

Abstract
Objective
To investigate the molecular mechanism for biased interleukin (IL-17) production by DBA/1 CD4+ T cells upon T cell receptor (TCR) and transforming growth factor (TGF)-β stimulation.
Methods
Purified naïve CD4+ T cells were stimulated with anti-CD3/CD28 under Th1, Th2, Th17 and inducible T regulatory (iTreg) conditions. Cytokine production was assayed by intracellular staining and ELISA. Expression of transcription factors were determined by RT-PCR, flow cytometry and immunoblotting.
Results
Naïve CD4+ T cells from DBA/1 mice produced more IL-17 under Th17 conditions compared to C57BL/6 or BALB/c mice. Further investigation revealed no difference amongst strains in CD4+ T cell survival, upstream TCR signaling and CD69 expression; phosphorylation of signal transducers and activators of transcription (STAT)-3 and expression of suppressors of cytokine signaling (SOCS)-3 that positively or negatively regulate IL-17 production. However, DBA/1 CD4+ T cells expressed increased levels of retinoic acid receptor related orphan nuclear receptor (ROR)-γt. Furthermore, under iTreg polarizing condition, DBA/1 CD4+ T cells showed a strikingly reduced Foxp3 expression. When interferon (IFN)-γ and IL-4 were blocked, Foxp3 expression increased but remained lower in DBA/1 CD4+ T cells following exposure to TGF-β compared to B6 CD4+ T cells. Moreover, DBA/1 CD4+ T cells showed reduced phosphorylation of Smad2 and 3 in both Th17 and iTreg conditions.
Conclusion
These results indicate that naïve DBA/1 CD4+ T cells have a dichotomous response to TGF-β: enhanced RORγt yet reduced Foxp3 up-regulation. This observation may help elucidate the branch point of TGF-β signaling leading to skewed Th17, but reduced iTreg differentiation.
Keywords: Rodent, T cells, Rheumatoid Arthritis, Cytokines, Cell Differentiation
Introduction
Collagen-induced arthritis (CIA) is an animal model for human rheumatoid arthritis (RA). Mouse CIA is elicited by immunization with collagen-type II (CII) in complete Freund's adjuvant (CFA) in susceptible strains. Among the inbred strains, DBA/1 mice are highly susceptible to CIA while other strains are relatively resistant. The unusual susceptibility of DBA/1 mice has been largely attributed to the major histocompatibility (MHC) haplotype, H-2q (1). However, recent studies have demonstrated the importance of T cell cytokines in regulating the disease susceptibility to CIA and disease severity (2-6). For example, the normally resistant MHC H-2b bearing C57BL/6 (B6) mice become highly susceptible to CIA when interferon (IFN)-γ or IFN-γ receptor gene is deleted in transgenic mice (2, 3, 6). DBA/1 mice lacking IFN-γR have early onset of disease and arthritis is more severe (4, 5). Defects in IFN-γ signaling leads to an uncontrolled production of IL-17 (7, 8). IL-17 is a pro-inflammatory cytokine and is involved in many aspects of the pathogenesis from synovial inflammation to bone resorption in RA (reviewed in (9)). The critical role of IL-17 or Th17 has been well elucidated in CIA and several other mouse models of inflammatory arthritis (8, 10-12).
We previously showed that one mechanism responsible for DBA/1 susceptibility is a relatively high IL-17 response following immunization with CII in CFA (7). The goal of this study was to identify the mechanisms responsible for the differential cytokine production by CD4+ T cells in DBA/1 mice. It has been demonstrated that transforming growth factor (TGF)-β signaling helps to determine the naïve CD4+ T cell fate upon TCR activation (13-15). Naïve CD4+ T cells may differentiate into Th17 or inducible T regulatory (iTreg) cells under the influence of TGF-β depending on the presence or absence of proinflammatory cytokines (15). In the presence of IL-6, TGF-β induces RORβt expression and directs naïve CD4+ T cells to differentiate towards Th17 cells. On the other hand, in the absence of IL-6, TGF-β may convert Foxp3 negative naïve CD4+ T cells into Foxp3 positive iTreg cells (15). In this study we demonstrate that naïve DBA/1 CD4+ T cells have a dichotomous response to TGF- β signaling which results in the biased development of Th17 cells and yet defective development of iTreg cells. These results suggest an intrinsic deviation in naïve CD4+ T cells of DBA/1 mice to become the inflammatory Th17 cells caused by an alteration in TGF-β signal transduction.
