates were collected and stored in 1 ml aliquots at −80 °C in 20% glycerol or utilized fresh. Soluble GST-fused proteins were purified from crude bacterial lysates by incubation at 4 °C for 60 min with 50% slurry glutathione-sepharose beads (Glutathione-Sepharose 4B, Amersham). Collected beads were re-suspended in equal volume of GST-pull down buffer (10% glycerol, 50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1% NP-40, 2 mM DTT with proteases and phosphatases inhibitors). For the pull-down experiments, proteins were extracted from COS-7 cells grown on a 100-mm dish with 300 μl of GST-pull-down buffer and incubated with 40 μl of GST-conjugated beads for 60 min at 4 °C. The beads were collected by centrifugation and washed three times with GST-pull down buffer. After the final washing, the beads were resuspended in an equal volume of 2X Laemmli buffer and boiled. The extracted proteins were resolved by SDS-PAGE and used for immunoblotting.
2.7. Measurement of intracellular cAMP
Intracellular cAMP was measured using a commercial kit (Amersham-Biosciences) based on an immunoenzymatic assay (EIA), according to the manufacturer’s instructions. About 2.6×105 COS-7 cells were plated in 35-mm dishes the day before transfection. 24 h after plating, cells were transfected as indicated in figure legends, and 24 h later, serum starved overnight. Cells were then treated with the indicated receptor agonists. After stimulation, cells were placed on ice, washed twice in cold PBS pH 7.4, and cAMP was extracted with 2 ml of ice-cold 65% ethanol. Cellular debris was removed by centrifugation for 15 min at 2000 g at 4 °C. 1 ml aliquots of supernatants were vacuum-dried at 60 °C. Dried pellets were resuspended in 750 μl of assay buffer (see manufacturer’s instructions). Typically, under basal conditions, 1–2 pmol of cAMP were extracted from a 35-mm dish of confluent COS-7 cells. cAMP results are presented as fold change over basal.
2.8. Protein concentration estimation
Protein concentration was determined using the method of Bradford [14].
2.9. Replication of data and statistical analysis
All experiments were repeated 2–3 times and representative blots are shown. The data represent the means±SEM (standard error of mean) obtained from three independent experiments. Each experimental point was performed in triplicate. The means were compared using unpaired t-test. P values<0.05 were considered significant.
3. Results
We verified the interaction between Gαo and RGS20 by GST pull-down. For this, we expressed FLAG-tagged RGS20 in COS-7 cells and tested its interaction with wild-type Gαo or a constitutively active Gαo mutant (Gαo –Q/L). As shown in Fig. 1A, RGS20 interacted with both GST-Gαo-WT and GST-Gαo-Q/L (no interaction with GST alone). Gαo-Q/L, however, was able to pull down a greater amount of FLAG-tagged RGS20. Fig. 1B shows the input lysate used in the pull-down (Fig. 1A).
Fig. 1.

Direct interaction between Gαo and RGS20. (A) Pull-down of RGS20 by GST-Gαo-WT (wild-type) and GST-Gαo-Q/L (constitutively active mutant). COS-7 cells grown in 100 mm dishes were transfected with the pBK-FLAG-RGS20 construct. The extracted proteins were incubated with glutathione-Sepharose beads conjugated to GST protein alone or to the fusion proteins GST-Gαo-WTor GST-Gαo-Q/L. The proteins eluted out of the collected beads were separated by SDS-PAGE (12% acrylamide), and RGS20 was visualized by immunoblotting with M2-FLAG antibody. (B) The cell lysates containing the FLAG-RGS20 used as inputs for the GST pull down were probed with M2-FLAG antibodies. A non-specific band migrating below the 30 kDa marker is seen with the lot of M2-FLAG antibody used. (C) Anti-FLAG western blot in COS-7 cells transfected or not with FLAG-RGS20. COS-7 cells were transfected with the pBK-FLAG-RGS20 construct or the vector alone (pBK-FLAG). 20 μg of the extracted proteins were resolved by 12% SDS-PAGE, transferred to nitrocellulose paper and probed for the FLAG-tag with a M2-FLAG monoclonal antibody. (D) Pull-down of Gαo by GST-RGS20. COS-7 cells were transfected with different amounts (1, 0.5 or 0.1 μg) of expression constructs for Gαo-WTor Gαo-Q/L mutant. The extracted proteins were incubated with equal amounts of glutathione-sepharose beads conjugated to the fusion protein GST-RGS20, and the pulled-down Gαo was revealed by SDS-PAGE/WB with an anti-Gαo antibody. (E) Approximately 5% of the lysates used as pull-down input was probed by SDS-PAGE/WB for Gαo protein.
