

Abstract
Objective
To determine whether n-3 fatty acids (n-3) influence arterial cholesterol delivery and lipoprotein lipase (LpL) levels in insulin-resistant mice.
Methods and Results
Insulin resistance contributes to risk of cardiovascular disease. It was previously reported that saturated fat (SAT) diets increased, but n-3 diets decreased, arterial low-density lipoprotein (LDL) cholesterol deposition from LDL total and selective uptake; this was associated with increased or decreased arterial LpL, respectively. Insulin receptor transgenic knockout mice (L1) were fed a chow, SAT, or n-3 diet for 12 weeks. Double-fluorescent boron dipyrromethene (BODIPY)–cholesteryl ester (CE) and Alexa dye-labeled human LDL were injected to separately trace LDL-CE and LDL–apolipoprotein B whole particle uptake. In contrast to SAT, n-3 diets markedly reduced all plasma lipids, ameliorating progression of insulin resistance. As opposed to SAT, n-3 reduced arterial LDL uptake, CE deposition, and selective uptake. Disparate patterns of CE deposition between diets were comparable with arterial LpL distribution; SAT induced high LpL levels throughout aortic media; LpL was limited only to intima in n-3–fed mice.
Conclusion
n-3 diets diminish arterial LDL-cholesterol deposition in mice with insulin resistance, and this is associated with changes in arterial LpL levels and distribution.
Keywords: n-3 fatty acids, insulin resistance, LDL, lipoprotein lipase, atherosclerosis, diet, fish oils
Cardiovascular disease (CVD) remains the leading overall cause of death in the United States. Type 2 diabetes mellitus, one of the most common noncommunicable diseases, has emerged as a major CVD risk factor; patients with type 2 diabetes have a 2- to 4-fold greater risk than nondiabetic individuals of developing atherosclerosis that increases the prevalence of CVD morbidity and mortality.1 Insulin resistance is an important pathophysiological defect underlying the current epidemic of CVD and type 2 diabetes. Insulin resistance usually starts with impaired insulin action or sensitivity in the regulation of blood glucose and commonly leads to hyperinsulinemia, elevated plasma free fatty acid (FFA) levels, and, later, to type 2 diabetes.2
Lipoprotein lipase (LpL) has been postulated to be another important contributor to atherosclerosis promotion in diabetic subjects. Although low circulating LpL activity is observed in insulin resistance and type 2 diabetes and is linked to premature atherosclerosis,3 macrophage-derived LpL levels and activity are increased in animals and humans with insulin resistance and diabetes.4 Thus, it is likely that these differential effects on LpL levels in specific tissues and cells might lead to many proatherogenic events by promoting focal lipid deposition5 (eg, in the arterial wall) while reducing lipoprotein clearance in the circulation.
Dietary fatty acids have direct regulatory effects on LpL expression and activity. Recent studies showed that an n-3 fatty acid–enriched (n-3) diet markedly reduced arterial low-density lipoprotein (LDL) whole particle uptake and LDL–cholesteryl ester (CE) selective uptake (SU) relative to a saturated fat–enriched (SAT) diet in mice, independent of plasma cholesterol levels.5,6 Researchers have previously demonstrated that LDL core lipids, particularly CE, can be delivered to cells without concomitant cellular uptake and internalization of whole LDL particles.7,8 Unlike high-density lipoprotein SU, LDL SU was not mediated by class B scavenger receptor but can be markedly increased by LpL.9 Moreover, feeding with high SAT diets markedly increased the contribution of SU to total arterial LDL-CE deposition5; in apolipoprotein (apo) E–knockout mice, as much as 50% or more of total LDL-CE uptake was accounted for by SU. Thus, we suggest that this mechanism contributes to an additional pathway for CE delivery in LpL-expressing cells (eg, macrophages) and may contribute to pathological accumulation of CE. At the level of the arterial wall, we postulate that increased LpL levels by a SAT diet increase LpL “bridging” activity that associates with trapping more LDL and selective LDL-CE uptake.5,6 In contrast, n-3 decrease arterial LpL and arterial CE deposition with markedly reduced whole LDL particle uptake and essentially no arterial SU from LDL.
We questioned how n-3 influence arterial LDL-cholesterol delivery and LpL levels in an insulin-resistant model (ie, mice that lack insulin receptors in every tissue, except liver, pancreatic β cells, and some parts of the brain).10 Thus, these mice (referred to as L110) are a model of insulin resistance in all cell types of the arterial wall. The L1 mouse model was chosen because its phenotype is typical of insulin resistance and likely would respond with changes similar to other animal models and perhaps humans, to different dietary fatty acids. Also, the L1 mouse model overcomes issues that are usually associated with other models of insulin resistance (eg, issues of obesity-related leptin signaling defect or abnormalities in lipid metabolism induced by hepatic insulin resistance), allowing us to directly analyze the effects of impaired insulin signaling in vascular tissues. We selected this model to demonstrate the effects of dietary fatty acids, SAT versus n-3, on lipid uptake in the arterial wall. Our results show that L1 mice were prone to the accumulation of arterial LDL-CE via SU, and this was linked to the elevated arterial LpL levels. SAT diets induced more severe insulin resistance and markedly increased LDL whole particle uptake and SU, with increased arterial LpL levels in L1 mice. In contrast, n-3 ameliorated abnormal plasma lipid profiles in L1 mice and reduced arterial LpL levels and total LDL and selective CE uptake levels.
Methods
Materials and Animals
BODIPY-C12 and dyes (Alexa Fluor 488, 546, 568, and 647) were purchased from Molecular Probes (Invitrogen Corp, Carlsbad, Calif). Rat anti–CD31 antibody was purchased from BD Pharmingen, San Jose, Calif. Polyclonal rabbit anti–CD68 antibody was purchased from Santa Cruz Biotechnology, Inc, Santa Cruz, Calif. LpL antibodies were provided by Ira Goldberg, MD (Columbia University, New York, NY), or Gunilla Olivecrona, PhD (Umea University, Umea, Sweden), or purchased from Santa Cruz Biotechnology, Inc. L1 mice were described in a previous publication.10 These mice were generated by intercrossing mice bearing a transthyretin-driven human insulin receptor gene (INSR) cDNA with Insr heterozygous knockout mice; they lack endogenous Insr and express the human transgene in hepatocytes, pancreatic β cells, and selective brain cells. The genetic makeup of these mice contains chromosomes derived from 129SV, C57BL/6, and FVB genomes; and animals were maintained on a mixed background. All animals were kept in house at the Columbia University Medical Center Animal Facility. All experimental procedures were performed in accordance with Columbia University’s institutional guidelines for use of laboratory animals and were approved by Columbia University’s Institutional Animal Care and Use Committee.
