

Abstract
Mitochondrial (mt) function depends critically on optimal interactions between components encoded by mt and nuclear DNAs. mitochondrial DNA (mtDNA) inheritance (SMI) is thought to have evolved in animal species to maintain mito-nuclear complementarity by preventing the spread of selfish mt elements thus typically rendering mtDNA heteroplasmy evolutionarily ephemeral. Here, we show that mtDNA intraorganismal heteroplasmy can have deterministic underpinnings and persist for hundreds of millions of years. We demonstrate that the only exception to SMI in the animal kingdom, that is, the doubly uniparental mtDNA inheritance system in bivalves, with its three-way interactions among egg mt-, sperm mt- and nucleus-encoded gene products, is tightly associated with the maintenance of separate male and female sexes (dioecy) in freshwater mussels. Specifically, this mother-through-daughter and father-through-son mtDNA inheritance system, containing highly differentiated mt genomes, is found in all dioecious freshwater mussel species. Conversely, all hermaphroditic species lack the paternally transmitted mtDNA (=possess SMI) and have heterogeneous macromutations in the recently discovered, novel protein-coding gene (F-orf) in their maternally transmitted mt genomes. Using immunoelectron microscopy, we have localized the F-open reading frame (ORF) protein, likely involved in specifying separate sexes, in mitochondria and in the nucleus. Our results support the hypothesis that proteins coded by the highly divergent maternally and paternally transmitted mt genomes could be directly involved in sex determination in freshwater mussels. Concomitantly, our study demonstrates novel features for animal mt genomes: the existence of additional, lineage-specific, mtDNA-encoded proteins with functional significance and the involvement of mtDNA-encoded proteins in extra-mt functions. Our results open new avenues for the identification, characterization, and functional analyses of ORFs in the intergenic regions, previously defined as “noncoding,” found in a large proportion of animal mt genomes.
Keywords: mtDNA, doubly uniparental inheritance, open reading frame, sex determination, heteroplasmy, Bivalvia
Introduction
Mitochondria are crucial for life and death processes in almost all eukaryotic cells (Scheffler 2008). They provide the majority of the cellular energy via oxidative phosphorylation and are intimately involved in other vital cellular mechanisms such as reactive oxygen species production and regulation, ion homeostasis, and apoptosis (Scheffler 2008). Evidence suggests that mitochondrial (mt) dysfunction also plays a critical role in various metabolic and degenerative diseases, cancer, and aging (Wallace 2005). To function properly, mitochondria depend on the coordinated expression of nuclear and mt genes necessitating the coevolution of nuclear and mt genomes (Blier et al. 2001; Lane 2009). In contrast to the nuclear genome that is generally transmitted by both parents, mt genomes are strictly maternally inherited in animals (Birky 2001). Strict maternal mitochondrial DNA (mtDNA) inheritance (SMI) coupled with a substantial mt bottleneck that occurs early in oogenesis (Roze et al. 2005; White et al. 2008) result in an essentially homoplasmic state for mtDNA, that is, a situation where all copies of mtDNA in each cell have identical sequences. The most widely accepted hypothesis for the prevalence of SMI in animals is that this mode of transmission is the most effective means of preventing the spread of selfish mt elements and maintaining optimum interactions between components encoded by the mt and nuclear DNAs for mt function (Hurst 1992; Ballard and Whitlock 2004; Lane 2005). In other words, SMI was likely favored because it ensures efficient mt respiration, thereby avoiding apoptosis (Lane 2005). Consistent with this hypothesis, intraorganismal heteroplasmy, that is, the presence of different mtDNAs within an individual, is considered to be a relatively ephemeral phenomenon, generally resolved to homoplasmy within a few generations, during animal mt evolution (Doublet et al. 2008; White et al. 2008). For example, rare cases of evolutionary persistent and stable intraorganismal mt heteroplasmy have been reported in oniscid crustaceans (Doublet et al. 2008), in the spider mite Tetranychus urticae (Van Leeuwen et al. 2008) and in a mutant strain of Drosophila (Debise et al. 1993). Plausible explanations for these apparently counter-intuitive observations are 1) variant mtDNA molecules may persist via a replicative advantage, 2) functional reductions caused by variant mtDNAs may be compensated by “wild-type” mtDNA molecules, and 3) heteroplasmy may persist via balancing selection (Doublet et al. 2008; White et al. 2008).
Another major difference between the mt and the nuclear genomes is that unlike nuclear DNA which is linear and encodes thousands of genes, animal mt genomes are almost universally compact, circular molecules (∼16-kb long) encoding 13 core energetic proteins and 24 structural RNAs required for their expression (Scheffler 2008; Lane 2009). The complexity of mt functions contrasts with the relatively limited genetic repertoire of mt genomes. To our knowledge, very few studies have demonstrated the involvement of typical animal mtDNA–encoded proteins in functions other than energy production (Gingrich et al. 2004; Chakrabarti et al. 2007, 2009). For example, the mtDNA-encoded nicotinamide adenine dinucleotide reduced form (NADH) dehydrogenase subunit 2 protein (ND2) was shown to be essential for the regulation of receptor activity in the excitatory synapses of the human brain (Gingrich et al. 2004). Because of the rather limited size and genetic capacity of animal mtDNA, the presence of extra-mt protein-coding genes, that is, genes other than those in the typical set of 13, is quite rare (Maximov et al. 2002; Guo et al. 2003; Gissi et al. 2008). Extra-mt protein-coding genes (e.g., humanin, mutS, dnaB, atp9, tatC) have been identified and annotated in the mt genomes of humans (Maximov et al. 2002), corals and sea anemones (Cnidaria), and sponges (Porifera) (Gissi et al. 2008). A few other mt open reading frames (ORFs) have been detected in the same animal groups and in the Placozoa but their expression remains unconfirmed (Dellaporta et al. 2006; Gissi et al. 2008). Except for the few cases mentioned above, all mitochondrially encoded proteins in animals are usually involved in mt energy production, mtDNA replication and expression, mtDNA repair or mt protein import (Burger et al. 2003; Gissi et al. 2008). In contrast, several chimeric protein-coding genes associated with sex determination have been found in mt genomes of angiosperm plants, a phenomenon known as cytoplasmic male sterility (CMS) (Hanson and Bentolila 2004; Burt and Trivers 2006; Chase 2007). To date, however, there have been no reports of mtDNA-associated sex determination in animals. It is therefore of considerable interest that we recently demonstrated the expression of novel, sex-associated mtDNA-encoded proteins in freshwater mussels (Bivalvia: Unionoida) (Breton et al. 2009). These results are particularly significant because bivalves are the only known exception to SMI in the animal kingdom (Breton et al. 2007).
The exceptional doubly uniparental mtDNA inheritance system (DUI) has been found in 46 species with dioecious breeding systems (=separate male and female sexes) belonging to the bivalve orders Mytiloida, Unionoida, and Veneroida (Breton et al. 2007; Theologidis et al. 2008). Instead of transmitting their mtDNA via SMI, females pass on their mtDNA (the F genome) to all offspring but males also inherit a second mt genome (the M genome) from their fathers (Hoeh et al.1991; Skibinski et al. 1994; Zouros et al. 1994; Breton et al. 2007). Therefore, female offspring are typically homoplasmic and male offspring are heteroplasmic (Hoeh et al. 1991; Skibinski et al. 1994; Zouros et al. 1994). Male somatic tissues contain and express predominantly the F genome, but the male germ line contains and expresses predominantly the M genome, and sperm contain exclusively the M genome (Dalziel and Stewart 2002; Venetis et al. 2006). Remarkably, the long term (>200 My) stability of this singular animal three genomes system has produced highly distinct conspecific F and M mt genomes with more than 50% amino acid sequence divergence in freshwater mussels (Breton et al. 2007; Doucet-Beaupré et al. 2010). Heteroplasmic sequence divergences between F and M genomes within freshwater mussel males are the highest intraindividual values yet reported, even higher than values reported in classical model systems used for the study of mito-nuclear coevolution and for which disruption of mt respiratory chain function was observed (Breton et al. 2007). To date, the factors responsible for the maintenance of this non-SMI mt transmission system have not been elucidated.
Two nonexclusive hypotheses have been proposed to explain the function of the M genome in bivalves: 1) it could increase the fitness of sperm and/or 2) it could be involved in sex determination (Zouros 2000; Breton et al. 2007). Unfortunately, little is known about sex determination in bivalves with DUI except that sex is not determined by heteromorphic sex chromosomes (Breton et al. 2007). It has been recently stated that paternal mtDNA and maleness are coinherited but not causally linked in marine mussels Mytilus (Mytiloida) (Kenchington et al. 2009). However, it appears that the presence of the paternal M genome and maleness can be consistently decoupled, that is, the presence of the M mtDNA is not required for an embryo to develop into a male, only in progeny from hybrid and triploid crosses (Saavedra et al. 1997; Theologidis et al. 2007; Kenchington et al. 2009). These results suggest that hybridization and polyploidization might interfere with normal sex specification determined by both nuclear and mt gene products, and that DUI is tightly linked to the specification of males and females (dioecy). Our recent discovery of two evolutionarily persistent (for >200 My), novel sex-specific mtDNA-encoded proteins, that is, the F-ORF protein coded by all F genomes and the M-ORF protein coded by all M genomes in freshwater mussels (Breton et al. 2009), suggests that they could be part of a mechanism that determines sex in this group of bivalves (Saavedra et al. 1997; Zouros 2000; Breton et al. 2007). However, the linkage between DUI and sex determination has yet to be confirmed and the processes responsible for the long-term persistence of the unique mtDNA transmission system in bivalves remain an open question (Hoeh et al. 2002).
