

Abstract
DNA rearrangements are important in genome function and evolution. Genetic material can be rearranged inadvertently during processes such as DNA repair, or can be moved in a controlled manner by enzymes specifically dedicated to the task. DNA transposases comprise one class of such enzymes. These move DNA segments known as transposons to new locations, without the need for sequence homology between transposon and target site. Several biochemically distinct pathways have evolved for DNA transposition, and genetic and biochemical studies have provided valuable insights into many of these. However, structural information on transposases – particularly with DNA substrates – has proven elusive in most cases. On the other hand, large-scale genome sequencing projects have led to an explosion in the number of annotated prokaryotic and eukaryotic mobile elements. Here, we briefly review biochemical and mechanistic aspects of DNA transposition, and propose that integrating sequence information with structural information using bioinformatics tools such as secondary structure prediction and protein threading can lead not only to an additional level of understanding but possibly also to testable hypotheses regarding transposition mechanisms. Detailed understanding of transposition pathways is a prerequisite for the long-term goal of exploiting DNA transposons as genetic tools and as a basis for genetic medical applications.
Keywords: Insertion sequence, integrase, transposition, genome instability, V(D)J recombination
Introduction
DNA transposons are distinct segments of DNA that can move to new locations within a genome without the need for homology between the transposon sequence and the new DNA target site (reviewed in Curcio and Derbyshire, 2003). This distinguishes them from other types of mobile genetic elements such as bacteriophages or genomic islands which use DNA recombinases or resolvases and generally require a short region of sequence identity between the ends of the element and the target site. Another large group of mobile elements, the retrotransposons, are abundant inhabitants of many eukaryotic genomes but much less frequent in prokaryotes. These copy themselves to a new location using an RNA intermediate that is subsequently reverse transcribed into double-stranded DNA. DNA transposons have no such intervening RNA intermediate, and many proceed through a so-called cut-and-paste mechanism involving a double-strand DNA intermediate. Recently, a completely different mechanism has been discovered in which a single strand of DNA is excised from one genomic location and moved to another (Barabas et al., 2008; Guynet et al., 2008). For the remainder of this review, we will focus on those pathways that proceed using both dsDNA substrates and intermediates.
There is an enormous diversity even among the class of transposons which use dsDNA intermediates. Perhaps the simplest of the autonomous transposable elements of this type, and certainly the most numerous in prokaryotes, are the insertion sequences (ISs). Most autonomous DNA elements identified in eukaryotes are also quite similar to prokaryote ISs in mechanism as well as in size. The original distinction between transposons and ISs was that the former carry additional genes to those involved directly in transposition while the latter include only the gene for their cognate transposase. More recently, detailed analysis of ISs in various bacterial genomes has revealed close relatives that include passenger genes encoding antibiotic resistance, methyltransferase activity, transcriptional regulation functions and proteins with unknown function (Siguier et al., 2009), further obscuring the line separating insertion sequences and transposons.
Mechanisms of DNA transposition
Among the dsDNA transposons, the most prevalent use a transposase which carries a conserved triad of amino acids – Asp, Asp and Glu, the DDE motif – which we will discuss in detail in the next section. Retroviruses fall into this category since not only do they encode an enzyme (integrase, IN) closely related to bacterial transposases, but they integrate into the host genome using a double-stranded DNA copy of their RNA genome (reviewed in Craigie, 2001). Moreover, the V(D)J recombination system which generates diversity in immunoglobulins and T cell receptors also uses an enzyme with a DDE motif, RAG1, which promotes transposition-like chemistry and can, under certain circumstances, behave like a DNA transposon (Hiom et al., 1998). In this review we will further restrict ourselves to transposition pathways that proceed through dsDNA intermediates using DDE-type enzymes.
Identification of the DDE triad stemmed from remarkable similarities observed between prokaryotic transposases and retroviral integrases (Fayet et al., 1990; Kulkosky et al., 1992). The crucial role of these residues in catalysis has subsequently been demonstrated by mutagenesis in many transposition systems, and the overall catalytic mechanism of enzymes containing a DDE motif has been well characterized (Mizuuchi, 1992; Mizuuchi and Baker, 2002). They catalyze a single chemical reaction: nucleophilic cleavage of a single phosphodiester bond. The tremendous diversity in transposition mechanisms derives from the combinatorial effect of using different nucleophiles on different DNA substrates in varying combinations, and whether or not replication occurs at some stage during these consecutive recombination events.