Feeding Protocols
Four-week-old male L1 mice, weighing 15 to 18 g, were given a normal chow diet (45 g total fat/kg, Purina LabDiet-5053) or a high-fat diet enriched in either n-3 or SAT for 12 weeks (n>10 for each group).5 The high-fat diets were purchased from 2 different companies and contained 210 g of total fat/kg of diet or 184.5 g of total fat/kg of diet for n-3 or SAT diets, respectively. The fat content of the SAT diet consisted of 71% SAT from coconut oil, 19% monounsaturated fat from olive oil, and 9% polyunsaturated fats from safflower and corn oil (TD 97108, Teklad). In the n-3 diet, half of the total fat was from menhaden oil enriched in eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) (960165, MP Biomedicals) with SAT, monounsaturated fat, and polyunsaturated fat (8%, 8%, and 6%, respectively, wt/wt) to prevent essential fatty acid deficiency. Both the n-3 and SAT diets contained 2 g of cholesterol, whereas the chow diet contained 0.2 g of cholesterol per kilogram of diet. The fatty acid composition of each diet was detailed previously.6 During the feeding period, blood was collected weekly and assayed for FFAs (NEFA C kit), triglycerides (TGs), and total cholesterol (T-Chol), according to the manufacturer’s procedures, as previously detailed.9 Blood glucose and insulin levels were determined after mice were euthanized at the end of a feeding period, with 2-hour fasting as previously described.10
Lipoprotein Profiles
At the end of the feeding period, plasma from each mouse was pooled and analyzed for changes in lipoprotein profiles among different diet groups using fast-performance liquid chromatography.11 Pooled plasma injected was 500 μL for each run.
LDL Preparation and Labeling
LDL (d=1.025 to 1.055 g/mL) was isolated from normolipidemic humans by sequential ultracentrifugation.5 We separated LDL at the narrower density gradient range to minimize the confounding effects of LDL of different size and heterogeneity. This fraction contains no or minimal apoE, which can be present in lighter LDL fractions (d=1.019 to 1.025 g/mL). Also, use of a “constant” human LDL source limits variability in mouse LDL associated with diet feeding to the host animals.
Isolated LDL was labeled with fluorescent BODIPY-C12 and dye (Alexa 568) to independently trace LDL-CE and whole LDL particle uptake, respectively. Detailed procedures for labeling and characterization of fluorescent LDL were previously described.5,6 Previous studies showed that data obtained by assaying total and selective arterial LDL uptake via either radiolabeled or fluorescent-labeled LDL were comparable and similar.5 Our labeling procedures also did not modify LDL physical properties or induce LDL oxidation.5–8
LDL Uptake in the Aorta
At the end of the feeding period previously described, L1 mice were injected with 200 μg of double-fluorescent–labeled LDL (BODIPY-CE and dye [Alexa]–apoB). Eight hours after injection, mice were euthanized and extensively perfusion fixed with a 4% paraformaldehyde solution. Isolated aortas were sectioned (12 μm) with a cryomicrotome after being embedded in optimal cutting temperature (Tissue-Tek). BODIPY-C12 and dye (Alexa 568) fluorescence were recorded with a laser scanning confocal microscope (Zeiss LSM-510) equipped with an image-capture device at ×20 and ×40 magnification. The microscope settings were 488 and 543 nm (excitation wavelength) and 475 to 575 and 560 nm (emission wavelength) for BODIPY-C12 and dye (Alexa), respectively. All images were captured with the same laser intensity, gain, and exposure times. To determine BODIPY-C12/dye (Alexa) ratios, we measured fluorescence intensity in the intima-medial layers of the aortic arch using computer software (ImageJ version 1.31; National Institutes of Health; available online at: http://rsb.info.nih.gov/ij/). These quantification analyses were performed on at least 5 different sections of proximal aortas in each mouse (n=5 for each mouse group), and the mean of ratios normalized for unit area of artery sections was determined for each group. The patterns of dye (Alexa)/BODIPY dual-channel colocalization were analyzed using computer software (ImageJ 1.41) with the colocalization plug-ins.12 Consistent with previous studies, we found no contribution of autofluorescence at the same wavelengths used for analyses of either BODIPY or dye (Alexa).
Arterial LpL and Cell Localization and Quantification
Arterial LpL content was assayed using immunohistochemical or immunofluorescent staining. For all procedures, sections were washed and treated with 4% normal goat serum to block nonspecific background staining before antibody application.
Sectioned aortas from L1 and wild-type mice were analyzed for arterial LpL by immunohistochemistry. This was performed by using a rabbit polyclonal anti–human LpL antibody (1:20) with a biotinylated anti–rabbit secondary antibody, followed by alkaline phosphatase in situ for measuring substrate products (Sigma Rabbit ExtrAvidin Alkaline Phosphatase kit), as described previously.13 The microscope settings for LpL were the same as previously described.5 Captured images were measured for LpL staining intensity in the arterial wall using computer software (ImageJ), similarly as previously described for fluorescence intensity quantification. Results from at least 10 different sections of the same proximal aorta per mouse were averaged in each group (n=4).
In separate analyses, arterial LpL content was assayed by immunofluorescent staining. Before antibody application, aorta sections were processed as previously described. Sections were incubated with a primary anti–LpL antibody (raised in rabbit) for 24 hours at 4°C or for 2 hours if incubated at room temperature. Sections were then rinsed and incubated for 1 hour in goat anti–rabbit IgG conjugated with dye (Alexa Fluor 647), followed by incubation with a cell nucleus marker (SYTOX Green) for 30 minutes. After several washes in PBS, stained sections were mounted and cover slipped. Fluorescence in the arterial wall was analyzed using a laser scanning confocal microscope (LSM-510 META) equipped with an image-captured device at ×10 and ×40 magnifications, as previously described. Labeled nuclei and LpL were visualized by excitation at a wavelength of 488 nm (with a 505- to 550-nm bandpass emission filter) and by excitation at a wavelength of 633 nm (with a 638-nm low-pass emission filter), respectively.
To measure arterial immunostained LpL in each optical section, regions of interest were defined based primarily on the location of cell nucleus staining (SYTOX Green), which highlights arterial structure. All LpL markers within each region of interest were semiautomatically counted using computer software (ImageJ).14 Each optical section was adjusted for proper threshold and background subtractions for separating the correct signal from background. Particle sizes within 90 to 255 Gy were counted using a function (Analyze Particles) of the computer software (ImageJ). Each group consisted of 3 to 4 mice. For each artery sample, 10 to 20 sections were prepared and analyzed. Possible clusters of LpL signals are also determined by the same function (Analyze Particles) because it provides options for segmentation when studying the circularity and size of particles.