Materials and Methods
Mice
C57BL/6, DBA/1 and BALB/c mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and kept in a modified specific pathogen-free facility at University of Washington and Portland VA Medical Center. Mice were used at age 4-12 weeks under a protocol approved by the Institutional Animal Care and Use Committee of University of Washington and Portland VA Medical Center.
Cytokines, antibodies to cytokines and ELISA for cytokines
Recombinant cytokines and antibodies to cytokines for ELISA and in cell culture were obtained from the following sources: Recombinant mouse IL-17, purified rat anti-mouse IL-17, biotinylated goat anti-mouse IL-17 and recombinant TGF-β (R&D Systems, Minneapolis, MN); biotinylated anti-mouse IL-4, IL-6 and IL-12/IL-23(p40) (BioLegend, San Diego, CA); purified anti-IL-6 and recombinant IFN-γ (BD PharMingen, San Diego, CA); biotinylated anti-mouse IFN-γ (Caltag, South San Francisco, CA); anti-mouse IL-23(p19), purified anti-mouse IFN-γ, IL-4, IL-6, IL-12 and IL-23 (eBioscience, San Diego, CA); and anti-mouse IL-4 (National Cancer Institute Biological Resources Branch Preclinical Repository, Rockville, MD). Cytokines in supernatants or serum were quantified by ELISA according to the manufacturer's instruction.
Isolation of naive CD4+ T cells and culture condition
Naïve CD4+ T cells were purified from spleens of C57BL/6, DBA/1 or BALB/c mice by using the CD4+ CD62L+ T cell Isolation Kit II (Miltenyi Biotec, Bergisch-Gladbach, Germany). Cells were cultured in RPMI medium 1640 with 10% FCS, 100 μg/ml streptomycin and 100 units/mL penicillin G at 37°C and in 5% CO2. Naïve CD4+ T cells with >96% purity in all strains of mice were activated by plate-bound anti-mouse CD3e (5 μg/ml, clone 500A2) and soluble anti-mouse CD28 (0.5 μg/ml, clone 37.51) for indicated time (BD PharMingen, San Diego, CA) and cultured under Th1 (IL-12 20 ng/ml, anti-IL-4 5 μg/ml), Th2 (IL-4 20 ng/ml, anti-IFN-γ 5 μg/ml), Th17 (IL-6 20 ng/ml, TGF-β 10 ng/ml, IL-23 20 ng/ml, anti-IFN-γ 5 μg/ml, anti-IL-4 5 μg/ml) or iTreg (IL-2 20 ng/ml, TGF-β 20 ng/ml, anti-IL-6 10 μg/ml) polarizing conditions.
RT-PCR
Total RNA was extracted by TRIzol Reagent (Invitrogen, Carlsbad, CA) and assayed by real time Q-PCR. cDNA was synthesized with First Strand cDNA Synthesis Kit (Fermentas, Glen Burnie, MD) by using oligo(dT)18 primer as according to the manufacturer's instruction. The specific primers were as fellows: Socs3, sense 5′-gagatttcgcttcgggacta-3′ and antisense 5′-aacttgctgtgggtgaccat-3′; Smad6, sense 5′-tgttcaggtctaaacgttcg-3′ and antisense 5′-tacagcgagtacgtgaccgt-3′; Smad7 sense 5′-ctcctgctgtgcaaagtgttc-3′ and antisense 5′-caggctccagaagaagttgg-3′; RORγt sense 5′-gacagggagccaagttctca-3′ and antisense 5′-gtgcaggagtaggccacatt-3′; GAPDH sense 5′-cttcttgtgcagtgccagcc-3′ and antisense 5′-gtgccgttgaatttgccgtg-3′. Reaction conditions consisted of a 30s denaturation step at 95°C, a 60s annealing step at 60°C and a 30s extension step at 72°Cfor 40 cycles.