Here, we address this fundamental question in a study designed to test the linkage between DUI and sex determination and to better understand the processes involved in the evolutionary persistence of DUI in bivalves. We demonstrate that DUI has been stably maintained in dioecious freshwater mussel species for over 200 My because the two mt genomes are most likely key participants in the sex determination system. Specifically, we demonstrate that the maintenance of dioecy in these bivalves is highly correlated with the presence of proteins coded by the highly divergent maternally and paternally transmitted mt genomes. Our results establish new features for animal mt genomes: the presence of novel mtDNA-encoded proteins with functional significance and the involvement of mtDNA-encoded proteins in extra-mt functions.
Materials and Methods
Testing for the Presence of an M Genome
Taxa sampled for the presence of an M genome included 14 species representing two freshwater mussel families (Unionidae and Margaritiferidae): the hermaphroditic Lasmigona compressa (n = 30) and L. subviridis (n = 10) and their dioecious congeners L. complanata (n = 12) and L. costata (n = 10); the hermaphroditic Margaritifera falcata (n = 18), its dioecious congener M. margaritifera (n = 11) and the dioecious, confamilial Cumberlandia monodonta (n = 11); the hermaphroditic Toxolasma parvum (n = 62) and its dioecious congeners T. glans (n = 14), T. minor (n = 28), and T. paulus (n = 16); and the hermaphroditic Utterbackia imbecillis (n = 58) and its dioecious congeners U. peggyae (n = 37) and U. peninsularis (n = 48) (see table 1). Sex was determined by microscopical examination of gonadal tissues. Purified mtDNA or total genomic DNA were, respectively, isolated from either mantle or testis tissue using a cesium chloride–gradient centrifugation technique (Utterbackia spp) (Hoeh et al. 1991) or a QIAGEN DNeasy animal kit (QIAGEN Inc., Valencia, CA) using the animal tissue protocol. For all individuals, an ∼700 bp fragment of cox1 and an ∼900 bp fragment of nad1 were amplified using modified versions of the universal cox1 primers (Folmer et al. 1994) (LCO22me2 5'-GGTCAACAAAYCATAARGATATTGG-3' and HCO700dy2 5'-TCAGGGTGACCAAAAAAYCA-3') (Walker et al. 2006, 2007) and freshwater mussel-specific nad1 primers FWMND1For (5'-AGAHMADTGCHYYAGRTTTAAGC-3') and FWMND1Rev (5'-TRCTTGGAAGGCARHTGTAC-3'). Polymerase chain reaction (PCR) conditions for cox1 and nad1 are presented in the subsection “Sequencing protocol for the mt genes Fcox1, Fnad1, F-orf and H-orf.” All sequences obtained from female or hermaphroditic individuals were confirmed as Fcox1 and Fnad1 via comparisons with GenBank F sequences. Some Fcox1 and Fnad1 sequences were used to generate the phylogeny. We were consistently able to obtain Mcox1 and Mnad1 sequences from male individuals of dioecious species but never from hermaphroditic individuals (see table 1).
Table 1.
Congeneric evaluations for the presence/absence of an M genome.
| Species (breeding system) | Mcox1 | Mnad1 | Mcox2–cox1a | Mcox2–cox1b | Mcox2–nad3 | M-orf | Mcox2e |
| Lasmigona compressa (h) | − | − | − | − | − | − | − |
| L. subviridis (h) | − | − | − | − | − | − | − |
| L. complanata (d) | − | − | + | − | − | + | + |
| L. costata (d) | − | − | + | − | − | + | + |
| Margaritifera falcata (h) | − | − | − | − | − | − | − |
| M. margaritifera (d) | + | − | + | − | + | − | + |
| Cumberlandia monodonta (d) | + | + | + | + | + | + | + |
| Toxolasma parvum (h) | − | − | − | − | − | − | − |
| T. glans (d) | + | − | + | + | + | + | + |
| T. minor (d) | − | − | + | + | + | + | + |
| T. paulus (d) | + | − | + | + | + | + | + |
| Utterbackia imbecillis (h) | − | − | − | − | − | − | − |
| U. peggyae (d) | − | − | − | + | + | + | − |
| U. peninsularis (d) | + | + | + | + | + | + | + |
Note.—(h), hermaphrodite; (d), dioecious; +, M genome amplification successful; −, M genome amplification failed;
“Mcox2–cox1 amplicon a” obtained using UNIOCOII.2 and HCO700dy2 primers (Curole 2004; Walker et al. 2006);
“Mcox2–cox1 amplicon b” obtained using MCOIIh35F and MCOI20R primers (Walker et al. 2007).
Consistent amplification of the M mtDNA is often difficult using universal primers due to the rapidly evolving nature of the M mtDNA and the presence of contaminating F mtDNA in DNA extracts from testes (Saavedra et al. 1997; Theologidis et al. 2007; Walker et al. 2007). Thus, to efficiently screen for the presence of the M genome, we designed (or used previously designed) (Curole 2004; Walker et al. 2007) “freshwater mussel-M-specific” and “freshwater mussel-genus-M-specific” mtDNA primers in locations where fixed nucleotide differences between freshwater mussel M and F sequences were noted or in locations exclusive to the freshwater mussel M genome (see supplementary fig. S1, Supplementary Material online).
(A) Mcox2–cox1 amplicon a: The “freshwater mussel-M-specific” primer UNIOCOII.2 (Curole 2004; Walker et al. 2006) was paired with HCO700dy2 (Walker et al. 2006) to amplify the cox2–cox1 fragment. These primers typically amplify approximately 1.1 kb of cox2–cox1 from F genomes and approximately 1.7 kb from M genomes because of the presence of the M genome-specific 3' extension of the cox2 gene in freshwater mussels (Walker et al. 2006). They have been previously shown to successfully amplify the Mcox2–cox1 fragment for >40 bivalve species from three different freshwater mussel families (Walker et al. 2006), including the Unionidae and Margaritiferidae. Amplifications for “Mcox2–cox1 amplicon a” were performed in 50 μl volumes of a solution containing 1X QIAGEN PCR buffer, 0.2 mM of each deoxynucleoside triphosphate (dNTP), 0.5 μM of each primer, 1U QIAGEN Taq, and ∼10–100 ng of template DNA. Reactions had an initial denaturing step of 95 °C for 1 min, followed by 40 cycles of denaturation at 94 °C for 60 s, annealing at 50 °C for 60 s, extension at 72 °C for 90 s, and a final extension at 72 °C for 10 min.
(B) Mcox2–cox1 amplicon b: The “freshwater mussel-M-specific” primer MCOIIh35F (Walker et al. 2007) was paired with the “freshwater mussel-M-specific” primer MCOI20R (Walker et al. 2007). These primers, which have been, respectively, designed in the 3' end of the Mcox2-homologous gene (Mcox2h) and in the 5' end of the Mcox1 gene, exclusively amplify the Mcox2 extension (Mcox2e) and do not produce any F amplicon. They have been previously successfully tested on >30 freshwater mussel species (Walker et al. 2007). PCR conditions were the same than for the “Mcox2–cox1 amplicon a,” except that the annealing temperature was 55 °C and that the extension time was 60 s.
(C) Mcox2–nad3 amplicon: The “freshwater mussel-M-specific” primer MCOIIh63F (Doucet-Beaupré et al. 2010) was paired with the freshwater mussel-specific primer UNIOND3-155F (5'-GTAAAACGACGGCCAGTGAG-3'). These primers, which have been, respectively, designed in the 3' end of the Mcox2h and in the 3' end of the nad3 gene, exclusively amplify a Mcox2h–nad3 fragment and do not produce any F amplicon. They have been successfully tested on >60 bivalve species from three different freshwater mussel families (i.e., Unionidae, Hyriidae, and Margaritiferidae) in our laboratory (Breton S, Hoeh WR, unpublished). PCR conditions were the same as for the “Mcox2–cox1 amplicon b,” except that the annealing temperature was 45 °C.
(D) M-orf amplicon: The “freshwater mussel-M-specific” primer MORFunioF (5'-AGYAAAABYCYMAAAAGVCTATG-3') was paired with the “freshwater mussel-M-specific” MORFunioR (5'-TGCCTCARYTTRGWCCKGTT-3'). These primers, which have been, respectively, designed in the 5' end of the Mnad4L gene and in the 3' end of the Matp8 gene, exclusively amplify the newly discovered male-specific M-orf gene and do not produce any F amplicon. They have been tested successfully on >18 species (families Unionidae and Margaritiferidae) in our laboratory (Breton S, Hoeh WR, unpublished). PCR conditions were the same as for the “Mcox2–cox1 amplicon b,” except that the annealing temperature was 45 °C.