The colocalization of arterial LpL with aortic cells was examined by immunofluorescence similar to the process previously described. Artery sections were coincubated with primary chicken anti-LpL, rat anti–endothelial cell (EC), and rabbit anti–macrophage antibodies for 24 hours at 4°C, followed by the 1-hour incubation of the mixture of corresponding secondary antibodies of dye (Alexa 647) anti–chicken IgG, dye (Alexa 488) anti–rat IgG, and dye (Alexa 546) anti–rabbit IgG. Fluorescence in the arterial wall was analyzed using a laser scanning confocal microscope (LSM-510 META). An argon laser (488 nm) and 2 helium-neon lasers (543 and 633 nm) were used for the excitation of dyes (Alexa 488, 546, and 647), respectively. The patterns of dye (Alexa 488, 546, and 647) 3-channel colocalization were analyzed by a threshold-based overlap analysis (ie, a binary test of whether the 3 signals occur in the same or in different regions using computer software [ImageJ 1.41] with colocalization).12 The percentage colocalization of aortic macrophages or ECs with LpL was measured as the percentage of macrophages or ECs containing pixels that also contained LpL signals using computer software (ImageJ).
Arteries from the aortic root to the tracheal artery from L1 mice and wild-type littermates were pooled and homogenized for Western blot analyses for LpL. Protein, 30 μg, from mouse artery homogenates (n=3 to 5) was separated with 8% nongradient SDS-PAGE and transferred to membranes (Duralon) for immunoblot analyses for LpL (1:500). After antibody applications, horseradish peroxidase–conjugated anti–rabbit IgG was detected using an enhanced chemilumescent substrate (SuperSignal) and exposed to X-ray films according to the manufacturer’s instructions. Three separate Western blots were performed from different animals. Bands corresponding to each protein were measured by densitometry and normalized for β-actin levels for each sample. Results were expressed as relative density units compared with the wild-type littermates.11
Statistical Analyses
Student t tests of group means were used for comparing end points, and ANOVA was used to evaluate potential interactions between diets. Statistical significance was determined at P<0.05.
Results
Diet Effects on Body Weight and Plasma Lipid Profiles
L1 mice were fed either a high-fat diet enriched in SAT or n-3 or a standard rodent chow diet as a control over a 12-week period. Mice fed the SAT or n-3 diet showed similar weight gain during the study, and both were greater than that of the chow-fed mice (P<0.01, Figure 1).
Figure 1.
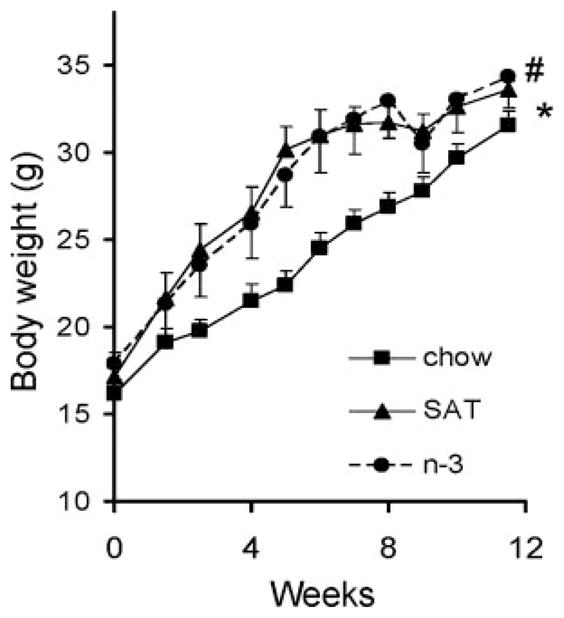
Effects of diet on mouse body weight in L1 mice fed a chow (■), SAT (▲), or n-3 (●) diet for 12 weeks. Each point represents the mean±SD of 10 mice. * and # denote significant differences between chow and SAT and between chow and n-3, respectively. P<0.001.
Despite the similar weight gain seen in SAT- and n-3–fed mice, the effects of diet on modulating plasma lipids were markedly different (Figure 2). After the 12-week feeding period, L1 mice receiving the chow diet were hyperlipidemic; plasma FFA and TG levels were significantly higher than those previously reported for a nondiabetic mouse model (ie, C57BL/6)6 (supplemental Table; available online at http://atvb.ahajournals.org). As shown in Figure 2, SAT diets further increased these already abnormal plasma lipid parameters in L1 mice; plasma FFA and TG levels were 3- and 6-fold elevated, respectively, when compared with the chow diet. On the other hand, n-3–fed L1 mice had no increase or much less of an increase in plasma FFA and TG levels compared with other groups throughout the study (Figure 2A and 2B) (P<0.05).
Figure 2.
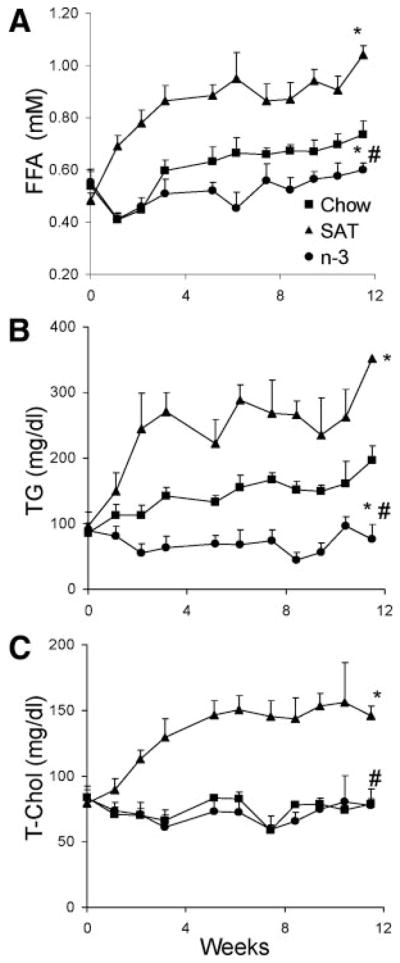
Plasma lipid profiles of L1 mice fed a chow (■), SAT (▲), or n-3 (●) diet. A through C, Blood samples were determined weekly for FFA (A), TG (B), and T-Chol (C) levels, as described in the Methods section. The results are expressed as the mean±SD (n>8). *P<0.05, chow vs SAT or n-3; #P<0.05, n-3 vs SAT.
SAT diet feeding led to a >2-fold increase in plasma T-Chol levels than did the chow diet (P<0.05), whereas cholesterol levels of n-3–fed mice remained constant toward the end of the feeding period and were similar to those of chow diet–fed mice (Figure 2C). These findings were of interest because changes in plasma T-Chol levels were similar between mice fed the n-3 diet containing high amounts of cholesterol (0.2%, wt/wt) and mice fed the chow diet containing much less cholesterol (0.02%, wt/wt). L1 mice receiving all diets had higher levels of all lipids compared with values previously reported in C57BL/6 mice6 (supplemental Table).