Flow cytometry
Cell surface markers were stained on freshly isolated lymphocytes with cocktails of antibodies to CD4, CD62L and CD44. Intracellular cytokine staining was performed using an intracellular staining kit (BD PharMingen). Purified naïve CD4+ T cells form normal mice or from CII-immunized mice were stimulated with phorbol myristate acetate (PMA) (50 ng/ml) and ionomycin (500 ng/ml) in the presence of GolgiStop solution (BD PharMingen) for 5 h and stained with allophycocyanin-conjugated anti-IFN-γ (clone XMG1.2; BioLegend), allophycocyanin-conjugated anti-IL-4 (clone 11B11; BioLegend), allophycocyanin-conjugated anti-CD69 (clone H1.2F3; BioLegend), phycoerythrin-conjugated anti-TGF-β (R&D systems) and phycoerythrin-conjugated anti-IL-17 (clone TC11-18H10.1; BD PharMingen) and analyzed on a FACSCanto (BD Biosciences, San Jose, CA). Foxp3 staining was performed using a Foxp3 staining kit (eBioscience) and allophycocyanin or fluoresceinisothiocyanate-conjugated anti-Foxp3 (clone FJK-16s; eBioscience). The analysis of phosphoorylated STAT3 was performed using 2% paraformaldehyde and Perm Buffer III (BD Bioscience), Alexa Fluor 488 labeled anti-STAT3 (pY705) (BD Bioscience).
Immunoblotting
Purified naïve CD4+ T cells were stimulated with plate-bound anti-mouse CD3e (5 μg/ml), soluble anti-mouse CD28 (0.5 μg/ml) and Th17 polarizing condition (IL-6 20 ng/ml, TGF-β 10 ng/ml, IL-23 20 ng/ml, anti-IFN-γ 5 μg/ml, anti-IL-4 5 μg/ml) for indicated time. Stimulation was terminated by washing in iced-cold PBS and lysing in detergent buffer containing protease inhibitors and sodium orthovanadate. Cell lysates were electrophoresed, transferred onto a nitrocellulose filter, and immunoblotted with antibodies against anti-phospho-Smad2 (Ser465/467) and anti-phospho-Smad3 (Ser423/425) (Cell Signaling).
Statistical analysis
All experiments were performed 2-5 times. Results were analyzed with Student's t-test.
Results
Heightened IL-17 production in DBA/1 mice is not restricted to CII
To determine whether biased IL-17 production in DBA/1 mice is restricted to CII, the major collagen type in articular cartilage, we immunized DBA/1, B6 and BALB/c mice intradermaly with 100 μg of bovine CII, or KLH or OVA emulsified in CFA. IFN-γ and IL-17 production by lymphocytes from draining lymph nodes (LN) and spleen were analyzed ex vivo. Compared to B6 and BALB/c, DBA/1 mice had a lower proportion of CD4+ T cells producing IFN-γ when analyzed ex vivo in all three Ag immunized groups. When in vivo primed lymphocytes were re-stimulated with Ag in vitro, DBA/1 mice showed substantially decreased IFN-γ but heightened IL-17 production to all three Ag compared to B6 and BALB/c mice (Supplementary Figure 1). This suggests that DBA/1 CD4+ T cells have a greater tendency to skew CD4+ Th cells to Th17 rather than Th1 following antigen stimulation.
Naïve CD4+ T cells from DBA/1 mice produce more IL-17 upon in vitro TCR activation
To test the possibility that DBA/1 T cells skew to Th17, we next purified naïve CD4+ T and stimulated them with anti-CD3/CD28 under Th17 polarizing conditions in vitro. The proportion of IL-17-producing CD4+ T cells in DBA/1 mice was 2-3 fold higher than that in B6 or BALB/c mice at each time point with the peak of intracellular IL-17 production at day 3 (Figure 1A). Consistent with this finding, IL-17 concentrations in DBA/1 culture supernatants were higher than those from B6 mice at all time points tested (Figure 1B). We next compared IFN-γ and IL-4 production between DBA/1 and B6 mice under Th1 and Th2 polarizing condition respectively. Surprisingly, CD4+ T cells from DBA/1 mice produced significantly more IFN-γ as well as IL-4 compared to B6 mice (Figure 1C). We further compared production of these cytokines by DBA/1 CD4+ T cells to BALB/c CD4+ T cells. DBA/1 CD4+ T cells also produced more IL-17, IFN-γ and IL-4 under these Th polarizing conditions compared to CD4+ T cells obtained from BALB/c mice.
Figure 1. Increased IL-17 production by DBA/1 Th17 cells.
