

Abstract
Chip-based high-throughput genotyping has facilitated genome-wide studies of genetic diversity. Many studies have utilized these large data sets to make inferences about the demographic history of human populations using measures of genetic differentiation such as FST or principal component analyses. However, the single nucleotide polymorphism (SNP) chip data suffer from ascertainment biases caused by the SNP discovery process in which a small number of individuals from selected populations are used as discovery panels. In this study, we investigate the effect of the ascertainment bias on inferences regarding genetic differentiation among populations in one of the common genome-wide genotyping platforms. We generate SNP genotyping data for individuals that previously have been subject to partial genome-wide Sanger sequencing and compare inferences based on genotyping data to inferences based on direct sequencing. In addition, we also analyze publicly available genome-wide data. We demonstrate that the ascertainment biases will distort measures of human diversity and possibly change conclusions drawn from these measures in some times unexpected ways. We also show that details of the genotyping calling algorithms can have a surprisingly large effect on population genetic inferences. We not only present a correction of the spectrum for the widely used Affymetrix SNP chips but also show that such corrections are difficult to generalize among studies.
Keywords: ascertainment bias, demography, single nucleotide polymorphisms, SNP chip data, population genetics
Introduction
Single nucleotide polymorphism (SNP) genotyping chips have been developed primarily for the use in association mapping, admixture mapping, identity by descent mapping, and other studies aimed at detecting phenotype/genotype associations (Hirschhorn and Daly 2005, Smith and O'Brien 2005, Purcell et al. 2007, Kingsmore et al. 2008, Albrechtsen et al. 2009). However, SNP genotyping chips have also recently been used in a number of population genetic studies (Jakobsson et al. 2008, Lao et al. 2008, Li et al. 2008, and other references below). Li et al. (2008) typed 650,000 SNPs in the Human Genome Diversity Panel (Cann et al. 2002) to analyze population structure. They performed analyses similar to the analyses implemented in the program structure (Pritchard et al. 2000), estimated population trees, performed principal component analyses (PCAs), and compared allele frequency spectra among populations. They discovered that the frequency spectrum was more skewed for some populations, such as Europeans and Africans, than for Asians and Native American populations. Differences in allele frequency spectra were interpreted as possible evidence of different demographic histories. Jakobsson et al. (2008) similarly genotyped 525,910 SNPs using the Human Genome Diversity Panel to analyze fine-scaled population structure. They also performed structure analyses and PCAs but focused more on copy number variants (CNVs) and did not directly interpret frequency spectra. Nelson et al. (2008) and Novembre et al. (2008) analyzed genotyping data from 500,568 SNPs from 3,192 European individuals and performed PCAs and structure analysis. The results from these papers have provided novel, and at times, surprising insights into human demography. For example, the first two principle components in the study by Novembre et al. (2008) described the geographic variation in Europe in two dimensions with remarkable accuracy. These studies serve as examples of the power and utility of the use of SNP genotyping chips in population genetics.
It is well known that SNP genotyping data may suffer from an ascertainment bias due to the procedure used to select SNPs (Nielsen 2000, Kuhner et al. 2000, Eller 2001, Wakeley et al. 2001, Nielsen and Signorovitch 2003, Nielsen 2004, Nielsen et al. 2004, Clark et al. 2005, Foll et al. 2008, Guillot and Foll 2009). The common SNP genotyping platforms include SNPs, which previously were discovered by sequencing. Such SNPs tend to be of higher frequency than random SNPs and may not be geographically representative. The degree of ascertainment bias depends on the size of the ascertainment panel used to select the SNPs and not the size of the sample under consideration (Nielsen et al. 2004). The consequence of the ascertainment bias is that the frequency spectrum tends to become biased toward common alleles, and various estimators of population genetic parameters such as measures of variability, population subdivision, and recombination may be biased. However, this effect has not been quantified in real SNP chip data, and it is still unclear to which extent previous analyses have been affected by these biases.
In this study, we analyze 39 individuals that previously have been analyzed using high-quality direct sequencing (Celera data) and genotype these individuals using a 500k Affymetrix SNP chip set. We compare analyses based on the directly sequenced data and the SNP genotyping data and use the comparison to quantify the effect of the ascertainment bias on population genetic analyses. In addition, we analyze SNP chip and sequencing data from the original HapMap individuals and sequencing data from the National Institute of Environmental Health Sciences (NIEHS) SNPs sequencing project (Livingston et al. 2004). We also show that using sequencing data from the same population, it is possible to correct the ascertainment bias and infer the correct frequency spectrum from the SNP chips.
Materials and Methods
Frequency Spectrum
In this paper, we will assume that all SNPs are diallelic and, without loss a generality, denote the genotypes for the jth SNP in the ith individual by gij∈{0,1,2}, where 0 is a homozygous genotype for the derived allele, 1 is the heterozygous genotype, etc. If we assume that there are no missing genotypes, then we can write the SNP type x as the total number of ancestral alleles for that SNP xj = ∑i = 1ngij, where n is the number of individuals. For m markers typed in n individuals, the unfolded frequency spectrum s = {s0,s1,…,s2n} is given by
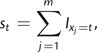 |
(1) |
where I is an indicator function.
For the sake of generality, we assume the existence of a s0 class. However, for all analysis in this paper, we only consider sites that are variable in the sample. This is appropriately conditioned on all analysis. The expected frequency spectrum in a sample of n individuals is given from the density function of the allele frequencies f in the population:
| (2) |
We assume that the count x of an allele, with frequency fx in the population, follows a binomial distribution so that x|fx∼B(2n,fx), where 2n is the number of chromosomes in the sample.