A high-fat diet can induce insulin resistance in mice.15 To distinguish between the effects of specific dietary fatty acids on the markers of insulin resistance, the SAT and n-3 diets used in our studies were isocaloric but had a higher fat content (42% of total calories) than normal rodent chow (21% of total calories). SAT-fed L1 mice had marked hyperglycemia and hyperinsulinemia (609±98 mg/dL and 10.4±3.1 ng/dL for glucose and insulin levels, respectively), whereas n-3–fed L1 mice had glucose and insulin levels of 419±37 mg/dL and 20.4±2.7 ng/dL, respectively. In keeping with insulin resistance and similar to what was previously reported,10 L1 chow-fed mice had plasma glucose and insulin levels of 351±114 mg/dL and 20.0±2.5 ng/dL, respectively. These findings suggest that qualitative, rather than measurement, differences in dietary fatty acids are more important in influencing blood glucose levels, in addition to plasma lipid levels.
To further characterize plasma lipoprotein profiles in these L1 mice after feedings with the different diets, changes in plasma lipoprotein distributions were analyzed using fast-performance liquid chromatography (Figure 3). In accordance with the increase in plasma TG levels, very low-density lipoprotein (VLDL) fractions were increased substantially in SAT-fed mice, 4-fold greater than the chow-fed group (Figure 3A). In contrast, n-3–fed mice had lower VLDL fractions. SAT diets also significantly increased L1 mouse plasma LDL fractions when compared with the chow or n-3 diet (Figure 3B).
Figure 3.
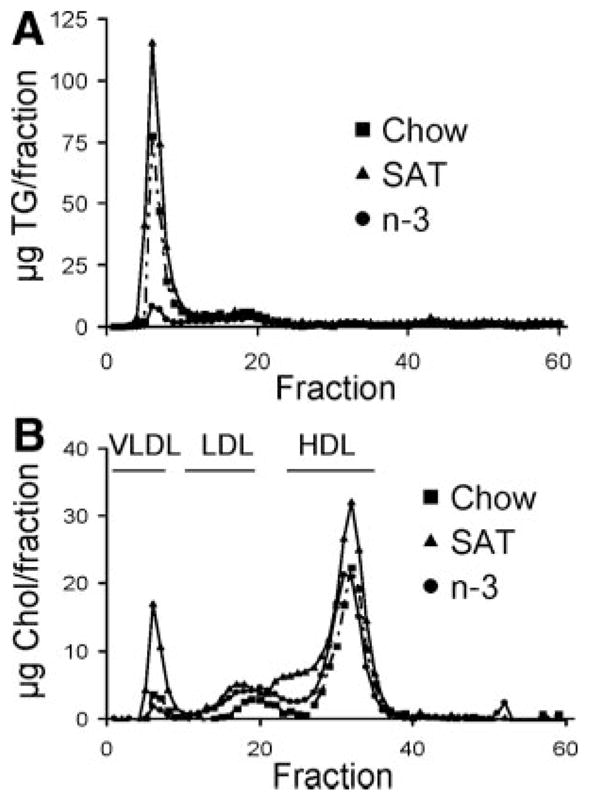
Plasma lipoprotein profiles in L1 mice fed a chow, SAT, or n-3 diet. A and B, TG (A) and Chol (B) levels were determined for each fraction after separating by fast-performance liquid chromatography. Elution fractions 2 to 10, 12 to 22, and 25 to 35 represent elution zones for very LDL, LDL, and high-density lipoprotein, respectively.
Diet Effects on Arterial LDL Uptake
We examined the effects of these dietary fatty acids on arterial LDL uptake and SU in the L1 model. We used fluorescent-labeled LDL to directly examine LDL uptake and LDL-CE deposition in the arterial wall. At baseline, before introduction of any experimental diets, L1 mice exhibited higher levels of LDL-CE uptake than their wild-type littermates (Figure 4A). A higher SU was indicated by the accumulation of LDL-CE tracer (ie, green fluorescent BODIPY in the arterial wall) and by more green fluorescence in the overlay when compared with wild-type littermates (P<0.05, Figure 4B).
Figure 4.
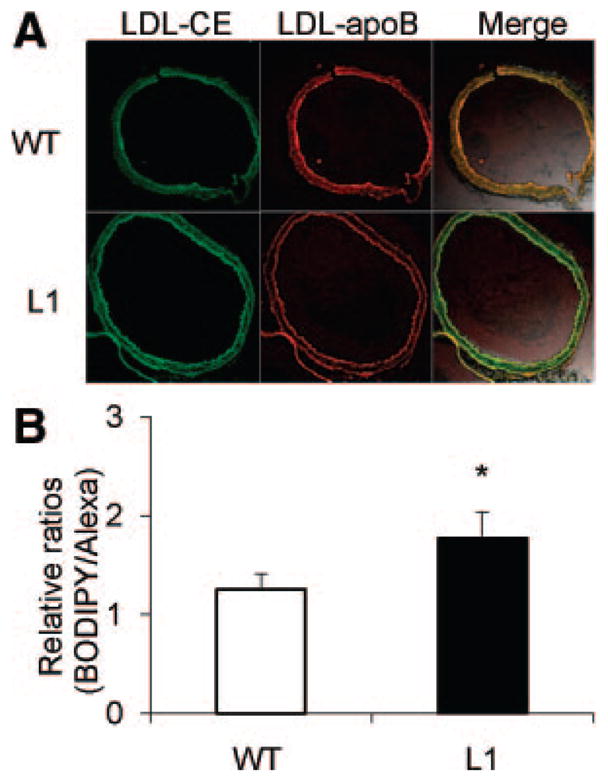
A, Comparison of arterial LDL-CE (BODIPY) and LDL-apoB (Alexa) uptakes in L1 mice and wild-type (WT) littermates. Merged 2-channel images (right) were used to assess colocalization of dye (Alexa) and BODIPY, with yellow fluorescence indicating colocalization (magnification ×20). B, Ratios of arterial uptake of total LDL-CE to whole LDL particle uptake determined for L1 mice and WT littermates, as described in the Methods section. Data are given as the mean±SE. *P<0.05.
Figure 5 shows the effects of different dietary fatty acids on LDL uptake and deposition in L1 mouse aortas. Compared with chow-fed L1 mice, the overlay image of SAT-fed mice showed disproportionably higher LDL-CE delivery relative to apoB, indicating SU. In contrast, with the reduced uptakes of LDL-apoB and LDL-CE, a similar amount of apoB and LDL-CE localized in the luminal side in n-3–fed mice. Moreover, there was a reduced infiltration of LDL in n-3–fed mice compared with chow- or SAT-fed mice (Figure 5A). These findings are consistent with previous reports on C57BL/6 mice6; SAT diets increased LDL uptake and SU (P<0.05), whereas n-3 diets consistently abolished arterial LDL SU, and the effects were pronounced in L1 mice (P<0.05, Figure 5B).