In general, transposition occurs first by cleavage of one strand at each transposon end. The nucleophile is generally H2O (Figure 1), and this reaction produces a 3′OH. For retroviruses, this is called “3′processing”. The two 3′OH groups (one at each end of the mobile element) then serve in a second chemical step as nucleophiles in a concerted attack on both strands of the target DNA (called “strand transfer”). The two strand transfer reactions occur a few base pairs apart on the target DNA, the precise distance between them being characteristic of the particular transposase. The breaks in the target DNA are repaired by host cell enzymes, a process that duplicates the target site on both flanks of the transposon.
Figure 1.

Dealing with the second strand. The color code is as follows: transposon DNA (green); flanking donor DNA (blue); target phosphates destined to be removed from the final liberated transposon (filled blue circles with a white “P”); phosphates destined to remain as 5′ transposon ends (open blue circles); the preferred stereoisomer, Sp or Rp, where known, is indicated within the circles; liberated 3′OH groups involved in strand joining reactions (open red circles); 3′OH destined to be removed from the liberated transposon (filled red circles); H2O is the attacking nucleophile in the hydrolysis reactions. (a) The Mu and Tn3 cleavage reactions. Note that the preferred stereoisomer has been demonstrated only for Mu and not for Tn3. (b) Tn7 cleavage reactions. Cleavage of the transferred strand (top of panel) is shown occurring prior to cleavage of the non-transferred strand (middle) leading to liberation of the transposon from flanking donor DNA (bottom of panel), although this order of cleavage reactions has not been demonstrated experimentally. The two types of cleavage are catalyzed by different enzymes. (c) Retroviral “processing” reaction, equivalent to cleavage of the transferred strand. An initial transcription step from the integrated provirus is indicated. The RNA genome is then encapsidated with a second copy and undergoes reverse transcription following infection to generate the double strand DNA integration intermediate. The intermediate is flanked by only short fragments of donor material and does not require second strand processing for insertion. (d) Transposition by the members of the IS630 family and the Tc1/Mariner superfamily is initiated by cleavage of the non-transferred strand (top of panel) at several bases within the transposon end (middle) leaving these bases attached to the liberated flanks following cleavage of the transferred strand (bottom). (e) For IS911, IS2, IS3 and other members of the IS3 family, single-end hydrolysis occurs (top). The liberated 3′OH then directs a strand transfer reaction to the same strand several bases 5′ to the other end of the element. This results in the formation of a single-strand circle which is then resolved into a transposon circle by replication from the free 3′OH (filled red circle). Single-strand hydrolysis at each 3′ end within the circle generates a linear transposon which can then undergo integration. (f ) The IS4 family and piggyBac have similar mechanisms. Following initial nucleophilic attack on the Rp target phosphate, the liberated 3′OH attacks an Sp phosphate in a trans-strand transfer reaction to generate a hairpin intermediate, liberating the transposon from its flanking donor DNA and inverting the target phosphate to its Rp configuration. These then become the substrates for a second hydrolysis. Note that the stereochemistry has been analyzed only in the case of Tn10. (g) Hermes and V(D)J transposition occur by initial cleavage of the non-transferred strand (top). The liberated 3′OH on the donor flank then attacks the opposite strand (middle) to generate hairpin structure on the donor flank (bottom). The stereochemistry has been analyzed for V(D)J only. Modified and reprinted from Turlan and Chandler (2000), with permission from Elsevier.