For the genotype data and the sequencing data, we do not observe the ancestral state of the alleles. This state can be inferred by using the alleles of close relatives, that is, the chimpanzee. To avoid misspecification problems, we choose to work on the folded frequency spectrum that is observed by counting the number of minor alleles. However, for the sake of simplicity, the models in the following sections are explained only for the unfolded frequency spectrum.
Comparing Frequency Spectra
In order to compare the frequency spectra for samples of different sizes, we project the spectrum down to a size, S, corresponding to a lower number of individuals. By subsampling S number of alleles, the probability (density) of an SNP of type t can be written as follows:
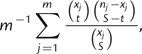 |
(3) |
where m is the number of SNPs, xj is the counts of the minor alleles at SNP j, and nj is the number of alleles at SNP j in the sample (Nielsen et al. 2004). This also allows us to use the sites with a low number of missing data by replacing nj with the number of nonmissing alleles. When comparing frequency spectra using subsamples, we use all sites with a number of nonmissing alleles equal to or greater than S.
Fitting the Frequency Spectrum
The underlying density function of the allele frequency for polymorphic sites is unknown. It is a function of the mutation rate, the effect of random drift, and demographic factors such as population size and changes, migration rates, and to a lesser extent selection and recombination. We do not attempt to model this complicated process here but will instead model the frequency spectrum using some simple standard statistical distributions:
Sawyer/Hartl
truncated normal
beta distribution
exponential mixture
where c is a normalizing constant and tNorm is a truncated normal distribution. The Sawyer/Hartl distribution is motivated by an infinite-sites model with selection (Sawyer and Hartl 1992). The second is the density function used by Nicholson et al. (2002), and the third is the stationary distribution in an equilibrium model with recurrent mutation (Wright 1931). The mixture of exponentials has no theoretical foundation, but as we will show, it provides a good fit to the observed distribution.
We estimate the parameters of the models using maximum likelihood shown here for the mixture of exponentials as an example.
Using the frequency spectrum of the sequencing data s, we can obtain a maximum-likelihood estimates for α1, α2, and σ. Parameter estimates are obtained from the sequencing data s by maximizing
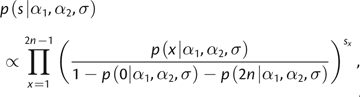 |
(4) |
where
Note that only the polymorphic sites are used in the estimation.
Ascertainment Bias
The ascertainment bias for SNP chip data is introduced when the SNPs are selected from a small panel of individuals. The SNPs will often be discovered in a panel of size d chromosomes by resequencing genomic regions of interest. When small panels are used, the chance of finding a common SNP with a high minor allele frequency is much higher than finding an SNP of low minor allele frequency. If the discovered SNPs are genotyped in larger samples, the frequency spectrum will have more SNP with higher minor allele frequencies than if the larger sample had been resequenced. Thus, the frequency spectrum for the chip data c is expected to be more screwed toward higher minor allele frequencies. We will denote the SNP discovery process as the ascertainment scheme, Asc, and further explanations will be given in the sections to follow.
Size of the Ascertainment Panel
The size of the ascertainment panel d used is estimated by maximizing the likelihood of observing the frequency spectrum for the SNP chip
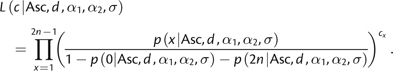 |
The probability of observing an SNP of type x can be written as the product of the probability of ascertaining such a type and the probability of the type
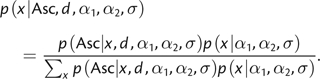 |
(5) |
The probability of ascertainment depends on the size of the ascertainment panel and the frequency of the SNP in the population
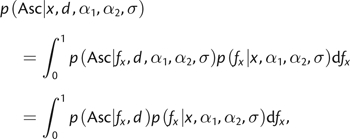 |
(6) |
where
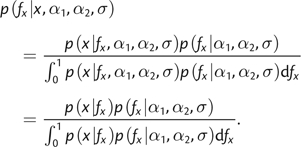 |
Notice that we have here assumed independence among SNPs. If SNPs are linked, this assumption is violated. The likelihood function as specified above then forms a composite likelihood function that should not be interpreted as a real likelihood function. However, estimates based on maximizing this function are still expected to be consistent (see, e.g., Wiuf 2006).
The Ascertainment Scheme
The simplest ascertainment scheme Asc is to only include a locus in the ascertained sample if the locus is polymorphic in a sample of size d. The probability of ascertaining an SNP with frequency fx is
| (7) |
| Another ascertainment scheme could be that a locus is only ascertained if both alleles are observed at least k times. The probability is then |
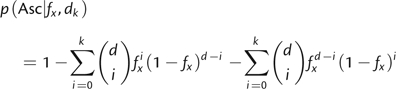 |
for k < d/2. We allow d to take on real values by using gamma functions for calculating the binomial coefficient.
Multiple Ascertainment Schemes
For SNP chip data, it is expected that multiple ascertainment schemes are used in multiple discovery panels. We choose to describe the multiple ascertainment schemes using a mixture model with the likelihood
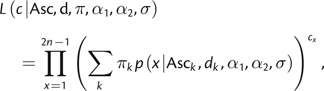 |
where πk is the mixture proportion for ascertainment panel k with sample size dk.