The RGS20 protein, when exogenously expressed in COS-7 cells, is detected by western blot as a doublet of bands migrating extremely close to one another in 12% SDS-PAGE, both above the 30 kDa molecular weight marker utilized in Fig. 1B. Because of the great similarity in the migration rate of the two bands of RGS20, it was not always possible to obtain a clear resolution of the two bands by SDS-PAGE. Furthermore, a non-specific band was detected with one of the batches of M2-FLAG antibody used, migrating at approximately 29 kDa, very close to the specific RGS20 bands, in Figs. 1B, C, 4D, and 6A. The non-specificity of this band is demonstrated by its presence in western blots for FLAG-RGS20 in lysates from untransfected COS-7 cells (Fig. 1C), and by its absence in the anti-FLAG western blots after pull-down of RGS20 (Fig. 1A).
Fig. 4.

Gαo stimulates the ubiquitination of RGS20. (A) COS-7 cells were transfected with pBK-FLAG-RGS20 with and without pcDNA3.1-Gαo-Q/L and, after 24 h, treated with 10 μM MG132 or vehicle (0.1% DMSO) for 10 h. The harvested cell pellets were lysed in denaturing conditions at 95 °C in 2% SDS to suppress deubiquitinating and proteolytic activities. RGS20 was immunoprecipitated from 0.5–1 mg of extracted proteins with 4 μg of M2-FLAG antibody and 30 μl of protein-G-agarose. Immunoprecipitated proteins were separated by SDS-PAGE with 4–12% gradient gel and ubiquitinated RGS20 was detected by Western blot with a monoclonal antibody anti-ubiquitin. (B) COS-7 cells were transfected with pBK-FLAG-RGS20 with and without pcDNA3.1-Gαo-Q/L and, after 24 h, treated with 10 μM MG132 or vehicle (0.1% DMSO) for 10 h. Cell lysis and immunoprecipitation of RGS20 were carried out as in Panel A. Immunoprecipitated proteins were separated by SDS-PAGE with 4–12% gradient gel and RGS20 was detected by Western blot with M2-FLAG antibody. (C) COS-7 cells were transfected with either pBK-FLAG-RGS20 or pcDNA3.1-FLAG-Traf2, in the presence or absence of pcDNA3.1-Gαo-Q/L, and all in the presence of pcDNA3.1-HA-Ubiquitin. After 24 h, cells were treated with 10 μM MG132 or vehicle (0.1% DMSO) for 10 h. The harvested cells were lysed in denaturing conditions at 95 °C in 2% SDS. RGS20 or Traf2 were immunoprecipitated from 0.5–1 mg of extracted proteins with 4 μg of M2-FLAG antibody and 30 μl of protein-G-agarose. Immunoprecipitated proteins were separated by SDS-PAGE in a 4–12% gradient gel and ubiquitinated species were detected by Western blot with anti-HA antibody (for HA-ubiquitin). (D) Corresponding whole cell lysates from C, probed for Gαo (top panel) and FLAG (bottom panel).
Fig. 6.