There are no covalent enzyme-substrate intermediates formed during this process, distinguishing it from reactions catalyzed by the ubiquitous serine and tyrosine recombinases (Grindley et al., 2006). Instead DNA transposition involves a series of single-step, direct in-line nucleophilic attacks. This mechanism of direct transesterification was firmly established by observations that the phosphate of the target scissile phosphodiester bond undergoes chiral inversion (i.e., an Rp diastereomer undergoes inversion to its Sp form) when chirality is imposed by replacement of a non-bridging oxygen by a sulfur atom. This has been observed to occur during several of the steps catalyzed by various systems including bacteriophage Mu, HIV integrase, Tn10, and V(D)J recombination (Mizuuchi and Adzuma, 1991; van Gent et al., 1996; Mizuuchi, 1997; Gerton et al., 1999; Kennedy et al., 2000).
In spite of their shared transposition chemistry, different DDE-type transposons have developed different variations in the steps leading to formation of a unique insertion intermediate (Figure 1). These differences reflect the way in which the second (non-transferred) strand is processed (Turlan and Chandler, 2000).
A first distinction is whether the second strand is cleaved at all. The simplest scenario is that used in generating the dsDNA intermediate seen during retroviral integration. Retroviral integrase, IN, removes two bases from the transferred strand at each end (Figure 1c). As neither strand was ever joined to anything, this leaves recessed ends with the characteristic 3′OH nucleophile ready for the strand transfer step (Craigie, 2001). In the cases of bacteriophage Mu, transposon Tn3 (and other members of this large family) and the IS6 family of prokaryotic insertion sequence, the non-transferred strand remains intact (Figure 1a) and strand transfer fuses both donor and target DNA molecules to form a so-called Shapiro intermediate (Shapiro, 1979). The branched fusion molecule, in which each transposon end is joined to the donor by one strand and the target by the other, is resolved by replication of the transposon. If both donor and target DNA are circular replicons (such as plasmids or chromosomes), this generates a cointegrate where donor and target DNA are joined and delimited at each junction by a single transposon copy. Transposition is then completed by recombination between the directly repeated transposons either using a host recombination system or as, for example with Tn3, using dedicated site-specific recombination systems encoded by the transposon itself (Grindley, 2002).
For all other known elements, cleavage of the second strand does occur, liberating the transposon from flanking DNA at the donor site. For the transposon Tn7 (Figure 1b), two different enzymes are used to generate an excised intermediate: TnsB catalyzes cleavage of the transferred strand and is predicted to contain a DDE motif, and TnsA catalyzes cleavage of the non-transferred strand and leaves a three base 5′ extension which is presumably removed during the DNA repair steps which must follow integration (Sarnovsky et al., 1996). Tn7 transposition requires the presence of target DNA to activate cleavage and transfer (reviewed in Craig, 2002; Parks and Peters, 2009). For members of the IS630 family and the related Tc1/mariner eukaryotic transposons, cleavage of the non-transferred strand occurs prior to cleavage of the transferred strand and at several bases within the transposon (Plasterk et al., 1999; Feng and Colloms, 2007; Figure 1d).
Members of the large IS3 family of prokaryotic ISs have developed yet another way of resolving the second-strand cleavage problem (Figure 1e; reviewed in Rousseau et al., 2002). Here, the transposase catalyzes cleavage at one end only. The resulting 3′OH is then used to attack the same strand three or four nucleotides within the DNA flank at the opposite end, thus creating a single-strand bridge between the two ends and a 3′OH positioned in the flanking donor DNA. This can act as a primer for DNA replication. Replication then produces a free covalently-closed circular IS in which the ends are abutted but separated by three or four base pairs (bp) of the original flanking DNA, and regenerates the original donor molecule. In the integration step, transposase catalyzes appropriate cleavages at each end to yield an integration intermediate with 3′OH ends.
Another way of ensuring second strand cleavage has been adopted by members of the IS4 family, Tn5 (IS50) and Tn10 (IS10) (Figure 1f; reviewed in Haniford, 2006; Reznikoff, 2008) and the related eukaryotic piggyBac transposon superfamily. Here the 3′OH generated at the end of the transferred strand is used to attack the opposite (non-transferred) strand to generate a hairpin. The DNA at the hairpin end is then cleaved, again using H2O as the nucleophile to create a typical insertion intermediate. In the case of piggyBac, the geometry of cleavages is such that the excised intermediate carries a 5′ tetranucleotide extension consisting of flanking DNA on both non-transferred strands (Mitra et al., 2008).