Estimates of FST
We estimated pairwise FST using the method by Weir and Cockerham (1984) and included only SNPs without missing data. We estimated standard errors of the FST estimates using 10,000 bootstrap samples where the ascertained SNPs were randomly sampled (with replacement). Because of linkage disequilibrium, SNPs within each gene will be correlated, violating the assumption of the bootstrap procedure. Therefore, we also estimated the standard error using a gene-wise sampling procedure. We randomly sampled the genes (with replacement) and estimated FST using the SNPs located in the sampled genes. This gives a conservative estimate of the standard error that is robust to within gene linkage disequilibrium. For testing the difference in FST estimates between the SNP chips and the resequencing data, we simulated the empirical null distribution of the differences by randomly permuting the SNPs between the chip and the resequencing data. This was done 1,000 times.
Principal Component Analysis
There are many ways to perform PCA on SNP data. Some suggest performing PCA on the `identical by state' matrix between individuals (Purcell et al. 2007). This ignores the allele frequencies so we choose to perform PCA using the method used in the Eigensoft software (Patterson et al. 2006). This method normalizes the genotypes so that each SNPs has a mean genotype value of 0 and similar variances. From the normalized data, the covariance matrix between individuals is approximated. PCA is performed on this matrix. We only included SNPs without missing genotypes and all nonpolymorphic SNPs were also removed. Only the first two principal components are shown and analyzed. We measure the mean pairwise Euclidian distance between and within populations based on the two first principal components. The standard errors are estimated using both a standard bootstrap and a gene-wise bootstrap as described for FST.
Simulation of Ascertainment Bias
We used the NIEHS resequencing data to simulate ascertainment bias. This was done by picking an ascertainment panel with a fixed number of individuals from one of the populations. Only SNPs polymorphic in the ascertainment panel are then genotyped. The individuals in the sample are not completely homogenous. For example, it has been shown that some of the African Americans in the Celera data (see below) have highly admixed genomes (Nielsen et al. 2009). This implies that the effect of ascertainment bias can be very different depending on the individuals used for the ascertainment. Therefore, we randomly permuted the genotypes within each population. This was done both for the ascertainment panel and for the individuals used for PCA and for estimating FST. This will neither affect the frequency spectrum nor the FST estimates but the effect of sampling, of individuals within populations, on the PCA will be greatly reduced.
SNP data
Celera Genomics
We genotyped 19 African Americans and 20 European Americans for approximately 500,000 SNPs using the Affymetrix Nsp and Sty SNP chips set. The genotype calling was performed using the BRLMM algorithm from the Affymetrix GTYPE software. We called an SNP if the confidence score was lower than 0.5 (default).
The same individuals have previously been sequenced by Celera genomics and are described in Bustamante et al. (2005) for the coding variants. More than 11,000 genes were sequenced including some of the introns and the regions adjacent to the genes. For this article, we used only the 20,893 synonymous diallelic SNPs that were also used in Lohmueller et al. (2008). This was done because the synonymous SNPs are less affected by selection so that these SNPs might better represent the rest of the genome.
NIEHS SNPs
We obtained direct sequencing data for multiple populations from the NIEHS Environmental Genome Project (Livingston et al. 2004). We focused on their Panel 2 that includes 95 individuals with know ethnicities. These individuals (downloaded 28 September 2008) had 38,295 diallelic SNPs in 251 genes. The populations are listed in supplementary table S1.
Seattle SNPs
Seattle SNPs (http://pga.gs.washington.edu/) is another resequencing project that is part of the National Heart Lung and Blood Institute's Program for Genomic Applications. This project focuses on candidate genes for inflammatory responses in humans. We used the 47 individuals from their Panel 2. These individuals are also part of the HapMap project and SNP chip data using the Affymetrix 500k platform (see below). The populations are listed in supplementary table S2.
HapMap SNP Chip Data
We used the SNP chip data from the original four HapMap populations. They were genotyped using the Affymetrix 500k Sty and Nsp chip set. The genotype calling was performed using the BRLMM algorithm and the genotype calls were available from the Affymetrix Web site (http://www.affymetrix.com/support/technical/sample_data/500k_hapmap_genotype.data.affx).
To show the effect of the choice of confidence score cutoff, we separated the SNPs into five categories based on their confidence scores from the BRLMM base-calling algorithm. The highest (worst) score from each genotype was used to label the SNPs.
Results
Frequency Spectrum for Affymetrix SNP Chip Data and Sequencing Data
Nineteen African Americans and 20 European Americans have previously been genotyped by direct Sanger sequencing for more than 11,000 genes by Celera Genomics (Bustamante et al. 2005, Nielsen et al. 2009). We genotyped the same individuals using the 500k Affymetrix SNP chip set. In our analysis, we only used the synonymous SNPs from the Celera resequencing data because the allele frequency distributions of these synonymous SNPs will show closer resemblance to the rest of the genome than the nonsynonymous SNPs. For both the SNP chip data and the resequencing data, we use only the diallelic SNPs without missing data.
As expected, the frequency spectrum for both the African Americans and the European Americans differs greatly between the SNP chip and the resequencing data. As shown in figures 1 and 2, the folded frequency spectrum is much more uniform for the SNP chip. Due to the ascertainment bias, there is a relative excess of SNPs with intermediate frequency and a deficiency of SNPs of high and low frequencies. As observed in other studies, the African-American individuals have more singletons than the European Americans in resequencing data (Boyko et al. 2008). This is also observed when the frequency spectrum for the 20 European Americans is projected down to 19 individuals (not shown). Another interesting observation is that the SNP chip frequency spectrum for the African Americans is not monotonously decreasing like the frequency spectrum for European Americans. Even more surprising is that the frequency of singletons for the SNP chip data in the European Americans is greater than that of the African Americans. Both these observations are also true for the Yorubans and Centre d'Etude du Polymorphisme Humain (CEPH) individuals in the HapMap SNP chip data (not shown).