RGS20 degradation regulates interactions between the Gs and Gi pathways. (A) Agonist-dependent degradation of RGS20. COS-7 cells were transiently transfected with RGS20, with and without Gαo, with and without 5-HT1A serotonin receptor (0.2 μg). After 24 h, cells were serum-starved in 0.1% BSA for ~16 h and stimulated with or without 10 μM 8-OH-DPAT for 2, 10 or 15 min. 10 μg of extracted total proteins were resolved by 12% SDS-PAGE and RGS20 was detected by immunoblotting with M2-FLAG antibody. A non specific band migrating at ~29 kDa is seen with the lot of M2-FLAG antibody used. (B) RGS20 blocks the inhibitory effect of 8-OH-DPAT on isoproterenol-stimulated synthesis of cAMP. COS-7 cells were transiently transfected with 5-HT1A receptor and Gαo without (white bars) or with RGS20 (dark bars). 24 h after transfection, cells were serum-starved in 0.1% BSA for ~16 h and then treated with or without 10−7 M isoproterenol (β2 adrenergic receptor agonist), 10−7 M 8-OH-DPAT (5-HT1A receptor agonist) or both for 20 min and cAMP levels were measured. cAMP values are presented as fold change over basal (no stimulation). Results are means±SEM, performed in triplicate, n=3. (C) Degradation of RGS20 induced by 8-OH-DPAT-dependent activation of Gαo restores the inhibitory effect of Gi activation on isoproterenol-stimulated cAMP synthesis. COS-7 cells, transiently co-transfected with 5-HT1A receptor, Gαo and RGS20, were serum-starved for ~16 h in 0.1% BSA, and pre-stimulated without (white bars) or with 10 μM 8-OH-DPAT (dark bars) for 4 h during the serum starvation period. After washing off the media, cells were stimulated with the indicated receptor agonists (see also Panel B legend). At the end of the stimulation, cellular cAMP levels were measured. (D) Proteasome inhibition opposes the ability of 8-OH-DPAT pre-treatment to rescue the effect of Gi activation on isoproterenol-stimulated cAMP synthesis. COS-7 cells, transfected as in Panel C, were serum-starved and treated for ~12 h without (white bars) and with 10 μM Lactacystin (dark bars). After pre-stimulating with 10 μM 8-OH-DPAT for 4 h and washing off the media, the cells were exposed to the indicated receptor agonists (see also Panel B legend) and cAMP levels were measured. (E) No effect of proteasome inhibition, in the absence of RGS20 expression, on the ability of 8-OH-DPAT pre-treatment to rescue the effect of Gi activation on isoproterenol-stimulated cAMP synthesis. COS-7 cells were treated as in Panel D, but were not transfected with RGS20. Results are means±SEM, performed in triplicate, for three separate experiments. For the sake of clarity the statistical significance of only the Gi inhibition of cAMP is shown.
We also tested if GST-RGS20 could interact with Gαo-WT and Gαo-Q/L. This experiment was done at three different amounts of transfected constructs. GST-RGS20 was able to interact with Gαo-Q/L more efficiently than with Gαo-WT (Fig. 1D), as it is demonstrated by the higher amount of Gαo-Q/L pulled down in comparison to Gαo-WT, revealed particularly when less Gαo was expressing (0.5 μg DNA), although equal amounts of Gαo proteins were expressed (Fig. 1E). These data suggest that RGS20 has higher affinity for the GTP-bound form of Gαo as compared to the GDP-bound form of the Gα subunit. When the FLAG-tagged RGS20 protein was co-expressed with other activated Gα subunits, remarkably lower levels of RGS20 were found in the presence of Gαo and Gαi2 (Fig. 2A), despite the loading of even amounts of proteins on gel. On the contrary, co-expression with WT-Gα subunits did not affect the levels of RGS20 (Fig. 2B).
Fig. 2.