Finally, to complete our list of known ways by which dsDNA transposons are mobilized, both the V(D)J system (reviewed in Jones and Gellert, 2004) and the members of the hAT family of eukaryotic transposons such as Hermes (Warren et al., 1994; Zhou et al., 2004) have adopted yet another pathway for second strand cleavage (Figure 1g). Here, cleavage of the non-transferred strand creates a 3′OH on the DNA flank. This then attacks the phosphodiester bond on the transferred strand generating the appropriate 3′OH necessary for strand transfer, and in so doing, creates a hairpin structure in the DNA flanks.
The fundamental building block of DNA transposases: the catalytic core
The common structural features of RNase H-like catalytic domains
The ability of transposases containing a DDE triad to catalyze nucleophilic cleavage of a single phosphodiester bond is a direct result of the role of these three acidic residues. The two Asp residues and a third (which is usually Glu but sometimes Asp, hence the broader DDE/D shorthand that we will use from now on) coordinate two divalent metal ions. These metal ions, most likely Mg2+ in vivo, are essential cofactors for both the DNA strand cleavage and strand transfer steps. The two metal ion catalytic mechanism was first described by Beese and Steitz (1991) for the exonuclease domain of the Klenow fragment, a domain that is topologically related to the transposase catalytic domains. The validity of the two metal ion mechanism for transposases was later confirmed by the crystal structures of the IS50 transposase (referred to as the “Tn5 transposase”; Davies et al. 2000; Steiniger-White et al., 2002), and was further extended to the topologically-related RNase H (Nowotny et al., 2005).
The striking feature of transposases which have catalytic domains containing the DDE/D triad is their topological similarity, despite the often negligible and therefore unrecognizable sequence similarity between them. There is also significant variation in the size of the domains, a consequence of variations in the lengths of the loops connecting the secondary structure elements, the lengths of the secondary structure elements themselves, and – as we shall see – extra domains inserted into the catalytic domain. Figure 2 shows the three-dimensional structures of a representative set of DDE/D catalytic domains that are involved either in transposition or retroviral integration. The conserved core of the domain is a mixed alpha-beta fold, β1-β2-β3-α1-β4-α2/3-β5-α4-α5 (Figure 3), that was first seen for RNase H (Yang et al. 1990); therefore, it is probably most accurate to refer to it as the “RNase H-like fold” or “ribonuclease H-like fold” as this is the terminology that has been adopted by the protein fold classification database SCOP (http://scop.mrc-lmb.cam.ac.uk/scop/). More precisely, it is a three-layered α/β/α domain with a central, mixed five-stranded β-sheet with a characteristic strand order of 32145, where strand 2 is antiparallel to the rest (Figure 2). In the literature, the fold is sometimes referred to as the “retroviral integrase fold”.
Figure 2.