FIG 1.

Folded frequency spectrum for 19 African Americans for synonymous sequencing data and for SNP chip data. Only SNPs without missing data are included.
FIG 2.

Folded frequency spectrum for 20 European Americans for synonymous sequencing data and for SNP chip data. Only SNPs without missing data are included.
The 2D frequency spectra in figure 3 show, just like the 1D, that the frequency is more even for the SNP chip data, whereas most of the density mass for the resequencing data is concentrated around the rare alleles. Both the 2D frequency spectrum for the SNP chip data and for the resequencing data show that the African Americans have more private SNPs than the European Americans.
FIG 3.

The 2D Celera frequency spectra (density) for 20 European Americans and 19 African Americans for the SNP chip data (left) and the synonymous SNPs from the resequencing data (right).
Ascertainment Bias and Geographic Variation
The effect of the ascertainment bias on the frequency spectrum also depends on the population where the ascertainment (SNP discovery) was performed. To illustrate this, we used data from the NIEHS SNPs sequencing project. This project examined individuals from five populations as follows: 14 African Americans, 12 Africans, 22 Europeans, 22 Hispanics, and 24 Asians. We simulated ascertainment using individuals from one of the five NIEHS populations. The results shown in figure 4 are the frequency spectra projected down to ten individuals that allow us to make proper comparisons among populations with different sample sizes. The frequency spectrum without ascertainment shows that the African and the African-American populations have the highest proportion of rare alleles compared with the other populations, whereas the Asian and Europeans have the lowest fraction of rare alleles. However, when ascertainment is performed in either of the five populations, the relative difference in frequency spectra between populations changes dramatically. For example, if ascertainment is performed in the Asian population, the frequency spectrum in the four other populations becomes very similar, but if the ascertainment is performed in the African population, the Hispanic population has the most rare alleles. Clearly, inferences based on allele frequencies must take the geographic or ethnic makeup of the ascertainment sample into account. Unfortunately, most SNP genotyping in humans is based on SNP selection procedures using data from dbSNP, which consists of an unspecified distribution of different ethnic groups and different sample size. For practical applications, it is therefore very difficult to adequately take the mixture of ethnicities in the ascertainment sample into account.
FIG 4.
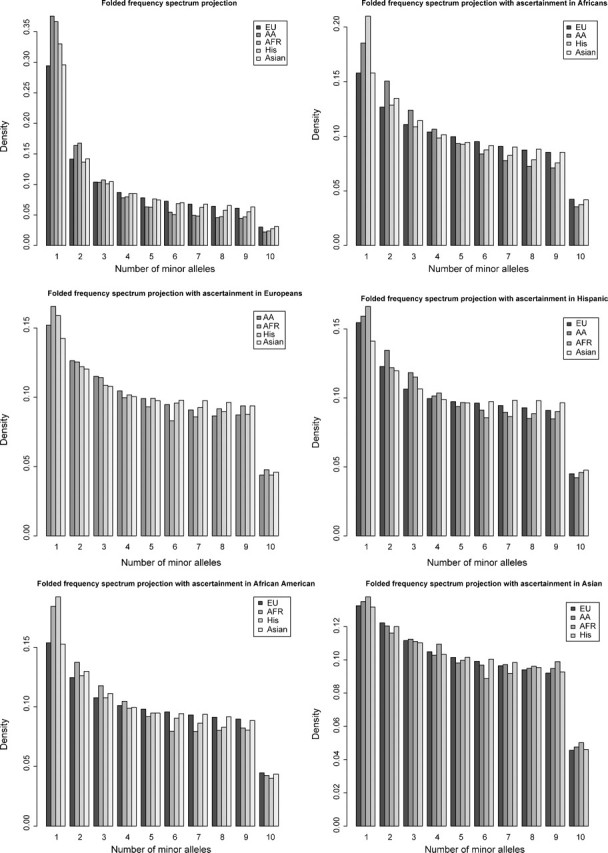
The effect of ascertainment bias on the frequency spectrum. The frequency spectrum is projected down to ten individuals for all populations for the NIEHS data. We simulated ascertainment in each of the five populations with an ascertainment sample of ten individuals.
Biases due to SNP Calling Procedures
Another concern when using SNP chip data for population genetic analyses is the effect on allele frequencies from the choice of the cutoff needed to call an SNP. The commonly used BRLMM algorithm for the Affymetrix 500k SNP chips provides a confidence score, and only genotypes under a certain fixed value of this score are called. Employing a lower cutoff results in a higher number of missing genotypes but a lower error rate (Affymetrix 2006). The default Affymetrix threshold is a confidence score of 0.5, which was used by Lao et al. (2008), whereas Novembre et al. (2008) used a threshold of 0.3 (Nelson et al. 2008). If SNPs with missing data are discarded, the maximum confidence score for an SNP determines whether an SNP is included in the analysis or not. The choice of cutoff will cause a bias in the frequency spectrum. This effect was clear in our own data (not shown), and to illustrate this effect also exists in other data, we reanalyzed publicly available Affymetrix data from the HapMap project (The International HapMap Consortium 2007). We stratify the HapMap data according to the confidence score for each SNP. The most striking difference in frequency spectra between SNPs of different maximal confidence scores is observed for the Japanese population (see fig. 5). SNPs with a higher maximum confidence score have a much more skewed frequency spectrum. Clearly, the choice of confidence score will greatly affect the frequency spectrum, and data from different analyses based on different confidence scores should not be combined. Likewise, if different samples or populations systematically differ in quality of DNA or in preparation, this can lead to artifactual systematic differences in allele frequencies. In real data, confidence scores might differ between populations for a variety of reasons leading to artifactual differences between populations. For example, the confidence scores differs between the different populations in the HapMap data as seen in Supplementary figure S1, Supplementary Material online. Note that the number of SNPs in the different confidence score bins differ greatly between the Japanese and the Chinese samples, even though they contain the same number of individuals. Thus, an observed difference between the frequency spectra of the two populations could be due to different SNP quality.