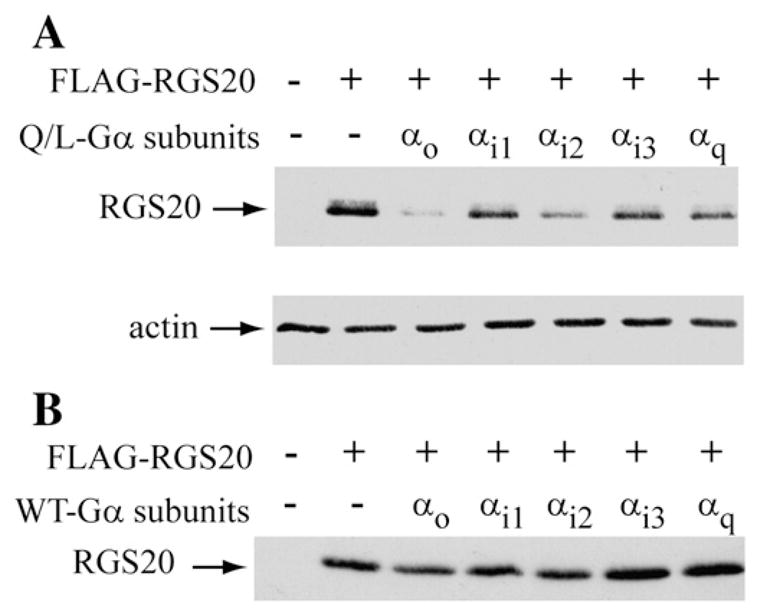
Activation of Gαo and Gαi2 results in alteration of protein levels of RGS20. (A) Coexpression of RGS20 and active mutants of Gα subunits. COS-7 cells were transiently cotransfected with pBK-FLAG-RGS20 with and without constitutively active (Q/L) mutant forms of several different α subunits of heterotrimeric G proteins. 20 μg of extracted proteins were loaded on SDS–10% polyacrylamide gel, separated by electrophoresis and analyzed by western blot with M2-FLAG antibody. The protein extracts were probed for actin as a loading control. (B): Coexpression of RGS20 and wild-type Gα subunits. COS-7 cells were transiently cotransfected with pBK-FLAG-RGS20 without or with the wild-type forms of the same α subunits of Panel A. 20 μg of extracted proteins were analyzed as in Panel A.
The decrease in levels of RGS20 upon co-expression with activated Gαo/i subunits was very reminiscent of the effect of Gαo co-expression with Rap1GAPII [10]. In the case of Rap1GAPII we had found Gαo stimulated ubiquitination and proteasomal degradation of Rap1GAPII. Hence, we determined if the proteasomal inhibitors MG132 and lactacystin blocked the Gαo-Q/L-induced decrease in levels of RGS20. We found that treatment with either inhibitor blocked the Gαo-Q/L-induced decrease of RGS20 (Fig. 3A, B and C). The time course of the protective effects of these inhibitors was similar to that seen with well established substrates for the proteasome such as p27 (Fig. 3A) and β-catenin (Fig. 3B).
Fig. 3.

Gαo-induced decrease in RGS20 levels is blocked by proteasome inhibitors. COS-7 cells were co-transfected with pBK-FLAG-RGS20 with and without the constitutively active mutant of Gαo-Q/L in pcDNA3.1(+). 24–48 h after transfection cells were treated with either 10 μM MG132 (A) or 10 μM Lactacystin (B) or vehicle (DMSO) for the indicated times. 20 μg of extracted proteins were analyzed by SDS-PAGE/Western blot with anti-M2-FLAG, anti-p27kip1 (A) and anti-β-catenin antibodies (B). (C) COS-7 cells transfected with the indicated expression constructs, were exposed to 10 μM MG132 or vehicle (DMSO) for 8 h. The proteasome inhibitor MG132 affects the stability of RGS20, in presence and absence of constitutively active mutant Gαo-Q/L.