(E) Mcox2e amplicon: The “anodontine (= freshwater mussel subfamily)-M-specific” primer pair AnodonMcox2eF (5'-GCATARRYWGAAGCRTCAAYTGG-3') and AnodonMcox2eR (5'-AARGWKGTATGTVTGGTGRTG-3) was used to amplify the Mcox2 extension of the Lasmigona species. The “margaritiferid (=freshwater mussel family)-M-specific” primer pair MarCumbMcox2eF (5'-TTGAKTGGRGTTKRTGGYTT-3') and MarCumbMcox2eR (5'-CACGAWAMAACCGAACYATC-3') was used to amplify the Mcox2 extension of M. margaritifera and C. monodonta (a confamilial species). The “Toxolasma (=freshwater mussel genus)-M-specific” primer pair ToxoMcox2eF (5'-TTTGAGGGGCATTTGATCTC-3') and ToxoMcox2eR (5'-TCCAAACGCCTTTAACCAAC-3') was used to amplify the Mcox2 extension of the Toxolasma species. The “Utterbackia (=freshwater mussel genus)-M-specific” primer pair UtterMcox2eF (5'-CCGGGAATTCCATCTACCTT-3') and UtterMcox2eR (5'-TAGTGGGGAGGGGGAGATAG-3') was used to amplify the Mcox2 extension of the Utterbackia species. All these primer pairs have been designed in the M genome-specific 3' extension of the cox2 gene and exclusively amplify Mcox2e (i.e., they do not produce any F amplicon). All PCR conditions were the same as for the “Mcox2-cox1 amplicon b,” except that the annealing temperature was 50 °C.
Complete mt Genome Sequencing and Analyses
Complete mt genomes were amplified using the following primer pairs for the U. peninsularis M genome (the M-specific primer UtterMcox2eR with Ambl16SFor, and the M-specific primer UtterMcox2eF with Ambl16SRev) (Doucet-Beaupré et al. 2010), for the U. peninsularis F genome and the hermaphroditic U. imbecillis F-like genome (the F-specific primer 5'FCOIPygF 5'-GTAAAACGACGGCCAGTGATCAC-3' with Ambl16SFor, and the F-specific primer FCOI195 with Ambl16SRev) (Doucet-Beaupré et al. 2010), for the hermaphroditic L. compressa, M. falcata, and T. parvum F-like genomes (the F-specific primer FCOIIhFor with Ambl16SFor, and the F-specific primer FCOI195R with Ambl16SRev) (Doucet-Beaupré et al. 2010), and for the hermaphroditic L. subviridis F-like genome (the F-specific primer FCOIIhFor with ND1F (Palumbi 1996), and the F-specific primer FCOI195R with Ambl16SRev). Specifically, these primers amplified each mt genome in two large overlapping fragments that were ∼11- and ∼5.5-kb long (except for the nearly complete mt genome of L. subviridis, which has been amplified in two fragments ∼6-kb long). PCR amplifications were performed in 50 μl volumes containing ∼20 ng of total DNA, 0.4 μM of each primer, 1× QIAGEN LongRange PCR Buffer, 2.5 mM MgCl2, 500 μM of each dNTP, and 2 U QIAGEN LongRange PCR Enzyme Mix (QIAGEN Inc.). The thermal cycling parameters were as follows: 3 min at 93 °C then 35 cycles of: (93 °C, 15 s; 53 °C, 30 s; 72 °C, 6 –11 min depending on fragment size). PCR products were purified using a QIAquick PCR purification kit according to supplier's (QIAGEN Inc.) instructions. The purified PCR products were sequenced at the Sequencing Platform of McGill University (Montréal, Canada), using the Genome Sequencer FLX sequencing service.
Sequences were assembled using the MacVector Sequence Analysis Software version 10.0 (Accelrys Inc, San Diego, CA). Examination of all ORFs was performed with ORF Finder using the invertebrate mt genetic code and protein coding and ribosomal RNA genes were identified by performing sequence similarity searches in GenBank using Blast (Benson et al. 2004) or by comparison with freshwater mussel mt genomes previously published. The ends of rrnL and rrnS genes were assumed to extend to the boundaries of their flanking genes. Candidate transfer RNA (tRNA) genes were identified either using tRNAScan-SE version 1.21 (Lowe and Eddy 1997) or by comparison with positionally similar tRNAs of other freshwater mussel as previously published. For comparisons, complete genome sequences were obtained from F mtDNAs from the National Center for Biotechnology Information (NCBI) GenBank entries for Cristaria plicata (FJ986302; Jiang WP, Li JL, Zheng RL , unpublished data), Hyriopsis cumingii (FJ529186; Zheng RL, Li JL, unpublished data), Lampsilis ornata (AY365193; Serb and Lydeard 2003), Pyganodon grandis (FJ809754; Doucet-Beaupré et al. 2010), Quadrula quadrula (FJ809750; Doucet-Beaupré et al. 2010), and Venustaconcha ellipsiformis (FJ809753; Doucet-Beaupré et al. 2010). MUSCLE (Edgar 2004) was used to align sequences and MEGA 3.0 (Kumar et al. 2004) was used to calculate the percentage of amino acid difference between mt genes. ORFs were examined using Fickett's (1982) test code algorithm. Sequence similarity searches for all F-ORFs and H-ORFs (supplementary table S1, Supplementary Material online, see below) were performed in GenBank using BlastX and PSI-Blast (Altschul et al. 1997) against the following databases: 1) nonredundant protein sequences, 2) SWISSPROT, 3) protein data bank, and 4) environmental samples.
We also performed sequence similarity searches using BlastN against expressed sequence tags (ESTs others) (Benson et al. 2004). Protein structure and function prediction were examined with I-TASSER, a state-of the-art hierarchical protein structure modeling approach that is based on the secondary-structure enhanced profile–profile threading alignment (Zhang 2008). Transmembrane helices and other potential protein features of sex-specific ORFs were identified using ConPred II (Arai et al. 2004) and PredictProtein (Rost et al. 2004). Hydropathy profiles were calculated by the method of Kyte and Doolittle (1982). All mtDNA sequences are available via GenBank accession numbers HM856634–HM856640.
Sequencing Protocol for the mt Genes Fcox1, Fnad1, F-orf, and H-orf
Fcox1, Fnad1, and F-orf sequences were obtained from complete F mtDNAs from the NCBI GenBank entries for C. plicata (FJ986302; Jiang et al. unpublished data), H. cumingii (FJ529186; Zheng and Li unpublished data), L. ornata (AY365193; Serb and Lydeard 2003), P. grandis (FJ809754; Doucet-Beaupré et al. 2010), Q. quadrula (FJ809750; Doucet-Beaupré et al. 2010), and V. ellipsiformis (FJ809753; Doucet-Beaupré et al. 2010). Total DNA was extracted from female and hermaphrodite gonad or mantle tissues for 30 additional freshwater mussel species (see supplementary table S1, Supplementary Material online) using a QIAGEN DNeasy animal kit (QIAGEN Inc.) following the manufacturer’s animal tissue protocol. As mentioned above, purified mtDNA was obtained using a cesium chloride–gradient technique for the Utterbackia species (Hoeh et al. 1991). Partial sequences of the mt cytochrome c oxidase subunit I (Fcox1; 512 bp) and NADH dehydrogenase subunit 1 (Fnad1; 620 bp), and complete sequences of the F-orfs and H-orfs were obtained using the following primer pairs: LCO22e2 and HCO700dy2 (Fcox1) (Walker et al. 2006), FWMND1For and FWMND1Rev (Fnad1, see above), and FORFFor (5′-TTGAAAHYARAWARARARYGTTC-3′) and FORFRev (5′-TAYTTWRCYGYDGYTCTGA-3′) (for both the F- and H-orfs). All amplifications were performed in 50 μl volumes of a solution containing 1× QIAGEN PCR buffer, 0.2 mM of each dNTP, 0.5 μM of each primer, 1U QIAGEN Taq, and ∼10–100 ng of template DNA. Reactions had an initial denaturing step of 95 °C for 1 min, followed by 40 cycles of denaturation at 94 °C for 60 s, annealing at 40 °C (cox1) or 50 °C (nad1 and orfs) for 60 s, extension at 72 °C for 60 s, and a final extension at 72 °C for 10 min. The amplified products were purified using a QIAquick PCR purification kit and sequenced at Geneway Research (Hayward, CA).
Sequences were edited with the program BioEdit version 7.0.4.1 (Hall 1999) and alignments were generated with MUSCLE (Edgar 2004) using default parameters. Sequences were characterized after alignment and comparison with data from the NCBI databank. Mitochondrial sequences have been submitted to GenBank (accession numbers HM849059–HM849614).