FIG 5.

Folded frequency spectrum for 45 unrelated Japanese HapMap individuals. The individuals were genotyped using approximately 500,000 SNPs on the Affymetrix SNP 500k chip set. The frequency spectrum is shown for SNPs binned based on their maximum BRLMM confidence score.
We emphasize that the effect observed here is not a particular bias in the BRLMM algorithm but arises through a natural tradeoff between accuracy and bias. Rare SNPs will likely always be called with less accuracy. The confidence level chosen to call an SNP will, therefore, naturally affect the inferred distribution of allele frequencies. In the future, it might be possible to correct for this bias in the estimation of allele frequency spectra without sacrificing accuracy, by explicit statistical modeling of the effects of SNP calling algorithms on inferred allele frequency distributions.
It is not surprising that ascertainment biases (and SNP calling procedures) will affect the frequency spectrum when analyzing SNP genotyping data. However, a number of population genetic analyses might be somewhat robust to the effect of these ascertainment biases. For example, analyses based on measures of population subdivision, such as FST, or on PCAs, which are commonly applied in the analysis of SNP chip data, may not depend strongly on allele frequencies and may, therefore, not suffer much from ascertainment biases. In the next two sections, we will further investigate the possible robustness of F and PCAs to the ascertainment biases.
FST
One of the most frequently used measures of population differentiations is FST. Using the NIEHS data, we examined the effect of ascertainment bias on FST estimates between European Americans and Africans. We simulated ascertainment in the African-Americans populations as described in figure 6. There is a small but clear effect of the ascertainment bias that increases as the number of African Americans used in the ascertainment panel decreases. In this particular example, the ascertainment bias leads to an increase in FST. The largest increase occurs simply by removing SNPs invariable among all 15 African Americans. Even if the ascertainment sample size is very large, there is a small but noticeable bias. The size of the ascertainment sample has less of an effect as long as it is not very small ( < 4 chromosomes). FST can be defined as the variance among populations divided by the total variance. To further investigate how the ascertainment bias affects FST, we plotted the mean between population variance and the mean total variance for pairwise FST between all combinations of populations except the African Americans (who were used as the ascertainment panel). Figure 7 shows total and between populations mean variance components for the ascertained SNPs normalized by the mean variance from all the SNPs. Both the mean total and the mean between population variance increase as the ascertainment panel get smaller. This explains why FST is less affected by ascertainment bias than the frequency spectrum itself, even though FST is a function of the 2D frequency spectrum. However, for some of the populations, the mean between population variance component increases more than the mean total variance, explaining why a bias will be observed for some populations. The effect is especially strong when the African population is one of the two populations compared, whereas the increase in total and between population variance is similar when populations that are more distant to the ascertainment population are being compared.
FIG 6.

Pairwise FST between European Americans and Africans for different ascertainment schemes. Standard error bars were estimated using 1,000 bootstrap samples. For the black bars, the SNPs are sampled independently and for the gray bars, the SNPs are sampled genewise from the 251 genes. The ascertainment was performed in African Americans and a varying number of individuals were used for the ascertainment. NAsc is the number of ascertained SNPs. The FST estimate using all SNPs (no ascertainment bias) is labeled “None.”
FIG 7.

Mean pairwise FST components between pairs of populations using the NIEHS data. The ascertainment was performed in African Americans and a different number of individuals were used for the ascertainment. Each SNP gives an estimate for the total variance and the between population variance. The plot shows the normalized mean variances. We normalized by dividing by the mean total variance and mean between population variance, respectively, for all the SNPs in the sample (regardless of ascertainment).
For the Celera data, we have both resequencing and SNP chip data for the same individuals. In table 1, we compare the FST estimates from the resequencing data and the SNP chip data. We also included data from the Seattle SNPs study (Seattle SNPs 2009) of resequenced Yorubans and CEPH individuals from the HapMap project where SNP chip data are also available. For the Celera data, FST is slightly higher for SNP chip data than for the resequencing data. However, essentially identical estimates are obtained for the Seattle SNP data.
Table 1.
FST for SNP Chips and Resequencing Data
| FST for SNP Chip | FST for Resequencing | P Value | Populations | |
| Seattle SNPs | 0.157 | 0.158 | 0.6 | African versus European |
| Celera | 0.0864 | 0.0756 | 0.001 | African Americans versus European Americans |
Note.—Differences in FST estimates were tested by a two-sided permutation test. The SNPs were randomly permuted 1,000 times between the chip data and the resequencing data. The Seattle SNPs data had 4,187 SNPs without missing genotypes for the resequencing data and 388,596 for the chip data. The Celera data had 4,519 SNPs without missing genotypes for the resequencing data and 378,724 for the chip data.