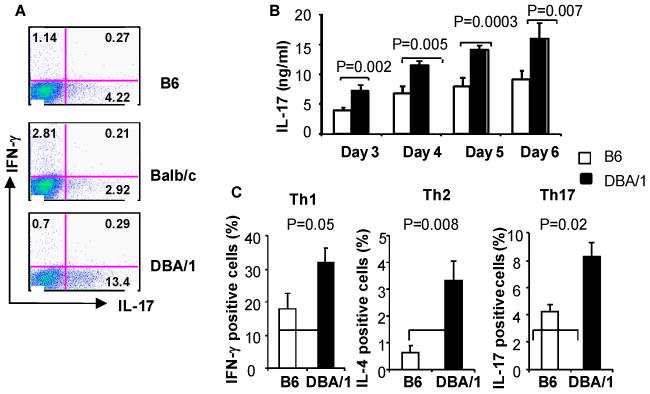
naïve CD4+ T cells were purified from B6, BALB/c and DBA/1 spleens. The cells were stimulated with 5 μg/ml of plate-bound anti-CD3 and 0.5 μg/ml of soluble anti-CD28 under Th1, Th2 or Th17 polarizing conditions as described in Methods. After 3 days in culture, the cells were stimulated with Phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) and ionomycin (500 ng/ml) and GolgiStop for 5 hours and intercellular cytokine expression determined by flow cytometry (A, C). Cytokine levels in supernatant were determined by ELISA (B). A: representative flow cytometry profile of 3 experiments. B, C: pooled data of three experiments (n=6 in each group).
DBA/1 Th17 T cells showed equivalent proliferation and cell death compared to B6
To determine whether the increased cytokine production by DBA/1 CD4+ T cells was the result of greater proliferation or reduced cell death, or both, we stimulated naïve CD4+ T cells under Th1, Th2 or Th17 polarizing conditions for 3 days, and counted total cell numbers (dead or dying cell numbers and live cell numbers) with trypan blue exclusion and with propidium iodide (PI) and Annexin V staining. The total numbers of live cells from DBA/1 were greater than those from B6 under Th1 and Th2 polarizing conditions (Figure 2A). However, the total number of live cells under Th17 polarizing condition was similar in DBA/1 and B6 mice (Figure 2A). Next, we examined cell proliferation using carboxyfluorecein diacetate succinimidyl ester (CFSE) dilution assay. Under Th1 and Th2, but not Th17 polarizing conditions, DBA/1 CD4+ T cells showed a modest increase of proliferation compared to B6 (Figure 2B). Increased cell proliferation and survival of B6 CD4+ T cells in Th1 and Th2 conditions may explain the increased production of IFN-γ and IL-4. However, under Th17 polarizing conditions, CD4+ T cell proliferation and survival were similar between these two strains suggesting that the increased IL-17 production by DBA/1 CD4+ T cells was not the result of increased proliferation (Figure 2A and B).
Figure 2. Survival and proliferation of CD4+ T cells under Th1, Th2 and Th17 polarizing conditions.

(A) Purified naïve CD4+ T cells were isolated and stimulated as in Figure 1. After 3 days incubation, live and dead CD4+ T cells were counted by trypan blue staining (pooled data of 3 experiments, n = 4 in each group). (B) Naïve CD4+ T cells were labeled with carboxyfluorecein diacetate succinimidyl ester (CFSE) and stimulated for 3 days (representative flow cytometry profile from one of two independent experiments, n=3 in each group).
Upstream signal transduction and STAT3 phosphorylation are similar between DBA/1 and B6 mice
To examine whether CD3 and CD28 signal transduction or PMA/ionomycin responses in DBA/1 CD4+ T cells were exaggerated, we investigated intracellular CD69 expression following ant-CD3 and CD28 signal or PMA and Ionomycin stimulation. CD69 expression is an early activation marker of T cell activation after influx of extracellular calcium (16). Three hours following PMA/Ionomycin stimulation, intracellular CD69 expression reached a peak in about 90% of CD4+ T cells from both DBA/1 and B6 mice (Figure 3A, B). On the other hand, following CD3 and CD28 stimulation, intracellular CD69 expression in DBA/1 CD4+ T cells was slightly lower than that observed in B6 cells at 3 and 5hr (Figure 3B). Direct quantification of calcium flux revealed no differences following TCR ligation (Supplementary Figure 2). Thus, upstream signaling of CD4+ naïve T cells in DBA/1 and B6 mice appeared equivalent. We also examined the base line expression of activation markers, CD44 and CD69 in freshly isolated naïve CD4+ T cells and found no difference between B6 and DBA/1 mice (Figure 3B). Similar experiments were performed from younger mice (4 and 5 weeks old) and expression of CD44 and CD69 by naïve CD4+ T cells was similar in both B6 and DBA/1 mice (Figure 3C).