Figure 5.

A, Effects of diet on fluorescent LDL-CE (BODIPY) and apoB (Alexa) uptakes in aortas. B, Ratios of arterial uptake of total LDL-CE to whole LDL particle uptake determined for each diet group. Data are given as the mean±SE. Ratios were normalized to the same unit area for each diet. A ratio >1 indicates SU (dotted line). *P<0.05, chow vs SAT or n-3; #P<0.05, SAT vs n-3.
Dietary Fat Changes Arterial LpL Levels and Distribution
Researchers have demonstrated that LpL associates the binding of lipoproteins to cell surface proteoglycans, especially heparan sulfate proteoglycans; this accelerates cellular uptake of LDL and other lipoproteins.7,16,17 Previous studies showed that increased LpL in the arterial wall was linked to increased arterial LDL uptake and SU in C57BL/6 and apoE-knockout mice.5
Therefore, we examined arterial LpL expression levels by immunohistochemistry and immunofluorescence in our insulin-resistant L1 mouse model. There was a marked increase in arterial LpL levels, as indicated by more reddish brown staining (Figure 6A) in L1 mice when compared with wild-type littermates. Similar findings of increased LpL were observed by Western blot analysis of pooled artery homogenates (Figure 6B), in which densitometric analyses of arterial LpL normalized for β-actin showed an approximate 20% increase in L1 compared with wild-type mice when both mouse groups were fed the chow diets (L1 versus wild type, 1.28±0.03 versus 1.07±0.03 arbitraty units; n=3 to 5; P<0.05).
Figure 6.
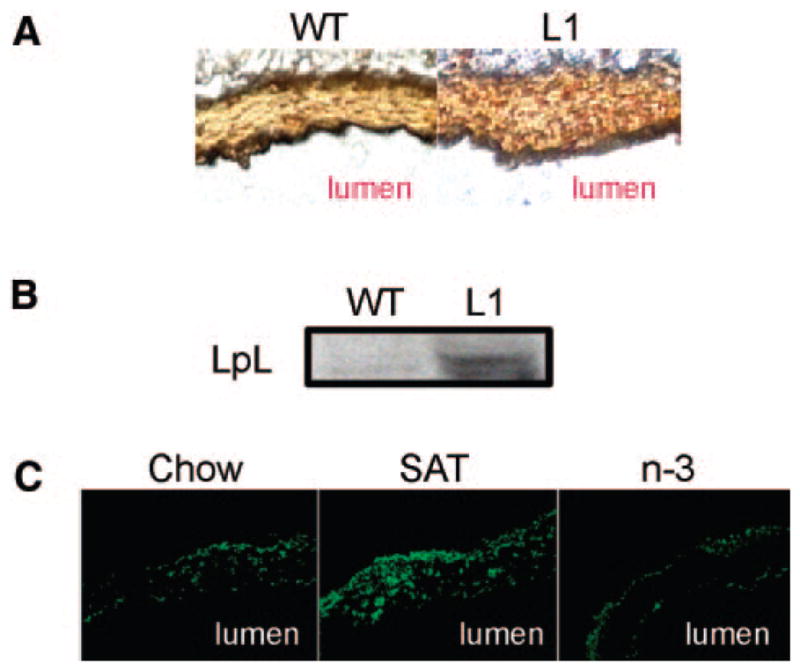
Effects of insulin resistance and diet on arterial LpL levels. A, Immunohistochemical staining of arterial LpL. Reddish brown staining shows the presence of LpL. B, Representative images of a Western blot for LpL from pooled mouse artery homogenates (n=3). C, Immunofluorescence of arterial LpL from mice fed a chow, SAT, or n-3 diet. Magnifications are ×20 (A) and ×40 (C). WT indicates wild type.
The effects of diet on arterial LpL levels in L1 mice were also examined (Figure 6C). Immunofluorescent staining of arterial LpL was markedly increased in SAT-fed mice when compared with chow-fed mice (SAT/chow ratio, 1.39±0.06; n=3; P<0.05). In contrast, n-3–fed mice showed low levels of arterial LpL (Figure 6C), paralleling the decrease of LDL-cholesterol deposition in the arterial wall.
Colocalization of LpL and Aortic Macrophages
Previous studies have shown that dietary saturated fatty acids increase macrophage-derived LpL mass, activity, and mRNA levels.18,19 Preliminary data (not shown) on C57BL/6 mice show that SAT, but not n-3, diets increased the number of arterial macrophages and that this could contribute to increased LpL levels. Thus, correlations of arterial LpL levels and macrophages were investigated in the L1 mouse model by immunofluorescence (Figure 7A). Counterstaining of LpL, ECs, and macrophages showed that the presence of LpL was linked to arterial medial macrophages (Figure 7B). More than 60% of LpL and macrophages colocalized in SAT-fed L1 mice (Figure 7C). In n-3–fed mice, there was almost no staining for LpL or macrophages in the arterial media; LpL was mostly at the intima, with some in the adventitial regions (Figure 7C). Thus, increases in arterial LpL link to increases of medial macrophages in the arterial wall.
Figure 7.
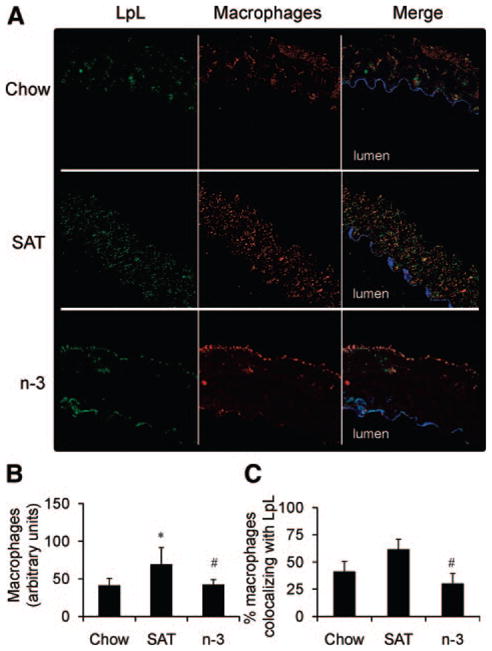
Arterial macrophages and LpL in the aorta. A, Representative images of chow-, SAT-, or n-3–fed L1 mouse aortas stained for ECs (blue), LpL (green), and macrophages (red) (magnification ×40). B, Immunofluorescence of macrophages measured for each group (n>3). C, Percentage of macrophages colocalizing with LpL determined for each group (n>3), as described in the Methods section. *P<0.05, SAT vs chow; #P<0.05, SAT vs n-3.