Principal Component Analysis
PCA is a very useful tool for capturing patterns in high-dimensional data and projecting them down to a low dimension. It is a technique that has been used with great success in a number of recent population genetic studies (Jakobsson et al. 2008, Lao et al. 2008, Li et al. 2008, Novembre et al. 2008). PCA is used to characterize how different multiple populations are, often using only the two first principal components. To illustrate the importance of the ethnicity of the ascertainment population, we performed PCA on the NIEHS data using different ascertainment populations. We used the genotype data for the African and European population for the PCA and simulated ascertainment using the Asian, Hispanic, or African-American populations. Using the two first principal components, we plotted the mean pairwise distance between and within the individuals from the two populations (see fig. 8). We did this for different sizes of the ascertainment panels and used standard bootstrap and a gene-wise bootstrap to estimate the standard errors.
FIG 8.

Mean pairwise distances within and between Africans and European Americans based on the two first principal components. Standard errors are estimated using bootstrap. The light colors are standard errors based on a gene-wise bootstrap and the dark colors are the standard bootstrap where the SNPs are sampled independently. Ascertainment, using an increasing number of individuals, was simulated using the Asian, the Hispanic, and the African-American population, respectively, from the NIEHS resequencing data. There are 5,948 SNPs without missing data in any population.
When the African Americans are used as the ascertainment panel, the distances appear to be the same regardless of the size of the ascertainment panel. However, when either the Hispanic or the Asian populations are used as ascertainment panel, the mean pairwise distances within the African and the European populations changes dramatically. When all SNPs are used, the Africans within population variation are much larger than the Europeans, whereas the opposite is true when the size of the ascertainment panel is small.
We also performed a PCA using four of the five populations from the NIEHS data and used the fifth populations as the ascertainment panel. In figure 9, we show the PCA plot without ascertainment and a plot where all the Asian individuals are used as the ascertainment panel. The figure shows that without ascertainment, the Africans and African Americans are further apart than the Europeans and Hispanics. When ascertainment is performed, the Africans and African Americans are close together and the Hispanic and Europeans are far from each other. This, however, could just be the eigenvalues changing order due to random chance. Therefore, we randomly sampled the same number of SNPs used in the PCA with the ascertained SNPs and performed PCA on these SNPs. We did 1,000 random samples and in none of these samples, the Hispanic and Europeans were further apart than the Africans and Africans Americans. Thus, the change in relative distances between populations is only due to the ascertainment. It is clear that ascertainment can have a substantial effect on PCAs, especially when the results of such analyses are represented graphically in terms of the leading principal components. PCA was also performed on the Celera individuals and the Seattle SNP individuals, both for the chip data and for the resequencing data (see Supplementary fig. S2, Supplementary Material online). There are some visible differences between the SNP chip data and the resequencing data but they are hard to quantify. And it should be noted that we expect that the Affymetrix SNP selection was performed based on SNP discovery studies involving both African/African Americans and individuals on European descent. If so then the effect of ascertainment bias on these individuals might be minor.
FIG 9.

PCA plot for the first two principal components for four populations from the NIEHS data. The left plot has no ascertainment bias. In the right plot, ascertainment was performed using all the 24 individuals from the Asian populations. All SNPs that were polymorphic in the four populations were used. Out of the 8,231 SNPs, 2,751 were ascertained in the left plot. We resampled 2,751 random SNPs without replacement from the 8,231 SNPs and performed PCA on them. None of the 1,000 resamples gave a higher distance between the European Americans than the distance between the African Americans and the Africans.
Ascertainment Correction
The observation that ascertainment can affect population genetic inferences suggests that it might be worthwhile to correct ascertainment biases statistically. This essentially involves a reverse engineering process estimating the distribution of ascertainment panels from the data. Unlike the analyses in Nielsen and Signorovitch (2003), we cannot for the Celera genotype data assume that the ascertainment sample is a subset of the genotyped sample. Ascertainment correction then requires modeling of the joint frequency spectrum of the unknown ascertainment sample and the known genotype sample. To do this modeling accurately, it is necessary to model the underlying distribution of allele frequencies in the population. We did this by fitting the distribution of allele frequencies to the Celera resequencing data, assuming a simple functional form for the distribution of allele frequencies without having to model the demographic histories of the populations. Three theoretically motivated distributions did not fit the data well (see Supplementary fig. S3, Supplementary Material online), but a simple mixture of exponentials provided a nice fit as seen in figure 10 for the synonymous Celera data. The mixture of exponentials also fitted the African-American data well but with different parameter estimates (not shown). Using this estimated allele frequency density, we then proceeded to estimate the ascertainment schemes that had been used to select the SNPs on the chip. Assuming a known number of ascertainment panels and a known and simple ascertainment scheme, we estimated the size of the ascertainment panels and the fraction of SNPs selected from each ascertainment panel as described in the Materials and Methods section. We did this for one or two ascertainment panels and using two ascertainment schemes. In one scheme, all SNPs were required to be polymorphic in the ascertainment panel and in the other, the minor allele in the ascertainment panel had to be observed twice. The results for the Celera synonymous SNP data for the European Americans (assuming that the ascertainment was also performed in the same population) are shown in table 2. Using the Akaike information criteria, the best model was the one where the ascertainment was performed in two ascertainment panels, where two minor alleles were needed in both panels. Using this estimated ascertainment scheme, we were able to correct the frequency spectrum for the chip data as shown in figure 11. Although one ascertainment scheme fitted the data best, all the ascertainment schemes improved the frequency spectrum greatly (see Supplementary fig. S4, Supplementary Material online). Similar ascertainment schemes were estimated when using the European individuals from the Seattle SNP data (not shown).
FIG 10.

We fitted the frequency spectrum of the Celera European Americans for the synonymous SNPs using a mixture of exponentials. The spectra for the observed and the fitted data are folded.