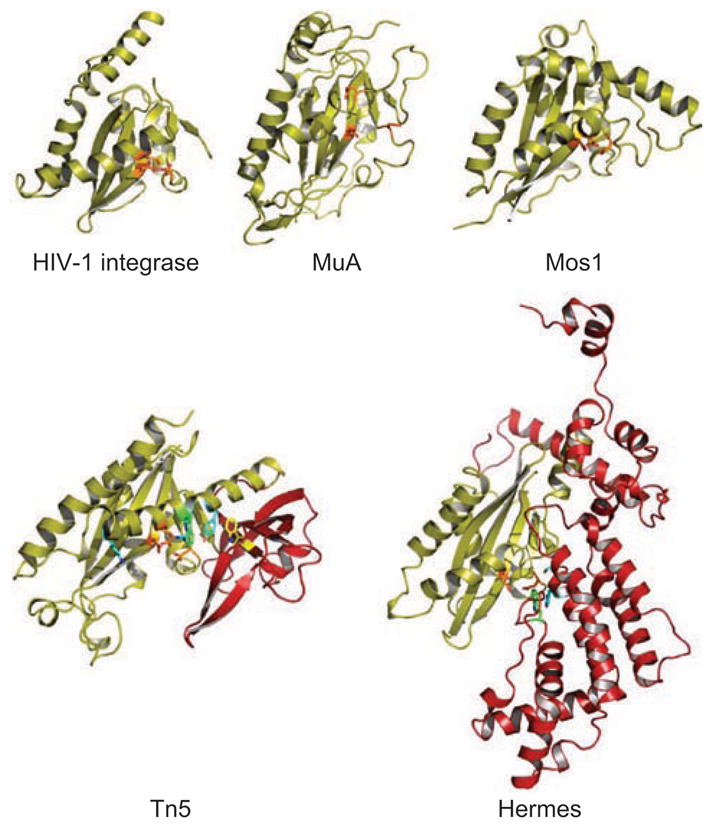
Ribbon diagrams of the aligned catalytic cores of four DNA transposases and of HIV-1 integrase. Residues shown in orange are the carboxylate active side residues, in green are the W residues of the Tn5 transposase and Hermes that are important in the reactions, in blue are the YRK residues of the YREK motif and in yellow is W298 of the Tn5 transposase. The insertion domains of the Tn5 transposase and Hermes are shown in red (note that there is a 15 amino acid loop from residues S481 to K496 that is disordered – and therefore not visible – in Hermes). The proteins are drawn to scale.
Figure 3.
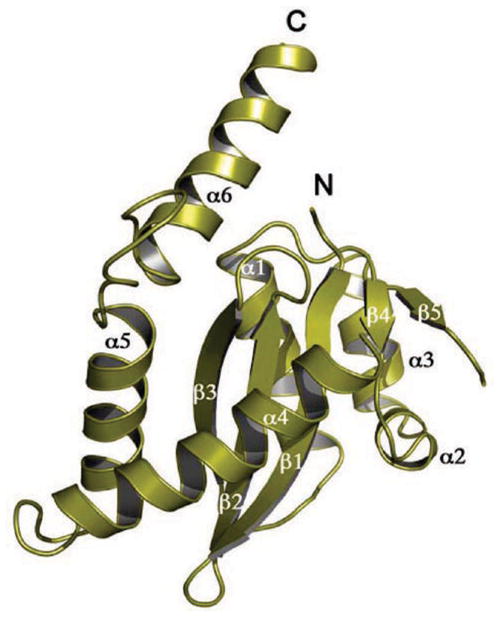
Ribbon diagram of the catalytic core domain of HIV-1 integrase, with the standard secondary structure elements highlighted.
The DDE/D catalytic triad of carboxylate side chains are always located on topologically equivalent secondary structure elements, with the first D always on β1, the second D always on or just after β4, and the E/D either on or just before α4. In crystal structures of uncomplexed (i.e., without DNA bound) transposases, the E/D residue is sometimes on a loop displaying some degree of disorder. For instance, in the crystal structure of the catalytic fragment of the bacteriophage Mu transposase (MuA) (Rice and Mizuuchi, 1995), the E residue (E392) was visible in the electron density but it was also clear that the observed configuration of the catalytic triad would be unable to coordinate two metal ion cofactors, indicating the need for a conformational change. In the first crystal structure of the catalytic domain of HIV-1 IN (Dyda et al., 1994; reviewed in Jaskolski et al., 2009), a 14 amino acid stretch that contains the catalytic E is completely disordered, without detectable electron density. It is quite possible that in many cases these observed disordered loops become ordered upon DNA binding, folding into a regular α-helical structure and becoming an upstream extension of α4. This means that, at least for some transposases, the active site assembles fully only after the transposon end DNA is captured and is ready for cleavage. This is certainly not always the case as comparison of the uncomplexed Tn5 transposase structure with that bound to transposon-end DNA shows little protein main chain movement, although there are some significant side chain movements (Davies et al., 1999; 2000).