We next determined if Gαo-Q/L stimulated the ubiquitination of RGS20. For this, we transfected COS-7 cells with FLAG-RGS20 with and without activated Gαo (Gαo-Q/L), and treated the cells with either MG132 or vehicle (DMSO). Cells were lysed, RGS20 was immunoprecipitated with the anti-FLAG antibody and the immunoprecipitate was probed by western blotting with anti-ubiquitin antibody. It was found that co-expression of Gαo increased the levels of ubiquitinated RGS20 (Fig. 4A). This increase was clearly visible even when the cells were not treated with MG132 (Compare lanes 1 and 3 in Fig. 4A). The detection of poly-ubiquitinated forms of RGS20 upon co-expression of activated Gαo, which is enhanced upon treatment with MG132, suggests that the Gαo-mediated decrease in RGS20 levels may be due to proteasomal-dependent degradation. The laddering effect that it is characteristic of poly-ubiquitinated species is also seen if the previous experiment is carried out by immunoprecipitating FLAG-tagged RGS20 using the M2-FLAG antibody and blotting with the same M2-FLAG antibody (Fig. 4B). With the latter antibody we can also detect the non-ubiquitinated form of RGS20 (doublet). In Gαo-Q/L expressing cells the relative amounts of the upper and lower band of the RGS20 doublet appear to be inverted. This is possibly due to a post-translational modification of RGS20 other than ubiquitination that is currently under investigation in our lab. The apparent different molecular weight of the doublet (relatively to previous experiments) can be explained by the use of different molecular weight markers. As a control compare, in the picture relative to a longer exposure to films of the same blot, the migration of the RGS20 doublet relatively to the non-specific band recognized by the anti-FLAG antibody.
We then determined if the effect of Gαo on the ubiquitination of RGS20 was specific by comparing the result of Gαo-Q/L expression on RGS20 to a well known ubiquitinated protein, Traf2 [15]. Under conditions where Gαo-Q/L stimulated the ubiquitination of RGS20 (Fig. 4C, second vs third lane from the left), the ubiquitination of Traf2 was unchanged (fourth vs fifth lane). Fig. 4D shows the corresponding whole cell lysate control blots.
We next tested if we could obtain receptor-dependent decrease in levels of endogenous RGS20. For this, we used Neuro2a cells, since we had previously been successful in using this cell line to study receptor-dependent degradation of Rap1GAPII. However, despite several attempts to raise antibodies against RGS20, we were unable to do so, preventing us from detecting native RGS20. We also obtained antibodies raised against human RGS20 [16] but were unable to detect the native RGS20 in either Neuro2a (a murine neuronal cell line) or COS-7 cells (a kidney cell line from monkeys), although we were able to detect the exogenously expressed protein. During the course of these experiments we tested whether several expressed Go/i-coupled receptors could reduce the levels of RGS20. Among the receptors that showed the most extensive decrease in RGS20 levels was the serotonergic 5HT1A receptor (data not shown). Since it was necessary the expression of Gαo to obtain the receptor effects, we co-expressed both the receptor and the G protein. Expression of RGS20 with Gαo and 5HT1A receptor induced the decrease in levels of RGS20 even without the addition of the agonist (8-OH-DPAT) (Fig. 5A). This receptor-induced decrease in the levels of RGS20 could be blocked by pretreatment with the proteasomal inhibitor lactacystin but not with the lysosomal inhibitor E64 (Fig. 5B). Since 5HT1A receptors are known to have substantial intrinsic activity [17], we used NAN-190, an inverse agonist for the 5HT1AR, to determine if receptor occupancy by the inverse agonist protected RGS20 against the decrease. As shown in the experiment in Fig. 5C, increasing concentrations of the inverse agonist blocked receptor-induced decrease in RGS20 levels.
Fig. 5.