Discussion
Atherosclerosis, a major contributor to CVD, can be modulated by dietary fat intake patterns, insulin resistance, and type 2 diabetes.20 Dietary SATs and insulin resistance both promote CVD; dietary n-3 prevent atherosclerosis and lower CVD morbidity and mortality.21–24 In the studies described herein, we examined the effects of dietary SAT or n-3 on plasma lipid, glucose, and insulin levels and on LDL cholesterol deposition in the arterial wall in L1 mice (an insulin-resistant mouse model). Although hepatic insulin signaling has not been disrupted in these L1 mice, compared with earlier studies in C57BL/6 mice,6 L1 mice exhibited abnormal plasma lipid profiles, including elevated plasma FFA and TG levels and different patterns of plasma lipoprotein distribution. These differences were further exacerbated by SAT feedings. On the other hand, the n-3 diets reduced all plasma lipids, including cholesterol, in L1 mice. Several findings from L1 mice in the current studies are different from the previous reports on nondiabetic C57BL/6 mice.6 Our diet studies on C57BL/6 mice showed similar increases of plasma T-Chol levels in both the SAT- and n-3–fed groups, presumably because of the high cholesterol content in both diets (0.2%, wt/wt, compared with 0.02%, wt/wt, in the chow diet).6 In L1 mice fed similar diets, plasma T-Chol levels were only significantly increased in SAT-fed, but not in n-3–fed, L1 mice. These results suggest that the n-3 diet is more effective in reducing plasma T-Chol levels in mice that have abnormal lipid profiles, such as L1 mice. Compared with previous studies using C57BL/6 mice, in which there was no VLDL fraction in chow-fed mice, L1 mice fed with the same chow diets had significant VLDL fractions. However, the VLDL fraction essentially disappeared in L1 mice that were fed the n-3 diet. Thus, reduced TG levels in n-3–fed L1 mice could result from the reduced plasma TG-rich VLDL pools and/or from inhibiting TG synthesis.25 Moreover, the results on weight change of L1 mice are different from the similar diet studies on C57BL/6 mice.6 SAT- and n-3–fed L1 mice had similar weight gain; however, substantially less weight gain was seen in C57BL/6 mice fed the n-3 diet compared with mice fed the SAT diet. Regardless of the fat content in each diet, our data indicate that L1 mice were more prone to diet-induced obesity.
Both the SAT- and n-3–fed groups had elevated plasma insulin levels, but the n-3–fed group had blood glucose levels similar to the chow-fed mice. We have not examined β-cell functions in the current studies; however, as characterized in previous studies,10 L1 mice have islets of normal size with normal architecture and a moderate reduction in β-cell mass. After 12 weeks of feeding, n-3–fed L1 mice displayed hyperinsulinemia, with relatively normal glucose levels. In contrast, SAT-fed mice had substantially higher glucose levels but lower insulin levels, suggesting the progressive deterioration of compensatory insulin secretion by pancreatic β cells. Thus, it is possible that the SAT-fed mice with persistent severe hyperinsulinemia and hyperglycemia have proceeded to β-cell failure. In contrast, n-3 may inhibit processes that are important to atherosclerosis progression even with ongoing hyperinsulinemia.
L1 mice do not develop accelerated atherosclerosis spontaneously, possibly consistent with the hypothesis that excessive, rather than reduced, insulin signaling is atherogenic.26,27 The alternative, but not mutually exclusive, explanation is that distinct signaling pathways mediate the effects of insulin on glucose versus lipid metabolism.28,29 More studies are needed to elucidate the underlying mechanisms.
Recent studies from Han et al27 reported that when L1 mice were backcrossed with LDL receptor–knockout mice on a uniform C57BL/6 genetic background, they were protected from atherosclerosis and showed reduced VLDL/LDL levels and hepatic VLDL secretion. Still, these mice were hyperinsulinemic. Furthermore, the reduced levels of hepatic insulin receptors found in these double-knockout mice led to the reduced insulin signaling via insulin receptor substrate 1/phosphatidylinositol 3-kinase and reduced levels of Akt phosphorylation and its downstream targets, including reduced expression of lipogenic genes and decreased VLDL secretion. This is of interest because our L1 mice have human insulin receptor transgene expressed in liver, similar to that found by Han et al. The distinguishing features between L1 mice used by Han et al and those in the present study are our findings of hyperglycemia and dyslipidemia, with significantly higher levels of VLDL and LDL. Our L1 mice were maintained on a mixed genetic background that might explain some of the differences in the predisposition to diet-induced hyperglycemia; their genetic makeup contains chromosomes derived from 129SV, C57BL/6, and FVB genomes. We have not characterized hepatic insulin signaling pathways in these L1 mice; however, we have found increased liver TGs in these L1 mice. When these mice were fed the SAT diets, the number and size of liver lipid droplets were further increased when compared with chow-fed L1 mice, but the n-3 diet markedly reduced hepatic lipid accumulation (data not shown).
The macrophage has emerged as an important player in the pathogenesis of both atherosclerosis and insulin resistance.18,19 Recent studies have suggested that saturated fatty acids induce macrophage inflammation through the activation of toll-like receptor/nuclear factor-κB–mediated signaling, whereas n-3 block these cascades.30–34 In addition, toll-like receptor 4–dependent activation of NADPH oxidase causes both inflammation and oxidative stress that contribute to arterial cell injury.35 Our preliminary data on C57BL/6 mice show that the SAT, but not the n-3, diets increased number of arterial macrophages (C.L.C., T.S., and R.J.D., unpublished data 2009–2010). The accumulation of macrophages in the arterial wall, triggered by SAT diets, can contribute to atherogenesis by secreting proinflammatory mediators. Furthermore, our in vitro studies show that a saturated fatty acid, palmitic acid, promoted inflammation in cultured murine macrophages by increasing mRNA gene expression levels of proinflammatory cytokines, such as interleukin 6, but that n-3 EPA decreased these levels (C.L.C., R.J.D., and U. J. Jung, unpublished data,). Furthermore, in our in vitro studies, LpL mRNA expression and protein levels were increased by saturated palmitic acid but attenuated by EPA.
In the current studies, the results of colocalization studies of LpL and macrophages in L1 mice with different diets suggest that increased LpL levels in SAT-fed mice were associated with increased aortic medial macrophages. Notably, LpL itself can augment vascular dysfunction in the setting of inflammation. LpL directly induces tumor necrosis factor α gene expression and acts synergistically with interferon γ in the stimulation of macrophage NO synthase expression and endothelial NADPH oxidase.36,37 LpL-induced tumor necrosis factor α expression can further increase intercellular adhesion molecule/vascular cell adhesion molecule, E-selectin, and expressions of other proinflammatory genes in vascular cells.38 In addition, LpL has been proposed to function as a monocyte adhesion protein on ECs and in subendothelial matrix.39,40 Taken together, we hypothesize that, although saturated fatty acids stimulate inflammation in vascular cells, n-3 suppress inflammatory pathways by inhibiting the accumulation of arterial macrophages and LpL and proinflammatory cytokine production in atherosclerosis-susceptible sites. Hyperglycemia, as documented in these L1 mice, was exacerbated by SAT, but not n-3, diets; this might lead to advanced glycation end products, which could stimulate macrophage LpL gene expression and secretion,41 another potential pathway that would contribute to the increased aortic LpL levels.