To date, there are only two transposases for which crystal structures have been determined in which both transposon DNA and the transposase catalytic domain are present. These are of the prokaryotic Tn5 transposase dimer bound to two transposon ends (Davies et al., 2000; reviewed in Steiniger-White et al., 2004 and Reznikoff, 2008) and the eukaryotic Mos1 DNA transposase dimer bound to two oligonucleotides representing the transposon Right End (Richardson et al., 2009). In both cases, the bound DNA approximates a version of an intermediate along the transposition pathway in which both strands have already been cleaved. An EM reconstruction is also available for the MuA transposase tetramer bound to two pre-cleaved 50-mer Right Ends (Yuan et al., 2005). Although not directly related to mobile DNA, crystal structures have been determined of RNase H bound to DNA–RNA hybrids (Nowotny et al., 2005), and insights from these structures as they relate to DNA transposition have recently been reviewed (Nowotny, 2009). In addition, an EM study of the RAG1/2 complex bound to recombination signal sequence (RSS) DNA has recently been reported (Grundy et al., 2009).
The catalytic cores of some DNA transposases are interrupted by insertion domains
Interestingly, the region of the catalytic core domain between β5 and α4 may have additional significance in the reaction chemistry. As observed in the HIV-1 IN, ASV IN, and MuA transposase structures, the RNase H-like fold generally has a short (7–15 amino acids) loop connecting β5 and α4 that is either disordered or only weakly ordered. In contrast, the Tn5 transposase has a 96 amino acid long domain inserted between β5 and α4 (Davies et al., 1999). We refer to this domain, or any other domain that is inserted at this topological location in the standard RNase H-like core, as an “insertion domain”. In the case of Tn5, the insertion domain is mostly β-stranded, containing only one short two-turn α-helix and four β-strands that extend the central β-sheet of the catalytic domain. This insertion domain performs a crucial function as it forms a number of intricate interactions with the DNA hairpin that is generated when the 3′OH group of the transferred strand (created at the first cleavage step) attacks the phosphate of the non-transferred strand (Davies et al., 2000; Figure 1f ).
A particularly interesting residue, W298, is found within the Tn5 transposase insertion domain. This stacks with the second base of the non-transferred strand which protrudes (is “flipped out”) from the DNA helix (see Figures 6 and 7 in Steiniger-White et al., 2004). This facilitates formation of the tight, short loop hairpin at the transposon end. Importantly, in those crystal structures of transposases where second strand cleavage does not proceed through a hairpin intermediate (MuA, Mos1, and HIV-1 IN) there is no insertion domain between β5 and α4. This raises the intriguing possibility that its presence or absence might correlate with the mechanism of second strand processing.
More recently, another type of insertion domain has also been seen – inserted at the same topological location of the standard RNase H-like fold – in the crystal structure of the eukaryotic Hermes transposase (Hickman et al., 2005). The long Hermes insertion domain (residues 265–552, counted from the end of β5 to the beginning of α4) is entirely α-helical and therefore bears no resemblance to the insertion domain of the Tn5 transposase. Indeed, both the Tn5 and the Hermes transposase insertion domains are structurally unique. Although, like Tn5, Hermes also forms hairpins during transposition, the hairpin is generated on the flanking DNA ends rather than on the transposon ends (Zhou et al., 2004). This is because the Hermes transposase first catalyzes cleavage of the non-transferred strand to generate a 3′OH group on the DNA flank; this then attacks the terminal phosphodiester bond on the opposite (transferred) strand to create a flanking DNA hairpin and a 3′OH group at each end on the transferred strand (Figure 1g).
The Hermes insertion domain also contains a conserved Trp residue, W319, shown to be important for one or more steps before strand transfer (Hickman et al., 2005). It presumably acts to facilitate hairpin formation as does the Tn5 transposase W298 residue. Consistent with the interpretation that W319 is therefore likely to be catalytically important, it is located close to the DDE side chains of the active site.
Interestingly, RAG1 is predicted to contain a large insertion domain between the second and third acidic residues of its identified DDE motif (264 residues, counting from D708 to E972), and secondary structure prediction clearly indicates it is likely to be entirely α-helical (Zhou et al., 2004) as is that of Hermes. It also contains a conserved Trp, W893, and mutation generates a serious defect in V(D)J recombination; however a role in hairpin formation/stabilization has not been demonstrated (Grundy et al., 2007). Since there is no high resolution three-dimensional structure of RAG1, it is not known if its insertion domain is structurally similar to that of Hermes, although it is certainly smaller (264 amino acids separate the second D and the third E in RAG1, whereas the separation is 324 amino acids in Hermes).