Receptor stimulated decrease in levels of RGS20. (A) Neuro2a cells were transiently co-transfected with pBK-FLAG-RGS20 and pcDNA3.1(+)-Gαo, with and without pcDNA3.1-5-HT1A-Receptor (1 μg cDNA). 24 h later, cells were washed twice in PBS, serum-starved in 0.5% FBS for ~16 h and stimulated with or without 10 μM 8-OH-DPAT (5-HT1A receptor agonist) for 2 h. 30 μg of extracted proteins were separated by 12% SDS-PAGE and RGS20 was detected by Western blotting with M2-FLAG antibody. (B) Neuro2a cells, transiently co-transfected as in Panel A, were serum-starved in 0.5% FBS and simultaneously treated with vehicle (DMSO), 10 μM E64 (inhibitor of lysosomal proteases) or 10 μM Lactacystin (proteasome inhibitor), for 20 h. 50 μg of extracted total proteins were resolved by 12% SDS-PAGE. RGS20 and Gαo were detected by Western blotting with M2-FLAG and anti-Gαo antibodies. (C) Neuro2a cells were transiently co-transfected with FLAG-RGS20, Gαo and the 5-HT1A serotonin receptor. After about 24 h, the cells were serum-starved in 0.5% FBS and simultaneously incubated for 18 h with the indicated concentrations of NAN-190 (a specific 5-HT1A receptor inverse agonist) or vehicle (DMSO, volume used corresponds to the highest concentration of NAN-190). The levels of RGS20 were determined by 12% SDS-PAGE/Western blotting on 60 μg of extracted total proteins.
We then titrated down the levels of the expressed 5HT1A receptor in both Neuro2a and COS-7 cells so that the receptor-dependent decrease in levels of RGS20 occurred in an agonist-dependent manner. A typical experiment is shown in Fig. 6A. It can be readily seen that although the receptor by itself induced some decrease in the levels of RGS20, addition of the agonist induces time-dependent decrease in the levels of RGS20 (compare lanes relative to treatment with 8-OH-DPAT for 2, 10 and 15 min). The band below the RGS20 doublet is a non specific band recognized in COS-7 cells by one of the lots of M2-FLAG antibody we used (please see Fig. 1C), which can also serve as a loading control. We used these conditions to determine the functional consequences of RGS20 in the integration of signals from Gs and Go/i-coupled receptors. In a first set of experiments we tested if the activation of 5HT1A receptor by the agonist 8-OH-DPAT resulted in receptor-induced inhibition of isoproterenol-stimulated cAMP levels, and if this inhibition would be relieved by expression of RGS20. The rationale for this experiment is that expression of RGS20, by stimulating the GTPase activity of Gαi, should reduce the steady-state level of activated Gαi and thus reduce the inhibition. This experiment is shown in Fig. 6B. As predicted, RGS20 blocks the effect of the activation of the Gi pathway. 8-OH-DPAT inhibits isoproterenol-stimulated cAMP levels (Fig. 6B, white bars), and this inhibition is relieved by the co-expression of RGS20 (Fig. 6B, black bars). In such a system, where RGS20 levels are decreased by stimulation of the 5HT1A receptor-Go/i pathway, if the cell was subjected first to stimulation of 5HT1A receptor pathway alone, followed by the subsequent simultaneous stimulation of β-adrenergic and 5HT1A receptors, the ability of the Go/i pathway to inhibit the Gs pathway-induced elevation of cAMP should be lost. In the experiment shown in Fig. 6C, cells expressing RGS20 were pre-treated without or with 8-OH-DPAT for 4 h (white and black bars, respectively). The cells were washed to remove 8-OH-DPAT. This was followed by treatment with and without isoproterenol in the presence and absence of 8-OH-DPAT for 20 min, and cAMP was measured. As expected, cells that were not pretreated with 8-OH-DPAT did not show any inhibition by the 5HT1A receptor stimulation, since the expressed RGS20 would block the effect of the Gαo/i (Fig. 6C, white bars). However, pretreatment with 8-OH-DPAT that should lead to the degradation of RGS20 should restore the Gi inhibition, and this is observed (Fig. 6C, dark bars). If the effect of pretreatment with 8-OH-DPAT is due to the proteasomal degradation of RGS20, then the inclusion of the proteasomal inhibitor during the pretreatment with 8-OH-DPAT should block the degradation of RGS20 and consequently allow for RGS20 to maintain its blockade of the Gαi inhibition