In the current studies, similar to others, we suggest that LDL-cholesterol accumulation is a key pathogenic event and requirement for lesion development in early atherosclerosis progression; however, there are other factors that can affect binding uptake and the flux of the lipoproteins to the artery. For example, the changes in lipid levels and lipoprotein profiles associated with the SAT diet could affect arterial LDL uptake and deposition (ie, it is possible that the increases of both VLDL and FFA accelerate LDL accumulation in the arterial wall). Another possibility that might influence LDL accumulation in the arterial wall is the change in vascular permeability.42
Feeding mice with high SAT diets may parallel conditions that increase arterial LDL delivery and CE deposition in humans. However, because SAT diets also enhance diabetes in mice, this could confound interpretation of some of our findings; the potential relative contributions of insulin resistance–induced hyperglycemia and dyslipidemia are not fully separated or defined from our studies. In mouse models (eg, LDL receptor–deficient mice), hyperlipidemia can be exacerbated by high SAT feedings with the onset of hyperglycemia and accelerated atherogenesis. Hyperglycemia may have an independent role in atherogenesis.43 Gerrity et al44 showed that fat-fed diabetic pigs had more atherosclerosis than similarly dyslipidemic fat-fed animals without diabetes. Consumption of a cholesterol-free diet by LDL receptor–knockout mice with diabetes induced by β-cell–directed viral antigen resulted in hyperglycemia without changes in plasma lipids and lipoproteins, but with more rapid atherosclerosis.43 Still, progression of atherosclerosis is mainly initiated by a high-fat diet with the addition of dietary cholesterol independent of hyperglycemia.15,45
In summary, total LDL uptake and SU can be important contributors to accelerated arterial LDL-CE uptake in insulin-resistant L1 mice. Moreover, in L1 mice, similar to other mouse models, cholesterol delivery to the arterial wall is modified by the type of dietary fat, in part, through changes in arterial LpL levels. Although SAT diet increased total LDL-CE uptake and SU, an n-3 diet inhibited these pathways important to atherosclerosis progression in L1 mice. We hypothesize that the effects of n-3 on decreasing arterial wall cholesterol delivery in insulin-resistant mice will be an important mechanism by which these bioactive n-3 could decrease risk of CVD in metabolic syndrome and type 2 diabetes.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported in part by grants HL087123 and DK58282 (Dr Accili) and HL40404 (Dr Deckelbaum) from the National Institutes of Health; the Columbia University Diabetes Endocrinology Research Center (DERC, DK63608); and National Institutes of Health training grant T32-DK007647 (Ms. Chang).
Footnotes
Disclosures
None.
References
- 1.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 2.Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27:242–259. doi: 10.1210/er.2005-0007. [DOI] [PubMed] [Google Scholar]
- 3.de Vries R, Borggreve SE, Dullaart RP. Role of lipases, lecithin:cholesterol acyltransferase and cholesteryl ester transfer protein in abnormal high density lipoprotein metabolism in insulin resistance and type 2 diabetes mellitus. Clin Lab. 2003;49:601–613. [PubMed] [Google Scholar]
- 4.Sartippour MR, Renier G. Upregulation of macrophage lipoprotein lipase in patients with type 2 diabetes: role of peripheral factors. Diabetes. 2000;49:597–602. doi: 10.2337/diabetes.49.4.597. [DOI] [PubMed] [Google Scholar]
- 5.Seo T, Qi K, Chang C, Liu Y, Worgall TS, Ramakrishnan R, Deckelbaum RJ. Saturated fat-rich diet enhances selective uptake of LDL cholesteryl esters in the arterial wall. J Clin Invest. 2005;115:2214–2222. doi: 10.1172/JCI24327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang CL, Seo T, Matsuzaki M, Worgall TS, Deckelbaum RJ. n-3 fatty acids reduce arterial LDL-cholesterol delivery and arterial lipoprotein lipase levels and lipase distribution. Arterioscler Thromb Vasc Biol. 2009;29:555–561. doi: 10.1161/ATVBAHA.108.182287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo T, Al-Haideri M, Treskova E, Worgall TS, Kako Y, Goldberg IJ, Deckelbaum RJ. Lipoprotein lipase-mediated selective uptake from low density lipoprotein requires cell surface proteoglycans and is independent of scavenger receptor class B type 1. J Biol Chem. 2000;275:30355–30362. doi: 10.1074/jbc.M910327199. [DOI] [PubMed] [Google Scholar]
- 8.Rinninger F, Brundert M, Jackle S, Kaiser T, Greten H. Selective uptake of low-density lipoprotein-associated cholesteryl esters by human fibroblasts, human HepG2 hepatoma cells and J774 macrophages in culture. Biochim Biophys Acta. 1995;1255:141–153. doi: 10.1016/0005-2760(94)00228-q. [DOI] [PubMed] [Google Scholar]
- 9.Seo T, Velez-Carrasco W, Qi K, Hall M, Worgall TS, Johnson RA, Deckelbaum RJ. Selective uptake from LDL is stimulated by unsaturated fatty acids and modulated by cholesterol content in the plasma membrane: role of plasma membrane composition in regulating non-SR-BI-mediated selective lipid transfer. Biochemistry. 2002;41:7885–7894. doi: 10.1021/bi011949g. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto H, Nakae J, Kitamura T, Park BC, Dragatsis I, Accili D. Transgenic rescue of insulin receptor-deficient mice. J Clin Invest. 2004;114:214–223. doi: 10.1172/JCI21645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lachmanovich E, Shvartsman DE, Malka Y, Botvin C, Henis YI, Weiss AM. Co-localization analysis of complex formation among membrane proteins by computerized fluorescence microscopy: application to immunofluorescence co-patching studies. J Microsc. 2003;212:122–131. doi: 10.1046/j.1365-2818.2003.01239.x. [DOI] [PubMed] [Google Scholar]
- 13.Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I. Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:10423–10428. doi: 10.1073/pnas.1732494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. J Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 15.Merat S, Casanada F, Sutphin M, Palinski W, Reaven PD. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose-rich diet. Arterioscler Thromb Vasc Biol. 1999;19:1223–1230. doi: 10.1161/01.atv.19.5.1223. [DOI] [PubMed] [Google Scholar]