Figure 3. CD69 expression in DBA/1 and B6 CD4+ T cells.

(A, B) Purified naive CD4+ T cells were stimulated with anti-CD3/anti-CD28 as above and then exposed to GolgiStop or PMA/ionomycin and GolgiStop for 3 or 5 hours respectively. Intracellular CD69 was quantified by flow cytometry (representative of three independent experiments, n=3 in each group). (C) Expression of CD69 in naïve CD4+ T cells. Lymphocytes from 4 week old B6 and DBA/1 mice were analyzed for CD69 expression in CD44lo and CD62Lhi population (pooled data of two experiments, n = 5 in each group).
Since IL-6 induced STAT3 phosphorylation plays an important role in Th17 differentiation (17), we compared total and phosphorylated STAT3 (pSTAT3) levels in naïve CD4 T cells under Th17 polarizing condition between DBA/1 and B6. Expression of total and phosphorylated STAT3 was similar between B6 and DBA/1 (supplementary Figure 3A and B). Since IL-23 also activates STAT3 (18), we examined STAT3 phosphorylation (pSTAT3) levels under Th17 polarizing condition with and without addition of IL-23 in the culture. However, addition of IL-23 in the culture did not affect the pSTAT3 expression in either B6 or DBA/1 CD4+ T cells. Since suppressors of cytokine signaling (SOCS3) suppresses STAT3 function, we, therefore, examined SOCS3 expression. The level of SOCS3 mRNA expressed by activated DBA/1 CD4+ T cells was similar to that in B6 (supplementary Figure 4). Autocrine IFN-γ is also known to be able to inhibit pSTAT3 via induction of SOCS3 expression (19). We therefore examined pSTAT3 under Th17 polarizing condition with addition of a neutralizing anti-IFN-γ antibody. pSTAT3 in DBA/1 CD4+ T cells did not differ from those in B6 mice throughout the 3 days of culture (not shown). These results suggest that IL-6/STAT3 signaling pathway in DBA/1 was not different from those in B6 mice.
DBA/1 CD4+ T cells showed increased RORγt and reduced Foxp3 expression under Th17 and iTreg polarizing conditions
In addition to IL-6 induced STAT3 phosphorylation, TGF-β signals play an essential role in mouse Th17 differentiation (20, 21). TGF-β can induce both RORγt and Foxp3 expression. We therefore examined expression of RORγt and Foxp3 under Th17 polarizing conditions. B6 CD4+ T cells showed little induction of RORγt until 72hr post stimulation, whereas RORγt was strongly expressed by DBA/1 CD4+ T cells (Figure 4). Next, we incubated naïve CD4+ T cells under either Th17 or iTreg polarizing conditions to measure Foxp3 expression. Surprisingly, DBA/1 CD4+ T cells showed reduced Foxp3 expression in response to TGF-β. In the presence of IL-6 (Th17 polarizing condition), TGF-β was able to induce significant levels of Foxp3 expression in B6 CD4+ T cells, but minimal expression in DBA/1 CD4+ T cells (Figure 5A). When no exogenous IL-6 was and added and endogenous IL-6 was neutralized by addition of a neutralizing anti-IL-6 antibody (iTreg polarizing conditions), Foxp3 expression was much enhanced in a dose dependent fashion to TGF-β in B6 but not in DBA/1 CD4+ T cells (Figure 5B). Both IFN-γ and IL-4 can inhibit Foxp3 expression. In order to examine the effect of IFN-γ and IL-4 on Foxp3 expression, we added anti-IFN-γ and anti-IL-4 antibodies in the iTreg culture. Blocking IFN-γ and IL-4 increased Foxp3 expression in both B6 and DBA/1 CD4+ T cells (Figure 5C), but the magnitude of Foxp3 expression remained lower in DBA/1 CD4+ T cells. These results suggest a divergent TGF-β signaling in DBA/1 CD4+ T cells in differentiation towards Th17 or iTreg.
Figure 4. Increased RORγt expression by DBA/1 CD4+ T cells.