of cAMP levels. To test this prediction we included lactacystin during the 8-OH-DPAT treatment of RGS20 expressing cells. This experiment is shown in Fig. 6D. Pretreatment with 8-OH-DPAT allows for inhibition by 8-OH-DPATof isoproterenol-stimulated cAMP levels to occur in RGS20 expressing cells (Fig. 6D, white bars). However, when lactacystin is included during the pretreatment with 8-OH-DPAT, the subsequent inhibition of isoproterenol-stimulated cAMP levels by the simultaneous addition of 8-OH-DPAT is lost (Fig. 6D, dark bars). Treatment of cells that were not transfected with RGS20 but treated with lactacystin, also showed that the inhibition by 8-OH-DPAT is lost (Fig. 6E), suggesting that the endogenous RGS in these cells may also be susceptible to proteasomal degradation. Taken together, the experiments in Fig. 6 show that changes in the levels of expressed RGS20 by prior exposure of cells to stimulation of Go/i pathway regulates the balance by the Gs and Gi pathways in controlling cellular cAMP levels when both pathways are simultaneously activated.
4. Discussion
This study demonstrates that by regulating the levels of RGS proteins we can regulate the co-incidence detection properties of the Gs and Gi-coupled receptor systems in intact cells. It has been known for some time now that signaling through the Gi pathway is regulated by RGS proteins, while the Gs pathway appear to be insensitive to the currently known 20 or more RGS proteins [18]. It is rather surprising that there are no well-established GAPs for Gs, but nevertheless this differential sensitivity of the two pathways for the modulation of signal flow by RGS proteins allows for the generation of regulatory capabilities such as that demonstrated in this study. Previous reports have shown that other RGS family members such as RGS4, RGS5 and RGS7 are degraded in an ubiquitin/proteasome-dependent pathway [19–21].
The potential implications of the current study are limited by our inability to detect native RGS20 and study its regulation. This could be due to two reasons. First, RGS20 is not particularly antigenic, thus limiting our ability to generate antibodies of sufficient sensitivities. Many components of G protein pathways, including many receptors, Gα subunits and effectors such as adenylyl cyclase, have proven difficult targets for the generation of antibodies. Second, it is possible that RGS20 is expressed only in a sub-set of neuronal cells and the cell lines we have tested do not express RGS20. Indeed, one of the original studies reporting the identification of RGS20 showed that the mRNA for RGS20 showed a differential distribution in various brain regions [11]. A study of the expression of the various RGS mRNAs in primary olfactory neurons indicated that there was a differential distribution of RGS20 and RGS9 which corresponded to certain odorant receptor zones [22]. Thus it may be necessary to identify precisely certain types of neurons where RGS20 is highly expressed and plays a major role in the native setting. Such precise identification of neurons is beyond the expertise of this laboratory and hence we conducted our study in an exogenously expressed system to describe the regulatory capabilities of this circuit.
The ability of receptor activation of the Gi pathway and of Gαo/i to target RGS20 for ubiquitination and degradation can be considered as a form of receptor-activated sensitization of this pathway. Thus, prior stimulation of the pathway may lead to subsequent enhanced response to stimulation due to the removal of a negative regulator. Since the output of this pathway is the inhibition of Gs stimulated cAMP, prior stimulation of the Gi pathway results in subsequent enhancement of the Gi effect when both the Gs and Gi pathways are simultaneously activated. This configuration of components thus provides for the organization of a regulatable co-incidence detector, which can alter its response depending on prior history. The central role of cAMP in cellular signal processing has been well characterized for many years. Mechanisms such as the one described in this study show how cellular cAMP responses can be controlled and thus allow cAMP levels to play a critical role in regulating many cellular processes.