Phylogenetic Analyses
Phylogenetic analyses were conducted on a concatenated two gene, 1,132-character data set (Fcox1 = 512 bp, and Fnad1 = 620 bp) using Bayesian inference (BI) via MrBayes (version 3.1.2) (Ronquist and Huelsenbeck 2003). The data set contained 188 terminals for which we generated sequences (supplementary table S1, Supplementary Material online). The Akaike information criterion, as implemented in JModelTest (Posada 2008) was used to determine the evolutionary model that best fit each gene partition. The general time reversible + G + I model (Rodriguez et al. 1990) was used in all BI analyses. Two independent simultaneous analyses were conducted for 18 million generations with four search chains each, and the DNA sequence data were partitioned by gene region and by codon position (two gene regions…three codon positions) yielding a total of six partitions and saving a total of 360,000 trees (one tree saved every 100 generations in each of the two analyses). To allow each partition to have its own set of parameter estimates, revmat, tratio, statefreq, shape, and pinvar were all unlinked during the analysis. To obtain the most accurate branch-length estimates possible, which is important for ancestral character state estimation with maximum likelihood (ML), the option prset ratepr = variable was employed as per the recommendations of Marshall et al. (2006). The standard deviation of the split frequencies for the two simultaneous analyses fell below 0.01 by six million generations and was <0.007 when the runs wereterminated (after 18 million generations). The 180,000 post-burn-in trees (determined by examination of the log probability of observing the data…generation plot) were used to calculate the BI majority rule consensus tree.
A best ML tree, based on analyses of the concatenated two gene matrix with no data partitioning, was generated using GARLI (v. 0.951; Zwickl 2006) (with default settings except for the following: autoterminate run 1,000,000 generations post last improved topology, lnL increase for significantly better topology = 0.0001 and score improvement threshold = 0.0005). A 1,000-replicate ML majority rule bootstrap tree (Felsenstein 1985) was generated using GARLI (with default settings except for the following: lnL increase for significantly better topology = 0.0001 and score improvement threshold = 0.0005). The nondefault GARLI settings significantly increased the length of the ML runs over those using the default settings but were implemented nevertheless because they yielded trees with higher log-likelihood scores. The topologies of the best BI and ML trees generated herein are consistent with previous estimates of freshwater mussel phylogeny (Hoeh et al. 2001; Roe and Hoeh 2003; Hoeh et al. 2009) and the nonmonophyly of the genus Lasmigona, as displayed in both the BI and the ML trees herein, has been previously noted (King et al. 1999).
The ML estimation of ancestral states, via Mesquite (Maddison WP and Maddison DR 2009), of breeding system (dioecy vs. hermaphroditism), orf type, and presence/absence of an M genome were identical because there is 100% concordance among the presence of dioecy, an F-orf type and an M genome. We optimized these characters on the Bayesian tree with maximum a posteriori probability tree (BI MAP), BI consensus tree and the best ML tree. In all three trees, the AsymmMK model of character evolution (Maddison 2006) was not significantly better than the Markov k-state 1 (MK1) parameter model (Lewis 2001), therefore the simpler MK1 model was preferred. All ancestral nodes on the three tree topologies (BI MAP and majority rule trees as well as the best ML tree) were significant for the presence of dioecy/F-orf /M genome except the last common ancestors to and within the four clades containing hermaphroditism/H-orf /no M genome, which were all significant for these character states. The proportional likelihoods (likelihoods of ancestral states scaled such that they add up to 1) for all the ancestral nodes on all three topologies were greater than 0.9, meaning that for each ancestral character state estimation, the probability that the majority state was correct was at least 10 times greater than that of the alternative state. Pairwise tests for correlated evolution (discrete) (Pagel 2000), as implemented in Mesquite, among breeding system type, F-genome orf type and presence/absence of an M genome were conducted on the three aforementioned topologies. The statistical significance of all test results was evaluated after applying the sequential Bonferroni test (Rice 1989).
Immunoelectron Microscopy
Venustaconcha ellipsiformis ovarian tissues were fixed in 1.0% glutaraldehyde and 4.0% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) for 4 h and washed three times for 10 min in buffer. Tissues were sectioned and prepared as in Chakrabarti et al. 2009. Tissue sections were incubated with the F-ORF primary antibody (1:100) in incubation buffer (10 mM phosphate buffer, 150 mM NaCl, pH 7.4 with 0.2% bovine serum albumin, and 15 mM NaN3) overnight at 4 °C. After six washes for 5 min in incubation buffer, grids were placed in gold solution (ultra small gold in goat antichicken Immunoglobulin Y serum, diluted 1:100 in incubation buffer, Electron Microscopy Sciences, Hatfield, PA) for 2 h. Six washes for 5 min in incubation buffer were followed by three 5 min phosphate buffer solution (PBS) washes. Sections were postfixed in 2.0% glutaraldehyde in PBS for 5 min, washed twice in PBS for 5 min and washed five times in dH2O for 2 min each. Silver enhancement (Electron Microscopy Sciences) was applied for 45 min followed by three 10 min washes in dH2O. Sections were poststained for 4 min in both uranyl acetate and lead citrate and viewed with a JEOL 1400 TEM (Transmission Electron Microscopy).
Results and Discussion
To evaluate the association of breeding system (dioecy vs. hermaphroditism [=combined sexes]) and type of mtDNA transmission system (DUI vs. SMI), we screened for the presence of M genomes in five hermaphroditic freshwater mussel species from two different taxonomic families (Unionidae and Margaritiferidae) whose closest relatives are dioecious species with DUI. Dioecy predominates in freshwater mussels with fewer than 3% of North American species being hermaphroditic (Hoeh et al. 1995). All PCR assays and subsequent sequencing analyses, using seven pairs of primers (including universal, freshwater mussel-specific, freshwater mussel-M genome-specific, and freshwater mussel genus-M genome-specific mtDNA primers), failed to produce M genome sequences from the five hermaphroditic species (table 1 and supplementary figs. S1 and S2, Supplementary Material online). In contrast, M genome sequences were always produced from their dioecious congeners (table 1 and supplementary figs. S1 and S2, Supplementary Material online). Our results thus indicate that hermaphroditic freshwater mussel species do not possess M genomes, that is, they lack DUI. Given the sensitivity and accuracy of PCR assays (Garrido-Ramos et al. 1998), consistent M-fragment amplification failures in all hermaphroditic species and successful amplifications in their dioecious close relatives strongly links dioecy to the presence of an M genome and hermaphroditism to the absence of M genomes in freshwater mussels.
To establish whether the loss of the M genome in hermaphroditic freshwater mussels is also accompanied by significant genic differences in their maternally inherited mt genomes when compared with the F genomes of dioecious species, we sequenced the complete mt genomes and/or the region comprising the newly discovered mtDNA-encoded protein gene F-orf from 40 freshwater mussel species (supplementary table S1, Supplementary Material online, and fig. 1). Remarkably, consistent breeding system–specific patterns of mtDNA macromutations were detected in the F-orf gene (supplementary table S1, Supplementary Material online and fig. 2), supporting the hypothesis that F genome sequences are also part of the mechanism specifying dioecy in freshwater bivalves. In addition to lacking an M genome, all five hermaphroditic species possess a highly divergent F-orf gene when compared with closely related dioecious species (supplementary table S1, Supplementary Material online and fig. 2). We refer to the divergent F-orf homologues in the hermaphroditic species as “H-orfs.” Based on our complete mt genomic sequences (fig. 1), the hypothesis that the hermaphrodites' F-like mt genomes possess a “normal” F-orf at a distinct locus can be rejected. Amino acid sequence comparisons of the F-ORF proteins in all the dioecious freshwater mussel species examined (N = 173 individuals from 35 species, 22 genera, and 3 families), representing >200 My of freshwater mussel evolution, reveal a single predicted transmembrane helix (TMH) in a conserved position, that is, in the N-terminus half of each F-ORF protein, followed by a relatively conserved 15 amino acid motif near the C-terminus, ([HY] [EPKNTS] [LVPI] [KNHVI] [NSKADP] S [SPK] [AG] [SHMEK] T [SDN] [VIL] [FCVNSTI] [DNQTK] [KDNTPV]). In stark contrast, the H-ORF proteins from the hermaphroditic species have the following characteristics: 1) they are typically longer than the F-ORF proteins from dioecious species, 2) they generally display surprisingly low levels of sequence similarity to F-ORF proteins from closely related dioecious species, 3) they typically contain repeat units not found in any of the F-ORF proteins from dioecious species, 4) they sometimes lack the predicted TMH (T. parvum) or the relatively conserved, near-C-terminus, 15 amino acid motif (U. imbecillis) seen in all F-ORF proteins characterized from dioecious species, and (v) they typically possess highly modified hydropathy profiles compared with those of F-ORF proteins in closely related, dioecious species (fig. 2, supplementary tables S1 and S2, Supplementary Material online). These distinctive features of the hermaphrodites' H-ORF proteins are consistent with a hypothesis of relaxed or diversifying selection and, at a minimum, indicate changes of function from that of the homologous F-ORF proteins in all dioecious, DUI containing freshwater mussel species.
FIG. 1.

Gene maps of the sex-associated and hermaphroditic “F-like” mt genomes of freshwater mussels. Gene identities: complex I in green; complex III in light blue; complex IV in blue; complex V in light purple; small and large ribosomal RNAs (in purple). Transfer RNAs (in gray) are depicted by one letter amino acid codes. F-ORF, F-specific open reading frame (orange); H-ORF, Hermaphroditic-specific open reading frame (pink); M-ORF, M-specific open reading frame (red). Genes positioned inside the circle are encoded on the light strand, and genes outside the circle are encoded on the heavy strand.