Purified naïve CD4+ T cells were cultured under Th17 polarizing conditions for 3 days as in Figure 6. RORγt mRNA expression at indicated time points were determined by quantitative PCR. Figure shows one of three independent experiments (n = 5 in each group).
Figure 5. Reduced expression of Foxp3 by DBA/1 CD4+ T cells.

Purified naïve CD4+ T cells obtained from B6 or DBA/1 mice were stimulated under Th17 or iTreg conditions and Foxp3 expression determined by intracellular staining after 3 days in culture. (A) Foxp3 expression by CD4+ T cells under Th17 or iTreg conditions. (B) Foxp3 expression with dose titration of TGF-β. (C) Foxp3 expression under iTreg conditions in the presence of IFN-γ or IL-4 neutralizing antibodies. Representative of three experiments, n = 4 in each group.
Smad2 and Smad3 phosphorylation was reduced in DBA/1 naïve CD4+ T cells
TGF-β signals are transduced through activation of Smad2/3. We examined induction of Smad2/3 phospholylation under Th17 or iTreg conditions by Western blot. Surprisingly, DBA/1 CD4+ T cells showed reduced phosphorylation of Smad2 and 3 (Figure 6A). Smad6/7 are inhibitory Smad family members and it is reported that Smad7 functions as an antagonist of TGF-β induced Foxp3 expression (22). We examined the Smad6 and 7 expression in Th17 and iTreg conditions. As shown in Figure 6B, the expression of both Smad6 and 7 was not significantly different between B6 and DBA/1 naïve CD4+ T cells at any of the time points examined (Figure 6B).
Figure 6. Decreased Smad2 and Smad3 phorsphorylation in DBA/1 CD4+ T cells.
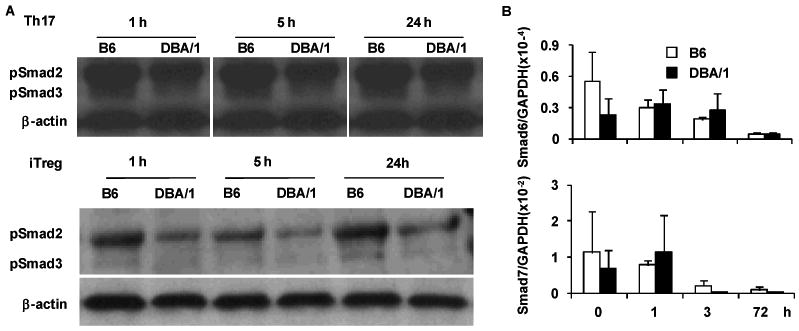
Purified naïve CD4+ T cells were cultured under iTreg and Th17 polarizing conditions for duration indicated. Cell lysates were obtained and (A) expression of phosphorylated Smad2 and Smad3 determined by western blotting (one of two independent experiments with similar results is shown); (B) expression of Smad6 and Smad7 was determined by quantitative PCR. No statistical significance was detected between DBA/1 and B6 mice at any time point (pooled data of two experiments, n = 4 in each group).
Discussion
Development of CIA in mice is influenced by many factors. The most notable one is the strong association of specific MHC H-2q expressed by the highly susceptible strain of mice, DBA/1 (1). However, recent studies have demonstrated the critical role of enhanced IL-17 production by DBA/1 mice in the development of arthritis (7). As demonstrated in this study, the biased IL-17 production is not restricted to response to CII, but to other antigens as well such as KLH and OVA. This suggests a global tendency of CD4+ T cells of DBA/1 to produce more IL-17 as compared with those in B6 and BALB/c mice – two strains known to be relatively resistant to CIA. The current study focused on signals that might influence naïve CD4 T cell differentiation into the Th17 subset. We found that TGF-β signaling in DBA/1 CD4+ T cells tilts towards Th17 with decreased iTreg formation.