- 16.Rumsey SC, Obunike JC, Arad Y, Deckelbaum RJ, Goldberg IJ. Lipoprotein lipase-mediated uptake and degradation of low density lipoproteins by fibroblasts and macrophages. J Clin Invest. 1992;90:1504–1512. doi: 10.1172/JCI116018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams KJ, Fless GM, Petrie KA, Snyder ML, Brocia RW, Swenson TL. Mechanisms by which lipoprotein lipase alters cellular metabolism of lipoprotein (a), low density lipoprotein, and nascent lipoproteins: roles for low density lipoprotein receptors and heparan sulfate proteoglycans. J Biol Chem. 1992;267:13284–13292. [PubMed] [Google Scholar]
- 18.Li L, Beauchamp MC, Renier G. Peroxisome proliferator-activated receptor alpha and gamma agonists upregulate human macrophage lipoprotein lipase expression. Atherosclerosis. 2002;165:101–110. doi: 10.1016/s0021-9150(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 19.Hayek T, Hussein K, Aviram M, Coleman R, Keidar S, Pavoltzky E, Kaplan M. Macrophage foam-cell formation in streptozotocin-induced diabetic mice: stimulatory effect of glucose. Atherosclerosis. 2005;183:25–33. doi: 10.1016/j.atherosclerosis.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 20.Hu FB, Cho E, Rexrode KM, Albert CM, Manson JE. Fish and long-chain omega-3 fatty acid intake and risk of coronary heart disease and total mortality in diabetic women. Circulation. 2003;107:1852–1857. doi: 10.1161/01.CIR.0000062644.42133.5F. [DOI] [PubMed] [Google Scholar]
- 21.Breslow JL. n-3 Fatty acids and cardiovascular disease. Am J Clin Nutr. 2006;83:1477S–1482S. doi: 10.1093/ajcn/83.6.1477S. [DOI] [PubMed] [Google Scholar]
- 22.Deckelbaum RJ, Worgall TS, Seo T. n-3 Fatty acids and gene expression. Am J Clin Nutr. 2006;83:1520S–1525S. doi: 10.1093/ajcn/83.6.1520S. [DOI] [PubMed] [Google Scholar]
- 23.Mozaffarian D, Rimm EB. Fish intake, contaminants, and human health: evaluating the risks and the benefits. JAMA. 2006;296:1885–1899. doi: 10.1001/jama.296.15.1885. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Harris WS, Chung M, Lichtenstein AH, Balk EM, Kupelnick B, Jordan HS, Lau J. n-3 Fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am J Clin Nutr. 2006;84:5–17. doi: 10.1093/ajcn/84.1.5. [DOI] [PubMed] [Google Scholar]
- 25.Harris WS. n-3 Fatty acids and serum lipoproteins: animal studies. Am J Clin Nutr. 1997;65:1611S–1616S. doi: 10.1093/ajcn/65.5.1611S. [DOI] [PubMed] [Google Scholar]
- 26.Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Han S, Liang CP, Westerterp M, Senokuchi T, Welch CL, Wang Q, Matsumoto M, Accili D, Tall AR. Hepatic insulin signaling regulates VLDL secretion and atherogenesis in mice. J Clin Invest. 2009;119:1029–1041. doi: 10.1172/JCI36523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, Kahn CR, Shulman GI. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105:1791–1797. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haeusler RA, Accili D. The double life of Irs. Cell Metab. 2008;8:7–9. doi: 10.1016/j.cmet.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 31.Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- 32.Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279:16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- 33.Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz EA, Zhang WY, Karnik SK, Borwege S, Anand VR, Laine PS, Su Y, Reaven PD. Nutrient modification of the innate immune response: a novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler Thromb Vasc Biol. 2010;30:802–808. doi: 10.1161/ATVBAHA.109.201681. [DOI] [PubMed] [Google Scholar]
- 35.Maloney E, Sweet IR, Hockenbery DM, Pham M, Rizzo NO, Tateya S, Handa P, Schwartz MW, Kim F. Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler Thromb Vasc Biol. 2009;29:1370–1375. doi: 10.1161/ATVBAHA.109.188813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Sapuri-Butti AR, Aung HH, Parikh AN, Rutledge JC. Triglyceride-rich lipoprotein lipolysis increases aggregation of endothelial cell membrane microdomains and produces reactive oxygen species. Am J Physiol Heart Circ Physiol. 2008;295:H237–H244. doi: 10.1152/ajpheart.01366.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renier G, Lambert A. Lipoprotein lipase synergizes with interferon gamma to induce macrophage nitric oxide synthetase mRNA expression and nitric oxide production. Arterioscler Thromb Vasc Biol. 1995;15:392–399. doi: 10.1161/01.atv.15.3.392. [DOI] [PubMed] [Google Scholar]
- 38.Kota RS, Ramana CV, Tenorio FA, Enelow RI, Rutledge JC. Differential effects of lipoprotein lipase on tumor necrosis factor-alpha and interferon-gamma-mediated gene expression in human endothelial cells. J Biol Chem. 2005;280:31076–31084. doi: 10.1074/jbc.M412189200. [DOI] [PubMed] [Google Scholar]
- 39.Fisher RM, Benhizia F, Schreiber R, Makoveichuk E, Putt W, Al-Haideri M, Deckelbaum RJ, Olivecrona G, Humphries SE, Talmud PJ. Enhanced bridging function and augmented monocyte adhesion by lipoprotein lipase N9: insights into increased risk of coronary artery disease in N9 carriers. Atherosclerosis. 2003;166:243–251. doi: 10.1016/s0021-9150(02)00337-4. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Renier G. Adipocyte-derived lipoprotein lipase induces macrophage activation and monocyte adhesion: role of fatty acids. Obesity (Silver Spring) 2007;15:2595–2604. doi: 10.1038/oby.2007.311. [DOI] [PubMed] [Google Scholar]
- 41.Beauchamp MC, Michaud SE, Li L, Sartippour MR, Renier G. Advanced glycation end products potentiate the stimulatory effect of glucose on macrophage lipoprotein lipase expression. J Lipid Res. 2004;45:1749–1757. doi: 10.1194/jlr.M400169-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Rutledge JC, Woo MM, Rezai AA, Curtiss LK, Goldberg IJ. Lipoprotein lipase increases lipoprotein binding to the artery wall and increases endothelial layer permeability by formation of lipolysis products. Circ Res. 1997;80:819–828. doi: 10.1161/01.res.80.6.819. [DOI] [PubMed] [Google Scholar]
- 43.Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, Chait A, Bornfeldt KE. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–668. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerrity RG, Natarajan R, Nadler JL, Kimsey T. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. 2001;50:1654–1665. doi: 10.2337/diabetes.50.7.1654. [DOI] [PubMed] [Google Scholar]
- 45.Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008;371:1800–1809. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.