FIG. 2.
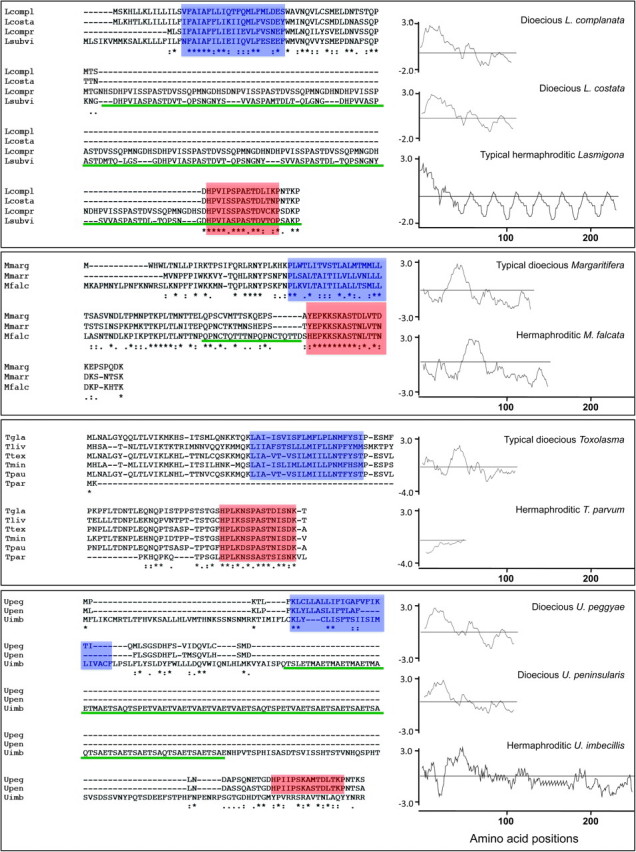
Primary and secondary structures of F-ORF and H-ORF amino acid sequences in dioecious and hermaphroditic freshwater mussel species, respectively. From above to below: Comparisons of dioecious Lasmigona spp. and hermaphroditic L. compressa and L. subviridis F- and H-ORFs. (Lcompl = L. complanata; Lcosta = L. costata; Lcompr = L. compressa; Lsubvi = L. subviridis). F-ORFs and H-ORF for Margaritifera margaritifera, M. marrianae and the hermaphroditic M. falcata. (Mmarg = M. margaritifera; Mmarr = M. marrianae; Mfalc = M. falcata). Comparisons of dioecious Toxolasma spp. and hermaphroditic T. parvum F-ORFs and H-ORF, respectively. (Tgla = T. glans; Tliv = T. lividus; Ttex = T. texasensis; Tmin = T. minor; Tpau = T. paulus; Tpar = T. parvum). F-ORFs and H-ORF for dioecious Utterbackia peggyae, U. peninsularis and hermaphroditic U. imbecillis. (Upeg = U. peggyae; Upen = U. peninsularis; Uimb = U. imbecillis). Alignment of amino acid sequences and secondary structures for the F- and H-ORFs are shown on the left. Color highlighting is as follows: blue box, the predicted transmembrane domain (TMH); reddish orange box, the relatively conserved, near-C-terminus, 15 amino acid motif; green, repetitive units. Hydropathy profiles are shown on the right. Numbers below profiles designate amino acid positions in each protein.
The rare hermaphroditic breeding system in five species, representing four distinct freshwater mussel genera, is always accompanied by the lack of an M genome, that is, all hermaphrodites display SMI, and the presence of an H-orf in their F-like mt genome. To test the hypothesis that this system demonstrates at least four independent transitions to the “hermaphroditic condition, SMI, and H-orf character states” from the “ancestral dioecious, DUI, and F-orf states” (Hoeh et al. 1995), estimates of phylogeny for the individuals examined herein were generated using ML and BI methods (BI MAP tree presented in fig. 3). Indeed, ML-based ancestral character state reconstructions on the best ML and BI MAP and majority rule trees all robustly indicate four independent dioecy-to-hermaphroditism, DUI-to-SMI and F-orf-to-H-orf transitions in the evolutionary history of these bivalves. As expected, because each of the four dioecy-to-hermaphroditism breeding system transitions was accompanied by the loss of the M genome and macromutational modifications to the F-orf gene, an ML-based analysis of character state correlation robustly rejects (P < 0.01) a model specifying the independent evolution of these three characters (i.e., pairwise tests for correlated evolution among breeding system type, F-genome orf type and presence/absence of an M genome all resulted in ∼zero probability (P < 0.01) that these three binary characters evolved independently). All test results remained significant after applying the sequential Bonferroni test (Rice 1989). Therefore, the absolute linkage of 1) hermaphroditic breeding system, 2) absence of M genome (=presence of SMI), and 3) highly modified F-orfs displayed in figure 3 strongly implicates both the F and M mt genomes as components of the sex determination system in freshwater mussels. Further, the fact that an F-ORF protein is always present with a set of M genome–encoded proteins and that every loss of the M genome is associated with macromutational modifications to the F-orf gene strongly argue for coordinated functional roles for the gene products of the F and M genomes, roles that must be concordantly modified to produce a hermaphroditic individual (Barker and Pagel 2005).
FIG. 3.
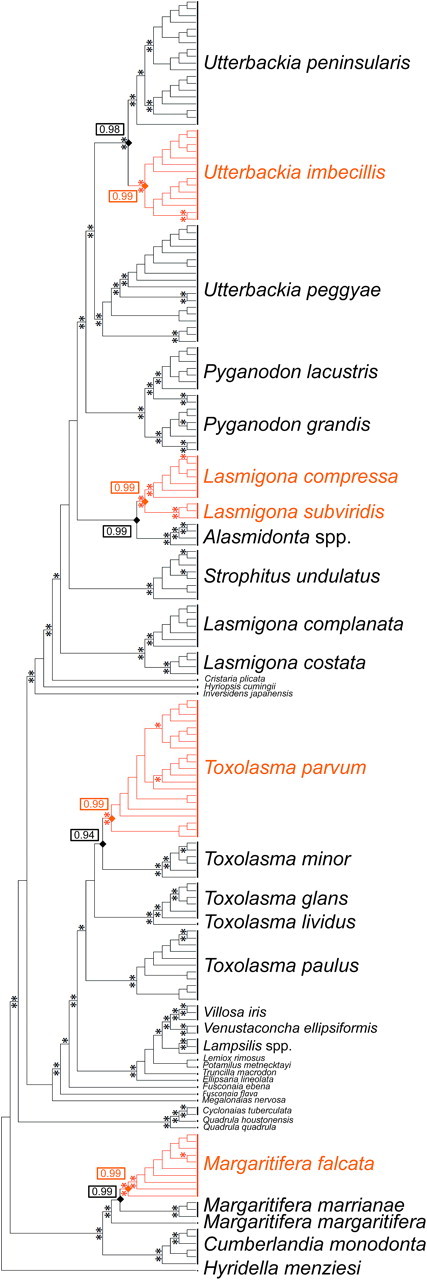
BI MAP tree from analysis of concatenated Fcox1/nd1 DNA sequences using Hyridella menziesi (Unionoida: Hyriidae) as the outgroup. An asterisk above a branch = BI posterior probability (×100) ≥ 95. An asterisk below a branch = ML BSP (1000×) ≥ 70. Black indicates lineages that are dioecious, contain an F-orf in the F mt genome and display DUI (=M mt genome present). Orange indicates lineages that are hermaphroditic, contain an H-orf in the F mt genome and display SMI (=M mt genome absent). Hermaphroditism, H-orfs and SMI concurrently evolved four distinct times. Proportional likelihood values (all nodes significant) are given in rectangles for the character state estimates at eight nodes (orange diamonds, character states = hermaphroditism, H-orf and M genome absent; black diamonds, character states = dioecy, F-orf, and M genome present, BSP = Bootstrap Support Percentages).
To date, the precise function(s) of the F-ORF and H-ORF proteins in freshwater mussels remain unclear. No significant amino acid sequence similarity with known proteins or no consistent results were detected using Blast tools. Interestingly, the estimated tertiary structure of the F-ORF protein from the dioecious freshwater mussel species V. ellipsiformis, indicated by an I-TASSER analysis (Zhang 2008), predicts involvement of this novel mt protein in DNA replication and/or DNA binding. Using immunoelectron microscopy to localize the F-ORF protein in mature ovarian eggs from V. ellipsiformis, we found that the F-ORF is present in mitochondria as expected but, more surprisingly, it is also present on the nuclear membrane and in the nucleoplasm (fig. 4). These extra-mt localizations represent additional evidence indicating nonoxidative phosphorylation functions for the F-ORF protein and support the hypothesis that this protein is involved in the genetic regulatory network specifying dioecy in freshwater mussels.
FIG. 4.