Table 2.
Likelihood Scores and Parameter Estimates for Different Ascertainment Schemes for the European American Celera Synonymous SNP Data
| Ascertainment Scheme | Likehood Score | d | π |
| 1 | 1,086,089 | 5.6 | 1 |
| 2 | 1,088,249 | 24.2 | 1 |
| 1, 2 | 1,085,690 | 2.5, 37.7 | 0.81, 0.19 |
| 1, 1 | 1,086,088 | 2.3, 6.4 | 0.35, 0.65 |
| 2, 2 | 1,085,505 | 4.5, 41.6 | 0.69, 0.31 |
Note.—d is the size of the ascertainment panel and π is the fraction of SNP selected from this panel. The ascertainment scheme is the number of minor alleles needed in the ascertainment panel required to ascertain the SNPs.
FIG 11.

The Celera European Americans synonymous frequency spectra for the resequencing data, the SNP chip data, and the corrected frequency spectrum. The correction assumed that two ascertainment panels were used and the SNPs were ascertained if both alleles were observed at least twice.
This analysis suggests that the real ascertainment in the Affymetrix SNP chip is complex and probably involves a scheme in which SNPs are preferred if both alleles have been observed at least twice. This would be a reasonable criterion for inclusion if there is a preference for verified SNPs that are less likely to be artifacts caused by sequencing errors. However, it causes an even stronger ascertainment bias than if ascertainment is just based on observing each allele at least once.
Discussion and Conclusions
Clark et al. (2005) demonstrated that data sets based on different ascertainment schemes give different patterns of FST. They also showed that both the estimates of the multiloci FST and the variation of single marker FST are affected. We observe an upward bias in FST and show that the effect of ascertainment bias strongly depends on how similar the populations that are being compared are to the population in the ascertainment panels. Although we have demonstrated that the bias will affect FST, we do not see a large difference in FST estimated from the SNP chip and resequencing data. In particular, the Seattle SNP data show no effect of the ascertainment bias on FST in the comparison between Africans and Europeans. The surprisingly robust performance of FST in this case may be due to the fact that ascertainment in part has been based on both Europeans and African Americans from the Perlegen data (Hinds et al. 2005).
We have shown that ascertainment biases can have a strong effect on the results of PCA. In light of this, we caution against simple interpretation of PCAs based on ascertained SNP data. Relative distances of the first two principal components in a PCA plot should not be used for conclusions regarding distances among populations or interpopulation variability. This is an important conclusion of this paper because PCA is rapidly becoming one of the preferred population genetic analyses tools for genome-wide data.
PCAs are frequently used to detect or correct for population stratification in association mapping studies. If the ascertainment bias affects all genomic regions equally and association mapping is based on the same markers that are used to control for stratification, there is no reason to suspect that the ascertainment bias will lead to false positives. However, if the employed ascertainment protocols used differ greatly among regions, it is possible that genomic control methods based on PCA or other techniques could be affected. Likewise, if SNP genotyping data are used for genomic control, but association mapping is carried out using resequencing of particular candidate genes, ascertainment might also affect the conclusions. The degree to which any studies might be affected by this is presently unknown.
We have shown that the fundamental nature of the SNP calling procedure by itself may induce a bias in the frequency spectrum by giving higher confidence scores to the SNP with lower minor allele frequencies. Thus, the threshold chosen for SNP calling will affect the frequency spectrum by itself. This may greatly affect population genetic analyses if the distribution of confidence scores differs among populations. Additionally, if SNPs with missing data are excluded from analyses, the frequency spectrum will be more even for populations with larger sample sizes. This makes it difficult to infer the ascertainment scheme used for the SNP chips and even more difficult to transfer any correction to other samples.
There are several issues we have not covered in this article that also may have an effect on genotyping data. One important issue is genotyping errors. Random genotyping errors tend to increase the minor allele frequencies for the samples and reduce the difference between populations. However, if the genotyping errors are not random between population, then genotyping error will increase the distance between populations. Choosing a more strict threshold for the genotyping score decreases the genotyping error but will, as discussed, potentially have an even larger effect on the frequency spectrum.
Using resequencing data, we estimated the ascertainment scheme used for the Affymetrix 500k SNP chip set. The model used is of course a great simplification of the real ascertainment scheme. However, using the inferred ascertainment scheme, we could to a large extent reconstruct the frequency spectrum of the resequencing data. Unfortunately, for most populations, resequencing data are not available and as we have shown, using the ascertainment scheme inferred from a distant populations might not work that well. As such, there is little hope to use inferred ascertainment procedures in a correction on data from distant populations. However, in genomic analyses on the focal population, the correction can be used to analyze genome-wide patterns. But even in such cases, the correction may if the ascertainment protocol varies among regions. Another assumption in our ascertainment model is that only one population was used in the ascertainment process. This assumption is likely to be incorrect because some of the big resequencing projects, such as the Perlegen and the Celera projects, have included several populations. Both these included an African or an African-American population that most likely have been used in the SNP ascertainment. We see several indication of this. First of all, in the Celera data, the African Americans have more private SNPs than the European Americans. Another indication is that the difference in FST estimates between the SNP chip data and the resequence data is not that large. However, the fact that the European Americans have more singletons on the SNP chip than the African Americans seems to indicate that the Europeans have been the primary populations for the ascertainment.