Acknowledgments
This research was supported by NIH grant DK38761. SRN was supported by a predoctoral NRSA award (GM-65065). JDJ was supported by the Mount Sinai MSTP program and a pre-doctoral NRSA award (DA-05798) from NIDA. TN was supported by the MSTP program and a predoctoral training grant HL-007824 from NHLBI.
Abbreviations
- RGS20
regulator of G protein signaling 20
- GAP
GTPase activating protein
- 5HT1AR
5-Hydroxytryptamine receptor 1A
- iso
isoproterenol
References
- 1.Yamamura H, Lad PM, Rodbell M. J Biol Chem. 1977;252(22):7964. [PubMed] [Google Scholar]
- 2.Taussig R, Iniguez-Lluhi JA, Gilman AG. Science. 1993;261(5118):218. doi: 10.1126/science.8327893. [DOI] [PubMed] [Google Scholar]
- 3.Londos C, Cooper DM, Rodbell M. Adv Cycl Nucleotide Res. 1981;14:163. [PubMed] [Google Scholar]
- 4.Katada T, Northup JK, Bokoch GM, Ui M, Gilman AG. J Biol Chem. 1984;259(6):3578. [PubMed] [Google Scholar]
- 5.Chen J, Iyengar R. J Biol Chem. 1993;268(17):12253. [PubMed] [Google Scholar]
- 6.Tang WJ, Gilman AG. Science. 1991;254(5037):1500. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- 7.Iyengar R. Science. 1996;271(5248):461. doi: 10.1126/science.271.5248.461. [DOI] [PubMed] [Google Scholar]
- 8.Rich KA, Codina J, Floyd G, Sekura R, Hildebrandt JD, Iyengar R. J Biol Chem. 1984;259(12):7893. [PubMed] [Google Scholar]
- 9.Jordan JD, Carey KD, Stork PJ, Iyengar R. J Biol Chem. 1999;274(31):21507. doi: 10.1074/jbc.274.31.21507. [DOI] [PubMed] [Google Scholar]
- 10.Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Philips MR, Devi LA, Iyengar R. J Biol Chem. 2005;280(12):11413. doi: 10.1074/jbc.M411521200. [DOI] [PubMed] [Google Scholar]
- 11.Barker SA, Wang J, Sierra DA, Ross EM. Genomics. 2001;78(3):223. doi: 10.1006/geno.2001.6659. [DOI] [PubMed] [Google Scholar]
- 12.Glick JL, Meigs TE, Miron A, Casey PJ. J Biol Chem. 1998;273(40):26008. doi: 10.1074/jbc.273.40.26008. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Ho G, Zhang JJ, Nieuwenhuijsen B, Edris W, Chanda PK, Young KH. J Biol Chem. 2002;277(50):48325. doi: 10.1074/jbc.M206116200. [DOI] [PubMed] [Google Scholar]
- 14.Bradford MM. Anal Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Yang Y, Ashwell JD. Nature. 2002;416(6878):345. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- 16.Jordan JD. Ph D Thesis. New York: Mount Sinai School of Medicine; 2002. [Google Scholar]
- 17.Hollinger S, Hepler JR. Pharmacol Rev. 2002;54(3):527. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 18.Ross EM, Wilkie TM. Ann Rev Biochem. 2000;69(1):795. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 19.Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV, Kwon YT. Proc Natl Acad Sci U S A. 2005;102(42):15030. doi: 10.1073/pnas.0507533102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davydov IV, Varshavsky A. J Biol Chem. 2000;275(30):22931. doi: 10.1074/jbc.M001605200. [DOI] [PubMed] [Google Scholar]
- 21.Benzing T, Brandes R, Sellin L, Schermer B, Lecker S, Walz G, Kim E. Nat Med. 1999;5(8):913. doi: 10.1038/11354. [DOI] [PubMed] [Google Scholar]
- 22.Norlin EM, Berghard A. Mol Cell Neurosci. 2001;17(5):872. doi: 10.1006/mcne.2001.0976. [DOI] [PubMed] [Google Scholar]