Immunoelectron microscopy (IEM) shows the presence of the F-ORF protein in the eggs of the dioecious species V. ellipsiformis. (A and C) Female-transmitted F-ORF is localized to the cytoplasm (mitochondria). (C) Female-transmitted F-ORF is localized to the nuclear membrane and nucleoplasm. (B and D) negative control IEM photomicrographs. (Note: black arrowheads in figure 4A indicate the presence of the F-ORF protein in mitochondria, small black arrow in figure 4B indicates a mitochondrion, small black arrow in figure 4D indicates the nuclear membrane). (E) Section of an egg showing egg plasma membrane (pl.m), vitelline matrix (v.m), and vitelline envelope (v.e).
Mitochondrial genes are known to play a role in sex determination in angiosperm plant groups exhibiting CMS (Hanson and Bentolila 2004; Burt and Trivers 2006; Chase 2007). These chimeric mt protein-coding genes disrupt pollen development and have been shown to contribute to reproductive isolation and speciation in plants (Hanson and Bentolila 2004; Chase 2007; Rieseberg and Blackman 2010). Unlike plants, mtDNA-based reproductive isolation appears to be much rarer in animals (but see Gershoni et al. 2009; Lane 2009). For example, mt genes have been implicated in reduced hybrid viability in Tigriopus copepods (Burton et al. 2006). One important pattern that emerges from these studies is that, in both plants and animals, reproductive isolation was related to rapid mtDNA divergence and/or important mt rearrangements (Hanson and Bentolila 2004; Burton et al. 2006; Chase 2007). Previous studies on the F- and M-ORF proteins (Breton et al. 2009) and the M-specific extension of the COX2 protein (Chapman et al. 2008) provided evidence for an extremely high amino acid substitution rate for these proteins in freshwater mussels. The hypervariability of the freshwater mussel F-ORF protein (and H-ORF protein) is confirmed by an amino acid sequence divergence analysis of all mtDNA-encoded protein genes (fig. 5). Our findings, in conjunction with previous studies (Chapman et al. 2008; Breton et al. 2009), clearly show that 1) the F-ORF and M genome-encoded proteins are among the fastest evolving proteins coded by freshwater mussel mt genomes and 2) mt genomic transitions are absolutely linked to the establishment of hermaphroditic species in freshwater mussels (fig. 3). These observations are consistent with the hypothesis that the F-ORF and M genome-encoded proteins are important factors in establishing reproductive isolation in freshwater mussels, as CMS proteins are in plants.
FIG. 5.

The extent of amino acid divergence (% amino acid difference) for each mt protein coding gene between U. imbecillis and U. peninsularis (Uimb/Upen), U. imbecillis and U. peggyae (Uimb/Upeg), U. peninsularis and U. peggyae (Upen/Upeg), and among species of the subfamilies Ambleminae (i.e., H. cumingii, L. ornata, Q. quadrula, T. parvum, V. ellipsiformis), and Anodontinae (i.e., C. plicata, L. compressa, Pygadodon grandis, U. imbecillis, U. peninsularis, U. peggyae).*F-ORF/H-ORF comparisons. (H-ORF protein sequences have been excluded from the analyses of Ambleminae and Anodontinae).
Interestingly, many intriguing parallels exist between DUI and CMS. Most obvious is that both DUI in freshwater mussels and CMS are associated with novel, mitochondrially encoded genes. As is generally the case for plant mt genomes (Chase 2007), bivalve mtDNAs display dramatic variation in size (15–32 kb), gene arrangement, and gene number (Gissi et al. 2008; Breton et al. 2007, 2009). Furthermore, mtDNA recombination, which is the mechanism responsible for the creation of chimeric genes associated with CMS (Chase 2007), has been confirmed in several bivalve species with DUI (Breton et al. 2007). It has been hypothesized that DUI first arose in a hermaphroditic bivalve lineage (Davison 2006; Breton et al. 2007). The results presented herein are compatible with that hypothesis. The dioecious breeding systems observed in both freshwater mussels and multiple angiosperm groups are likely derived conditions that were initiated by “gain-of-function macromutations” in mtDNA, that is, the acquisitions of 1) novel functioning mt ORFs and the M genome in an ancestral freshwater mussel lineage and 2) mt CMS genes, that is, novel functioning mt ORFs in angiosperms. The four freshwater mussel lineages that independently lost both the M genome and a normally functioning F-orf gene likely reverted to the ancestral hermaphroditic breeding system as do plant lineages that either lose the CMS specifying mt gene or gain a nuclear restorer locus (Chase 2007).
In principle, CMS can select for a reversal of the very thing from which it springs: maternal inheritance. Because mitochondria in pollen are less likely to carry a CMS gene than mitochondria in ovules, nuclear genes could be selected to retain paternally derived mitochondria over maternally derived ones. The result could be a degree of paternal “leakage” or even outright paternal inheritance (Burt and Trivers 2006; Pearl et al. 2009). A similar scenario could explain the origin of DUI in freshwater mussels: paternally transmitted mtDNAs were promoted because they were less likely to facilitate femaleness and could participate in the maintenance of ∼50:50 sex ratios in mussel populations (Passamonti and Ghiselli 2009). The relative stability of the DUI system in freshwater mussels for >200 My can be explained by 1) the F and M mt genomes' potential participation in the genetic regulatory network specifying dioecy and 2) because the mother-through-daughter and father-through-son mtDNA inheritance pattern provides a mechanism for lineage-specific containment of selfish mtDNA genes (Zouros et al. 1994). Given their reproductive tracts (Hoeh et al. 1995), it is likely difficult for hermaphroditic freshwater mussels to avoid self-fertilization thus mechanisms that promote dioecy could have been selected, over the long-term, because they limit inbreeding. Inbreeding depression is likely a causal factor underlying the rarity of hermaphroditic freshwater mussel species (fig. 3; Hoeh et al. 1995).
It is thus conceivable that DUI first emerged in an ancestral hermaphroditic species (Davison 2006, Breton et al. 2007), which tolerated any detrimental effects of biparental inheritance of mtDNA genomes (Hurst 1992; Hurst and Hoekstra 1994; Reboud and Zeyl 1994; Ballard and Whitlock 2004; Lane 2005). These harmful effects would likely be less “intense” in self-fertilizing species because organelles present in the sperm cell are the same as those in the female eggs, thus reducing potential conflict between mtDNAs of different origins. Consequently, under the hypothesis that only the nuclear genome would be responsible for mt inheritance, selection for such control should be relaxed and biparental inheritance should be more common in self-fertilizing species than in outcrossing species (Reboud and Zeyl 1994). However, if the egg- and sperm-transmitted mt genomes can play a role in their inheritance and modify the reproductive system (or sex allocation in the gonad) in their favor, then biparentally inherited mtDNAs are most likely to invade outcrossing populations (in self-fertilizing species, such mtDNAs would only replace other copies of themselves) (Reboud and Zeyl 1994). In principle, such a situation is inconsistent with both the selfish-conflict hypothesis, wherein the evolution of uniparental inheritance of mtDNA is postulated as the best means to eliminate the negative consequences of mt competition and within-individual intergenomic conflict (Hurst 1992; Ballard and Whitlock 2004; Lane 2005), and the hypothesis that natural selection favors uniparental inheritance because it enables the best match of nuclear and mt genomes for optimal mt function (Hurst 1992; Ballard and Whitlock 2004; Lane 2005). Theoretically, however, egg- and sperm-transmitted mt genomes that play a role in their inheritance could lead to the evolution of separate male and female sexes (e.g., Delph and Wolf 2005), and the strong link observed between the presence of female- and male-transmitted mtDNAs and the maintenance of dioecy in bivalve species with DUI is consistent with this scenario. The persistence and relative stability of DUI would be explained by the functional differentiation of the F and M mitochondria into two uniparental lineages to secure more reliable transmission and avoid the spread of selfish mt elements (Hurst and Hoekstra 1994; Zouros et al. 1994). Furthermore, given the extremely high levels of divergence between the proteins coded by the freshwater mussel F and M genomes (Doucet-Beaupré et al. 2010), we predict that distinct sets of nucleus-encoded, mt proteins exist to facilitate efficient oxidative phosphorylation.
The originality of our results is thus 3-fold. First, animal mt genomes are certainly less functionally limited than previously thought. A more generalized prediction that follows from this conclusion is that novel functional, potentially lineage-specific, mt ORFs are likely present in many animal species, including humans (Saccone et al. 1987; Maximov et al. 2002). Interestingly, there is increasing evidence from genomic and transcriptomic sequencing studies that “ORFs” or “novel genes of unknown function,” present in closely related species but for which homologues cannot be determined among more distantly related species, are often involved in important lineage-specific processes, such as sex determination and immune system functioning (Khalturin et al. 2009). Even very short mt ORFs should be thoroughly studied, and not automatically considered “noncoding,” because they could have functional significance. For example, the shortest functional ORFs described to date are 11-amino-acid-long peptides that have been shown to control gene expression and tissue folding in Drosophila (Galindo et al. 2007). Second, animal mt protein-coding genes can be exported outside mitochondria (e.g., to the nucleus) and be involved in extra-mt functions. More speculatively, animal mtDNA could be directly involved in sex determination, as for CMS in plants. Our results demonstrate a tight linkage between the presence of proteins coded by the highly divergent maternally and paternally transmitted mt genomes and the specification of dioecy in freshwater bivalves. With 20,000–30,000 living species (14 orders and ∼ 105 families; www.bivatol.org), bivalves constitute a highly diverse class within the Mollusca, which is the second largest animal phylum. Our current information regarding the taxonomic distribution of DUI is relatively rudimentary such that this mt inheritance/sex determination system association, with its unique and rapidly evolving genes that potentiate genetic divergence among populations, will eventually be shown to reside in a much larger number of bivalve species. Last, our results demonstrate that intraorganismal mt heteroplasmy has been and continues to be adaptive, over millions of years, in some animal species.