We performed a stepwise analysis to dissect the factors that might influence the biased IL-17 production by DBA/1 CD4+ T cells. We first examined T cell cytokine production in naïve CD4+ T cells under Th1, Th2 and Th17 polarizing conditions. Surprisingly, DBA/1 CD4+ T cells produced higher levels not only of IL-17, but also of IFN-γ and IL-4. This could have been due to a lower threshold of activation in DBA/1 T cells or enhanced activation of early events after calcium influx in DBA/1 CD4+ T cells. However, we observed no differences in calcium flux, nor in expression of the early activation marker, CD69. CD69 expression is dependent on external calcium entry and has been used to assess immediate events downstream of cell calcium influx (16). In fact, DBA/1 CD4+ T cells expressed less CD69 in response to CD3/CD28 stimulation. Further analysis demonstrated increased survival of DBA/1 CD4+ T cells under both Th1 and Th2 conditions, providing a likely explanation for the accumulation of Th1 and Th2 cells and cytokines. These results may be further explained by increased IL-2 receptor expression by DBA/1 CD4+ T cells at equal amounts of IL-2 production (data not shown). Significantly, when cells were cultured under Th17 conditions, there was a similar proliferation rate and survival between DBA/1 and B6 CD4+ T cells yet DBA/1 cells produced significantly more IL-17 than those of B6 mice.
To differentiate into Th17 cells upon TCR activation, naïve CD4+ T cells need signals from IL-6 together with TGF-β and then IL-23 is required to stabilize the Th17 clones (13, 14, 21). IL-6 activates STAT3 that presumably interacts with RORγt to induce transcription of the IL-17 gene promoter. However, analysis of total and phosphorylated STAT3 with and without addition of IL-23 did not reveal differences between DBA/1 and B6 CD4+ T cells suggesting that the IL-6 and IL-23 transduction pathways were normal in DBA/1 mice. SOCS3 is known to block STAT3 signaling and inhibit Th17 differentiation (19). Since we observed that this negative regulatory mechanism was also intact in DBA/1 CD4+ T cells, we focused on the effect of TGF-β on DBA/1 CD4+ T cells. We found that TGF-β-induced Foxp3 expression was reduced, yet TGF-β enhanced RORγt expression in DBA/1 CD4+ T cells. TGF-β can regulate both RORγt and Foxp3 expression, depending on dosage and concomitant presence of other cytokines. Low concentrations of TGF-β with IL-6 and with or without other pro-inflammatory cytokines promote Th17 differentiation through up-regulation of IL-23 receptor expression, while high concentrations of TGF-β without pro-inflammatory cytokines promotes Foxp3 expression and induces iTreg from naïve CD4+ T cells (13-15). TGF-β-induced Foxp3 directly interacts with RORγt on exon 2, and represses RORγt expression through this interaction (14, 23). Moreover, forced expression of Foxp3 suppress RORγt mediated IL-17 promoter activation. Therefore, the reduced TGF-β-mediated Foxp3 induction in DBA/1 CD4+ T cells leads to decreased iTreg cells but increased RORγt expression and IL-17 production by Th17 cells. The reciprocal relationship between iTreg and Th17 cells therefore skews the CD4+ T cell response to increased Th17 in DBA/1 mice.
TGF-β signals through both Smad-dependent and Smad-independent pathways (reviewed in (24), (25)). TGF-β induces phosphorylation of Smad2 and 3 which then binds to the common Smad, Smad4 and translocates to the nucleus to induce transcription of target genes. The TGF-β signaling pathway in induction of iTreg and Th17 cells is complex and the bifurcation is not fully understood (26), (25). Current evidence suggests both Smad-dependent and independent pathways are involved (27). Data from Smad-4 deficient CD4+ T cells suggest that Smad4 is required for iTreg but not Th17 development (26). We show here that under Th17 and iTreg polarizing conditions, phosphorylation of Smad2 and 3 in DBA/1 naïve CD4+ T cells was decreased compared with phosphorylation in B6 mice. These data are consistent with functional studies in Smad2/3-double knockout mice (25). Interestingly, both Smad2 and Smad3 are dispensable for TGF-β-induced RORγt expression, but are required for Foxp3 expression. These observations could explain how reduced Smad2/3 phosphorylation allows for increased TGF-β-mediated RORγt induction and concomitantly decreased Foxp3 induction. Clearly, further studies are required to examine the fine regulation of TGF-β signaling in DAB/1 CD4+ T cells beyond Smad signal pathways.