As the price of resequencing continuous to drop the use of SNP chips will probably decrease. Although resequencing data undoubtedly will pose major challenges by itself, due to sequencing errors and alignment errors, these problems seem small compared with the problems facing valid population genetic analyses of chip-based genotyping data. Given the sometimes severe problems associated with the use of genotyping SNPs for population genetic analyses, it might be worthwhile to wait for the results of large-scale resequencing studies of global human variation, rather than relying on temporary, and possibly flawed analyses using SNP genotyping data.
Supplementary Material
Supplementary figures S1–S4 and tables S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the Danish Research Council, the Center for Pharmocogenomics, and by National Institutes of Health grant NIGMS R01HG003229. Thanks to Kirk Lohmoeller for assistance with the Celera resequencing data.
References
- Affymetrix. 2006. BRLMM: an improved genotype calling method for the GeneChip Human Mapping 500K Array Set. Technical Report. Santa Clara (CA): Affymetrix, Inc. Available from: http://www.affymetrix.com/support/technical/whitepapers/brlmm_whitepaper.pdf. [Google Scholar]
- Albrechtsen A, Sand Korneliussen T, Moltke I, van Overseem Hansen T, Nielsen FC, Nielsen R. Relatedness mapping and tracts of relatedness for genome-wide data in the presence of linkage disequilibrium. Genet Epidemiol. 2009;33:266–274. doi: 10.1002/gepi.20378. [DOI] [PubMed] [Google Scholar]
- Boyko AR, Williamson SH, Indap AR, et al. (14 co-authors) Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008 doi: 10.1371/journal.pgen.1000083. 4:e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante CD, Fledel-Alon A, Williamson S, et al. (14 co-authors) Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, et al. (41 co-authors) A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller E. Effects of ascertainment bias on recovering human demographic history. Hum Biol. 2001;73:411–427. doi: 10.1353/hub.2001.0034. [DOI] [PubMed] [Google Scholar]
- Foll M, Beaumont MA, Gaggiotti O. An approximate Bayesian computation approach to overcome biases that arise when using amplified fragment length polymorphism markers to study population structure. Genetics. 2008;179:927–939. doi: 10.1534/genetics.107.084541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot G, Foll M. Correcting for ascertainment bias in the inference of population structure. Bioinformatics. 2009;25:552–554. doi: 10.1093/bioinformatics/btn665. [DOI] [PubMed] [Google Scholar]
- Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- Hirschhorn J, Daly M. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, et al. (24 co-authors) Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Kingsmore S, Lindquist I, Mudge J, Gessler D, Beavis W. Genome-wide association studies: progress and potential for drug discovery and development. Nat Rev Drug Discov. 2008;7:221–230. doi: 10.1038/nrd2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner MK, Beerli P, Yamato J, Felsenstein J. Usefulness of single nucleotide polymorphism data for estimating population parameters. Genetics. 2000;156:439–447. doi: 10.1093/genetics/156.1.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao O, Lu TT, Nothnagel M, et al. (33 co-authors) Correlation between genetic and geographic structure in Europe. Curr Biol. 2008;18:1241–1248. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, et al. (11 co-authors) Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Livingston RJ, von Niederhausern A, Jegga AG, Crawford DC, Carlson CS, Rieder MJ, Gowrisankar S, Aronow BJ, Weiss RB, Nickerson DA. Pattern of sequence variation across 213 environmental response genes. Genome Res. 2004;14:1821–1831. doi: 10.1101/gr.2730004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S, et al. (12 co-authors) Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Bryc K, King KS, et al. (23 co-authors) The Population Reference Sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet. 2008;83:347–358. doi: 10.1016/j.ajhg.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G, Smith AV, Jonsson F, Gustafsson O, Stefnsson K, Donnelly P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. J R Stat Soc Ser B. 2002;64:695–715. [Google Scholar]
- Nielsen R. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics. 2000;154:931–942. doi: 10.1093/genetics/154.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R. Population genetic analysis of ascertained SNP data. Hum Genomics. 2004;1:218–224. doi: 10.1186/1479-7364-1-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hubisz MJ, Clark AG. Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics. 2004;168:2373–2382. doi: 10.1534/genetics.104.031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hubisz MJ, Torgerson D, et al. (13 co-authors. Darwinian and demographic forces affecting human protein coding genes. Genome Res. 2009;19:838–849. doi: 10.1101/gr.088336.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Signorovitch J. Correcting for ascertainment biases when analyzing SNP data: applications to the estimation of linkage disequilibrium. Theor Popul Biol. 2003;63:245–255. doi: 10.1016/s0040-5809(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, et al. (12 co-authors) Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006 doi: 10.1371/journal.pgen.0020190. 2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. (11 co-authors) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SA, Hartl DL. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–1176. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seattle SNPs 2009. Available from: http://pga.gs.washington.edu/ [Google Scholar]
- Smith M, O'Brien S. Mapping by admixture linkage disequilibrium: advances, limitations and guidelines. Nat Rev Genet. 2005;6:623–632. doi: 10.1038/nrg1657. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J, Nielsen R, Liu-Cordero SN, Ardlie K. The discovery of single-nucleotide polymorphisms—and inferences about human demographic history. Am J Hum Genet. 2001;69:1332–1347. doi: 10.1086/324521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating f-statistics for the analysis of population structure. Evolution [Internet] 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. Available from: http://www.jstor.org/stable/2408641. [DOI] [PubMed] [Google Scholar]
- Wiuf C. Consistency of estimators of population scaled parameters using composite likelihood. J Math Biol. 2006;53:821–841. doi: 10.1007/s00285-006-0031-0. [DOI] [PubMed] [Google Scholar]
- Wright. Evolution in mendelian populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]