Conclusion
Most of the annotated animal mt genomes published to date contain intergenic regions that have been classified as “noncoding” (as were some F- and M-orf regions originally) because 1) all 37 typical mt genes were already annotated, 2) they do not contain ORFs or the ORFs are too short, and/or 3) the ORFs lack significant similarity to known proteins. Thus, the possibility of mt ORFs encoding peptides with key, nonoxidative phosphorylation functions has certainly been overlooked in animals. The potential role of freshwater mussel F and M mt genomes in specifying dioecy demonstrates that the genetic and functional repertoire of animal mtDNA is considerably greater than typically acknowledged. Our results are particularly significant because they 1) demonstrate the existence of novel functional, lineage-specific, mtDNA-encoded proteins in animal mt genomes, 2) display extra-mt localization and function for mtDNA-encoded proteins, and 3) give further credence to the hypothesis that the intraorganismal mtDNA heteroplasmy present in some species has deterministic underpinnings (Van Leeuwen et al. 2008; Pearl et al. 2009). In our opinion, novel mt ORFs, with key biological functions and great potential for involvement in the speciation process, await discovery in other animal species.
Supplementary Material
Supplementary figures S1 and S2 and tables SI and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank Andrea L. Case (Kent State University [KSU]) and Gertraud Burger (Université de Montréal) for helpful discussion and reviews of the manuscript and Arielle Zouela (KSU) and Shannon Rapovy (KSU) for their help in the lab. This research was supported by a National Science Foundation grant to W.R.H., a Natural Sciences and Engineering Research Council of Canada grant to D.T.S., a National Institutes of Health grant to H.P. (GM86782-01A1), and by research funds from the Friends of the North Carolina State Museum of Natural Sciences to A.E.B. S.B. was financially supported by a Natural Sciences and Engineering Research Council of Canada fellowship.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Mitsuke H, Ikeda M, Xia JX, Kikuchi T, Satake M, Shimizu T. ConPred II: a consensus prediction method for obtaining transmembrane topology models with high reliability. Nucleic Acids Res. 2004;32:W390–W393. doi: 10.1093/nar/gkh380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Barker D, Pagel M. Predicting functional gene links from phylogenetic-statistical analyses of whole genome. PLoS Comput Biol. 2005;1:e3. doi: 10.1371/journal.pcbi.0010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D, Karsch-Mizrachi I, Lipman D, Ostell J, Wheeler D. GenBank update. Nucleic Acids Res. 2004;32:D23–D26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky CW., Jr. The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu Rev Genet. 2001;35:125–148. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- Blier PU, Dufresne F, Burton RS. Natural selection and the evolution of mtDNA-encoded peptides: evidence for intergenomic co-adaptation. Trends Genet. 2001;17:400–406. doi: 10.1016/s0168-9525(01)02338-1. [DOI] [PubMed] [Google Scholar]
- Breton S, Doucet-Beaupré H, Stewart DT, Hoeh WR, Blier PU. The unusual system of doubly uniparental inheritance of mtDNA: isn't one enough? Trends Genet. 2007;23:465–474. doi: 10.1016/j.tig.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Breton S, Doucet-Beaupré H, Stewart DT, Piontkivska H, Karmakar M, Bogan AE, Blier PU, Hoeh WR. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: unionoida) with doubly uniparental inheritance of mtDNA: gender-specific Open Reading Frames (ORFs) and putative origins of replication. Genetics. 2009;183:1575–1589. doi: 10.1534/genetics.109.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Lang FB. Mitochondrial genomes: anything goes. Trends Genet. 2003;19:709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Burt A, Trivers R. Genes in conflict: the biology of selfish genetic elements. Cambridge (MA): Harvard University Press; 2006. [Google Scholar]
- Burton RS, Ellison CK, Harrison JS. The sorry state of F2 hybrids: consequences of rapid mitochondrial DNA evolution in allopatric populations. Am Nat. 2006;168:S14–S24. doi: 10.1086/509046. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, Shepardson S, Karmakar M, Trdan R, Walker J, Shandilya R, Stewart DT, Vijayaraghavan S, Hoeh WR. Extra-mitochondrial localization and likely reproductive function of a female-transmitted cytochrome c oxidase subunit II protein. Dev Growth Differ. 2009;51:511–519. doi: 10.1111/j.1440-169X.2009.01113.x. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, Walker JM, Chapman EG, Shepardson SP, Trdan RJ, Curole JP, Watters GT, Stewart DT, Vijayaraghavan S, Hoeh WR. Reproductive function for a C-terminus extended, male-transmitted cytochrome c oxidase subunit II protein expressed in both spermatozoa and eggs. FEBS Lett. 2007;581:5213–5219. doi: 10.1016/j.febslet.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman EG, Piontkivska H, Walker JM, Stewart DT, Curole JP, Hoeh WR. Extreme primary and secondary protein structure variability in the chimeric male-transmitted cytochrome c oxidase subunit II protein in freshwater mussels: evidence for an elevated amino acid substitution rate in the face of domain-specific purifying selection. BMC Evol Biol. 2008;8:165. doi: 10.1186/1471-2148-8-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase CD. Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007;23:81–90. doi: 10.1016/j.tig.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Curole JP. Universal primers for the specific amplification of the male mitotype of the Unionoidea (Bivalvia) Cons Genet. 2004;5:733–735. [Google Scholar]
- Dalziel AC, Stewart DT. Tissue-specific expression of male-transmitted mitochondrial DNA and its implications for rates of molecular evolution in Mytilus mussels (Bivalvia: mytilidae) Genome. 2002;45:348–355. doi: 10.1139/g01-159. [DOI] [PubMed] [Google Scholar]
- Davison A. The ovotestis: an underdeveloped organ of evolution. Bioessays. 2006;28:642–650. doi: 10.1002/bies.20424. [DOI] [PubMed] [Google Scholar]
- Debise R, Touraille S, Durand R, Alziari S. Biochemical consequences of a large deletion in the mitochondrial genome of a Drosophila subobscura strain. Biochem Biophys Res Commun. 1993;196:355–362. doi: 10.1006/bbrc.1993.2256. [DOI] [PubMed] [Google Scholar]
- Dellaporta SL, Xu A, Sagasser S, Jakob W, Moreno MA, Buss LW, Schierwater B. Mitochondrial genome of Trichoplax adhaerens supports Placozoa as the basal lower metazoan phylum. Proc Natl Acad Sci U S A. 2006;103:8751–8756. doi: 10.1073/pnas.0602076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delph LF, Wolf DE. Evolutionary consequences of gender plasticity in genetically dimorphic breeding systems. New Phytol. 2005;166:119–128. doi: 10.1111/j.1469-8137.2005.01339.x. [DOI] [PubMed] [Google Scholar]
- Doublet V, Souty-Grosset C, Bouchon D, Cordaux R, Marcadé I. A thirty million year-old inherited heteroplasmy. PLoS One. 2008;3:e2938. doi: 10.1371/journal.pone.0002938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet-Beaupré H, Breton S, Chapman EG, Blier PU, Bogan AE, Stewart DT, Hoeh WR. Mitochondrial phylogenomics of the Bivalvia (Mollusca): searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol Biol. 2010;10:50. doi: 10.1186/1471-2148-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Fickett JW. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 1982;10:5303–5318. doi: 10.1093/nar/10.17.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer O, Black M, Hoeh WR, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:1052–1062. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Ramos MA, Stewart DT, Sutherland DW, Zouros E. The distribution of male-transmitted and female-transmitted mitochondrial DNA types in somatic tissues of blue mussels: implications for the operation of doubly uniparental inheritance of mitochondrial DNA. Genome. 1998;41:818–824. [Google Scholar]
- Gershoni M, Templeton AR, Mishmar D. Mitochondrial bioenergetics as a major motive force of speciation. Bioessays. 2009;31:642–650. doi: 10.1002/bies.200800139. [DOI] [PubMed] [Google Scholar]
- Gingrich JR, Pelkey KA, Fam SR, Huang Y, Petralia RS, Wenthold RJ, Salter MW. Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proc Natl Acad Sci U S A. 2004;101:6237–6242. doi: 10.1073/pnas.0401413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:1–20. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hanson MR, Bentolila S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell. 2004;16:S154–S169. doi: 10.1105/tpc.015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeh WR, Blakley KH, Brown WM. Heteroplasmy suggests limited biparental inheritance of Mytilus mitochondrial DNA. Science. 1991;251:1488–1490. doi: 10.1126/science.1672472. [DOI] [PubMed] [Google Scholar]