The Hermes insertion domain also contributes to formation of the functional transposase oligomer, another contrast with the insertion domain of Tn5 which plays no apparent role in oligomerization. This additional functionality may partially explain why it is substantially larger than the 96-residue insertion domain of the Tn5 transposase. There is another complication with the reactions catalyzed by Hermes and the V(D)J recombination system, relative to that of Tn5, which may account for the substantial difference in sizes of the insertion domains. Whereas for the IS4 family member Tn10 – and therefore also very likely for Tn5 – both hairpin formation and the strand transfer reaction use the same 3′OH group of the transferred strand as a nucleophile (Figure 1f; Kennedy et al., 2000), the first cleavage of the Hermes and RAG1 reactions generates an 3′OH nucleophile on the non-transferred strand. This 3′OH group is used to generate the flanking end hairpin yet the nucleophile that is subsequently used for strand transfer is the 3′OH of the transferred strand created during hairpin formation (Figure 1g). The simplest way to imagine how this might happen is if the enzyme active site somehow switches from one DNA strand (that cleaved first) to the other (the transferred strand). This suggests either the need for major conformational rearrangements which are not necessary for the Tn5 transposase, or the use of multiple sites. It is quite possible that these mechanistic differences are reflected in the structural differences displayed by the different insertion domains.
While it is not an insertion domain, a structural motif of 23 amino acids is inserted between strands β1 and β2 in the standard RNase H-like fold in the Mos1 transposase. In the uncomplexed three-dimensional structure of Mos1, this segment is disordered (Richardson et al., 2009). On the other hand, in the structure of the Mos1 complex with cleaved transposon Right Ends (Richardson et al., 2009), it becomes ordered and appears to play a crucial role in transposase dimerization, making protein–protein interactions across the dimer interface and important protein–DNA contacts. The transposase dimer seen in the Mos1/DNA structure has both 3′ ends of two transferred strands in the two active sites, a configuration that appears to be stabilized to a large extent by these protein–protein and protein–DNA interactions. As this configuration captures a state in the reaction where the transferred strands have been cleaved (Figure 1d), the inserted segment therefore contributes to the proper execution of second strand processing, at least indirectly.
Some DNA transposases possess a YREK motif
In addition to the DDE/D motif, another notable sequence motif found in some dsDNA transposases is a Y-(2)-R-(3)-E-(6)-K motif, the so-called “YREK” motif originally identified in the IS4 family (Rezsöhazy et al., 1993). This motif is located on α4, where the E of the YREK motif is the same E as that of the catalytic DDE motif. As seen in the structure of the Tn5 transposase bound to DNA, α4 is wedged into the DNA minor groove immediately adjacent to the hairpin (see Figure 4A in Davies et al., 2000). In this location, the Y, R, and K side chains form a number of important contacts with the transposon end. Embedded in the YREK motif, just after R322, is residue W323 which is pushed into the DNA minor groove. It is likely that this interaction is crucial for hairpin formation as it may act to push the base to be flipped out whereas W298 appears to capture this base, probably by base stacking. It appears therefore that hairpin formation during Tn5 transposition is assisted by two Trp residues acting in a “push and pull” configuration (Bischerour and Chalmers, 2007). Interestingly, in the Tn10 transposase (also an IS4 family member) there is instead an M after the R of the YREK motif that may have a similar role in extruding the flipped-out base (Allingham et al., 2001; Bischerour and Chalmers, 2009).
Although it is tempting to generalize the need for two similarly placed Trp residues to all transposition systems that proceed through hairpin intermediates, it may be that the “push and pull” interactions observed for Tn5 system are only needed because the hairpin formed has a very short loop, i.e. the double-stranded break occurs at the very end of the transposon both for Tn5 and for Tn10. If the resulting hairpin has a longer and therefore presumably more flexible loop (more unpaired bases), flipping out a specific base to make room at the DNA end for the hairpin to form may not be as important as it is for the extremely short loop of Tn5 and Tn10.