The effects of TGF-β manipulation in arthritis susceptible rodent strains are complex. Intra-articular injection of TGF-β in Lewis rats actually caused inflammation and arthritis (28) although IL-17 was not known and was therefore not quantified at that time. Whereas TGF-β was also detected in the joints of DBA/1 mice, exogenous TGF-β suppressed development of arthritis when administered at the time of arthritis induction but was ineffective in suppressing the disease after arthritis onset (29). Neutralizing endogenous TGF-β by systemically administered anti-TGF-β antibody slightly increased the number of affected paws but this effect was diminished in the later phase of the disease (29). Based on the findings in our study, it will be informative for future research to re-examine the effects of exogenous TGF-β in rodent models of arthritis while simultaneously quantifying Th17 and iTreg cells. However, conditional manipulation will almost certainly be necessary to dissociate the effect of TGF-β on T cells versus APC. Regardless of the global effects of TGF-β manipulation, our studies suggest that delineation of the branch point between TGF-β promotion of RORγt versus Foxp3 expression will allow for more precise drug targeting with preservation of the beneficial effects of TGF-β.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Jeff Ledbetter and Dr. Ed Clark for comments on the manuscript.
Abbreviations used in this paper
- CIA
collagen-induced arthritis
- CII
collagen Type II
Footnotes
This work was supported by National Institutes of Health Grant, AR055254, and the Herndon and Esther Maury Fund
References
- 1.Wooley PH, Luthra HS, Stuart JM, David CS. Type II collagen-induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J Exp Med. 1981;154(3):688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu CQ, Song Z, Mayton L, Wu B, Wooley PH. IFNgamma deficient C57BL/6 (H-2b) mice develop collagen induced arthritis with predominant usage of T cell receptor Vbeta6 and Vbeta8 in arthritic joints. Ann Rheum Dis. 2003;62(10):983–90. doi: 10.1136/ard.62.10.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortmann RA, Shevach EM. Susceptibility to collagen-induced arthritis: cytokine-mediated regulation. Clin Immunol. 2001;98(1):109–18. doi: 10.1006/clim.2000.4961. [DOI] [PubMed] [Google Scholar]
- 4.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol. 1997;158(11):5507–13. [PubMed] [Google Scholar]
- 5.Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. 1997;158(11):5501–6. [PubMed] [Google Scholar]
- 6.Guedez YB, Whittington KB, Clayton JL, Joosten LA, van de Loo FA, van den Berg WB, et al. Genetic ablation of interferon-gamma up-regulates interleukin-1beta expression and enables the elicitation of collagen-induced arthritis in a nonsusceptible mouse strain. Arthritis Rheum. 2001;44(10):2413–24. doi: 10.1002/1529-0131(200110)44:10<2413::aid-art406>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 7.Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56(4):1145–51. doi: 10.1002/art.22453. [DOI] [PubMed] [Google Scholar]
- 8.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179(9):6228–36. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 9.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–98. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 10.Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002;46(3):802–5. doi: 10.1002/art.10173. [DOI] [PubMed] [Google Scholar]
- 11.Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. 2007;204(1):41–7. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006;176(9):5652–61. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8(9):967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 14.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453(7192):236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues Mascarenhas S, Echevarria-Lima J, Fernandes dos Santos N, Rumjanek VM. CD69 expression induced by thapsigargin, phorbol ester and ouabain on thymocytes is dependent on external Ca2+ entry. Life Sci. 2003;73(8):1037–51. doi: 10.1016/s0024-3205(03)00377-1. [DOI] [PubMed] [Google Scholar]
- 17.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179(7):4313–7. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 18.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168(11):5699–708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol. 2008;180(6):3746–56. doi: 10.4049/jimmunol.180.6.3746. [DOI] [PubMed] [Google Scholar]
- 20.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 21.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179(4):2041–5. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 23.Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem. 2008;283(25):17003–8. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- 24.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 25.Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem. 147(6):781–92. doi: 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29(1):44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu L, Wang J, Zhang F, Chai Y, Brand D, Wang X, et al. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J Immunol. 184(8):4295–306. doi: 10.4049/jimmunol.0903418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen JB, Manthey CL, Hand AR, Ohura K, Ellingsworth L, Wahl SM. Rapid onset synovial inflammation and hyperplasia induced by transforming growth factor beta. J Exp Med. 1990;171(1):231–47. doi: 10.1084/jem.171.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorbecke GJ, Shah R, Leu CH, Kuruvilla AP, Hardison AM, Palladino MA. Involvement of endogenous tumor necrosis factor alpha and transforming growth factor beta during induction of collagen type II arthritis in mice. Proc Natl Acad Sci U S A. 1992;89(16):7375–9. doi: 10.1073/pnas.89.16.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.