- Hoeh WR, Bogan AE, Heard WH. A phylogenetic perspective on the evolution of morphological and reproductive characteristics in the Unionoida. In: Bauer G, Wächlter K, editors. Ecology and evolution of the freshwater mussels Unionoida. Berlin: Springer-Verlag; 2001. pp. 257–280. [Google Scholar]
- Hoeh WR, Bogan AE, Heard WH, Chapman EG. Palaeoheterodont phylogeny, character evolution, diversity and phylogenetic classification: a reflection on methods of analysis. Malacologia. 2009;51:307–317. [Google Scholar]
- Hoeh WR, Frazer KS, Naranjo-Garcia E, Trdan RJ. A phylogenetic perspective on the evolution of simultaneous hermaphroditism in a freshwater mussel clade (Bivalvia: unionidae: utterbackia) Malacol Rev. 1995;28:43–60. [Google Scholar]
- Hoeh WR, Stewart DT, Guttman SI. High fidelity of mitochondrial genome transmission under the doubly uniparental mode of inheritance in freshwater mussels (Bivalvia: unionoidea) Evolution. 2002;56:2252–22561. doi: 10.1111/j.0014-3820.2002.tb00149.x. [DOI] [PubMed] [Google Scholar]
- Hurst LD. Intragenomic conflict as an evolutionary force. Proc R Soc Lond B. 1992;248:135–140. [Google Scholar]
- Hurst LD, Hoekstra RF. Shellfish genes kept in line. Nature. 1994;368:811–812. doi: 10.1038/368811a0. [DOI] [PubMed] [Google Scholar]
- Kenchington EL, Hamilton L, Cogswell A, Zouros E. Paternal mtDNA and maleness are co-inherited but not causally linked in mytilid mussels. PLoS One. 2009;4:e6976. doi: 10.1371/journal.pone.0006976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TCG. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 2009;25:404–413. doi: 10.1016/j.tig.2009.07.006. [DOI] [PubMed] [Google Scholar]
- King TL, Eackles MS, Gjetvaj B, Hoeh WR. Intraspecific phylogeography of Lasmigona subviridis (Bivalvia: unionidae): conservation implications of range discontinuity. Mol Ecol. 1999;8:S65–S78. doi: 10.1046/j.1365-294x.1999.00784.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA 3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lane N. Power, sex, suicide. Mitochondria and the meaning of life. New York: Oxford University Press; 2005. [Google Scholar]
- Lane N. On the origin of bar codes. Nature. 2009;462:272–274. doi: 10.1038/462272a. [DOI] [PubMed] [Google Scholar]
- Lewis PO. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol. 2001;50:913–925. doi: 10.1080/106351501753462876. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP. Confounding asymmetries in evolutionary diversification and character change. Evolution. 2006;60:1743–1746. [PubMed] [Google Scholar]
- Maddison WP, Maddison DR. Mesquite: a modular system for evolutionary analysis. 2009. Version 2.7 [cited 2009 Aug 27]. Available from: http://mesquiteproject.org. [Google Scholar]
- Marshall DC, Simon C, Buckley TR. Accurate branch length estimation in partitioned Bayesian analyses requires accommodation of among-partition rate variation and attention to branch length priors. Syst Biol. 2006;55:993–1003. doi: 10.1080/10635150601087641. [DOI] [PubMed] [Google Scholar]
- Maximov V, Martynenko A, Hunsmann G, Tarantul V. Mitochondrial 16S rRNA gene encodes a functional peptide, a potential drug for Alzheimer's disease and target for cancer therapy. Med Hypoth. 2002;59:670–673. doi: 10.1016/s0306-9877(02)00223-2. [DOI] [PubMed] [Google Scholar]
- Pagel M. Discrete. 2000. Version 4.0. [Google Scholar]
- Palumbi SR. Nucleic acids II: the polymerase chain reaction. In: Hillis DM, Moritz C, Mable BK, editors. Molecular systematics. Sunderland (MA): Sinauer Associates; 1996. pp. 205–247. [Google Scholar]
- Passamonti M, Ghiselli F. Doubly uniparental inheritance: two mitochondrial genomes, on precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 2009;28:79–89. doi: 10.1089/dna.2008.0807. [DOI] [PubMed] [Google Scholar]
- Pearl SA, Welch ME, McCauley DE. Mitochondrial heteroplasmy and paternal leakage in natural populations of Silene vulgaris, a gynodioecious plant. Mol Biol Evol. 2009;26:537–545. doi: 10.1093/molbev/msn273. [DOI] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Reboud X, Zeyl C. Organelle inheritance in plants. Heredity. 1994;72:132–140. [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Blackman BK. Speciation genes in plants. Ann Bot. 2010;106:439–455. doi: 10.1093/aob/mcq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F, Oliver JL, Marin A, Medina JR. The general stochastic-model of nucleotide substitution. J Theor Biol. 1990;142:485–501. doi: 10.1016/s0022-5193(05)80104-3. [DOI] [PubMed] [Google Scholar]
- Roe KJ, Hoeh WR. Systematics of freshwater mussels (Bivalvia: unionoida) In: Lydeard C, Lindberg DR, editors. Molecular systematics and phylogeography of mollusks. Washington (DC): Smithsonian Books; 2003. pp. 91–122. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rost B, Yachdav G, Liu J. The predictprotein server. Nucleic Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze D, Rousset F, Michalakis Y. Germline bottlenecks, biparental inheritance and selection on mitochondrial variants: a two-level selection model. Genetics. 2005;170:1385–1399. doi: 10.1534/genetics.104.039495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra C, Reyero M-I, Zouros E. Male-dependent doubly uniparental inheritance of mitochondrial DNA and female-dependent sex-ratio in the mussel Mytilus galloprovincialis. Genetics. 1997;145:1073–1082. doi: 10.1093/genetics/145.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone C, Attimonelli M, Sbisà E. Structural elements highly preserved during the evolution of the D-loop-containing region in vertebrate mitochondrial DNA. J Mol Evol. 1987;26:205–211. doi: 10.1007/BF02099853. [DOI] [PubMed] [Google Scholar]
- Scheffler IE. Hoboken (NJ): Wiley-Liss; 2008. Mitochondria. [Google Scholar]
- Serb JM, Lydeard C. Complete mtDNA sequence of the North American freshwater mussel, Lampsilis ornata (Unionidae): an examination of the evolution and phylogenetic utility of mitochondrial genome organization in Bivalvia (Mollusca) Mol Biol Evol. 2003;20:1854–1866. doi: 10.1093/molbev/msg218. [DOI] [PubMed] [Google Scholar]
- Skibinski DOF, Gallagher C, Beynon CM. Sex-limited mitochondrial DNA transmission in the marine mussel Mytilus edulis. Genetics. 1994;138:801–809. doi: 10.1093/genetics/138.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theologidis I, Fodelianakis S, Gaspar MB, Zouros E. Doubly uniparental inheritance (DUI) of mitochondrial DNA in Donax trunculus (Bivalvia: donacidae) and the problem of its sporadic detection in Bivalvia. Evolution. 2008;62:959–970. doi: 10.1111/j.1558-5646.2008.00329.x. [DOI] [PubMed] [Google Scholar]
- Theologidis I, Saavedra C, Zouros E. No evidence for absence of paternal mtDNA in male progeny from pair matings of the mussel Mytilus galloprovincialis. Genetics. 2007;176:1367–1369. doi: 10.1534/genetics.106.069930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen T, Vanholme B, Van Pottelberge S, Van Nieuwenhuyse P, Nauen R, Tirry L, Denholm I. Mitochondrial heteroplasmy and the evolution of insecticide resistance: non-mendelian inheritance in action. Proc Natl Acad Sci U S A. 2008;105:5980–5985. doi: 10.1073/pnas.0802224105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetis C, Theologidis I, Zouros E, Rodakis GC. No evidence for presence of maternal mitochondrial DNA in the sperm of Mytilus galloprovincialis males. Proc R Soc Lond B. 2006;273:2483–2489. doi: 10.1098/rspb.2006.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Bogan AE, Bonfiglio EA, Campbell DC, Christian AD, Curole JP, Harris JL, Wojtecki RJ, Hoeh WR. Primers for amplifying the hypervariable, male-transmitted COII-COI junction region in amblemine freshwater mussels (Bivalvia: unionoidea: ambleminae) Mol Ecol Notes. 2007;7:489–491. [Google Scholar]
- Walker JM, Curole JP, Wade DE, Chapman EG, Bogan AE, Watters GT, Hoeh WR. Taxonomic distribution and phylogenetic utility of gender-associated mitochondrial genomes in the Unionoida (Bivalvia) Malacologia. 2006;48:265–284. [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DJ, Wolff JN, Pierson M, Gemmell NJ. Revealing the hidden complexities of mtDNA inheritance. Mol Ecol. 2008;17:4925–4942. doi: 10.1111/j.1365-294X.2008.03982.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E. The exceptional mitochondrial DNA system of the mussel family Mytilidae. Genes Genet Syst. 2000;75:313–318. doi: 10.1266/ggs.75.313. [DOI] [PubMed] [Google Scholar]
- Zouros E, Ball AO, Saavedra C, Freeman KR. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci U S A. 1994;91:7463–7467. doi: 10.1073/pnas.91.16.7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl DJ. [Austin (TX)]: The University of Texas at Austin; 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. [PhD dissertation] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.