While the YREK motif is a feature of the IS4 family and may be indicative of a mechanism that proceeds through a hairpin intermediate on the transposon end (but not on the flanking end), the K/R residue that follows the catalytic E/D by about seven residues is present in many other families that do not possess a YREK motif (Mahillon and Chandler, 1998). This K/R seems likely to be catalytically important, as demonstrated for HIV-1 IN where mutation of K159 (which follows the catalytic E152) leads to a loss of in vitro cleavage and strand transfer activities (Jenkins et al., 1997), and for the IS1 transposase, where the similarly located R198A mutation has a serious in vivo transposition defect (Ton-Hoang et al., 2004).
RNase H-like DNA transposases are modular proteins
The overall properties of RNase H-like transposases do not rely on the catalytic domain alone. These proteins always carry additional domains such as helix-turn-helix (HTH) or winged-helix domains, zinc-binding domains (mostly with undefined functions), and domains required for multimerization.
While the RNase H-like catalytic domain itself must have some DNA binding activity, this is often non-specific and/or weak. Rather, sequence-specific DNA binding (such as binding transposon or viral DNA ends) is carried out by sequence-specific DNA binding domains of various kinds, typically located upstream of the catalytic domain. In this sense, DDE/D transposases are always modular proteins, with different functions distributed between different domains. The sequence-specific DNA binding function that allows the transposase to locate the ends of its mobile element can reside in a single domain or be distributed among several domains. For example, MuA transposase recognizes its transposon ends using a winged helix DNA binding domain followed by an HTH domain (reviewed in Chaconas and Harshey, 2002; Rice, 2005); members of the mariner family use two HTH domains (reviewed in Plasterk et al., 1999; van Pouderoyen et al., 1997; Watkins et al., 2004; Richardson et al., 2009); and OrfAB, the transposase of IS911, uses a single HTH domain (Rousseau et al., 2002).
Among the repertoire of site-specific DNA binding domains, unusual α-helical, non-standard fold domains are sometimes present, as in the case of Tn5 (Davies et al., 2000). The eukaryotic Hermes transposase has an idiosyncratic intertwined helical domain (Hickman et al., 2005), and a similar domain has recently been observed in RAG1 (Yin et al., 2009). Retroviral integrases contain an α-helical Zn2+ binding domain upstream from the catalytic domain that is very similar to HTH domains, but its role in site-specific binding of viral ends is not yet clear (Craigie, 2001).
There is even greater diversity in domains downstream of the RNase H-like catalytic domain. Although in some cases, such as many of the hAT family eukaryotic DNA transposases, there are no additional domains, when present, C-terminal domains can be involved in a number of functions: non-specific DNA binding, such as binding target DNA (which is generally, but certainly not always, non-specific), or in multimerization or protein–protein interactions with other components of the transpososome. An interesting variation appears with the IS110 family where the RNase H-like catalytic domain appears at the amino terminus of the transposase and is followed by an all-helical C-terminal domain with no obvious indication as to its function. This unusual arrangement is accompanied by the rather unusual property of strictly sequence-specific integration (Higgins et al., 2009).
RNase H-like DNA transposases need to multimerize
Transposase molecules must form multimeric complexes within the transpososome, and the currently available experimental three-dimensional structures allow us some new insights into how this is achieved. It is remarkable that despite the obvious structural homology of the catalytic domains, the mode of transposase oligomerization is very divergent, as each of the systems for which structural data are available form oligomers in their own unique way. Some DNA transposases such as MuA and Tn5 are monomers when not bound to DNA, and oligomerize upon binding; for instance Tn5 forms a dimer while MuA becomes tetrameric. Others are multimeric on their own, and it is not clear that the multimerization state changes upon DNA binding. For example, there is evidence that the hAT superfamily member Hermes is a hexamer even without bound DNA (Hickman et al., 2005) and HIV-1 IN forms tetramers both with and without DNA bound (Bao et al., 2003; Ren et al., 2007; Hare et al., 2009; Michel et al., 2009) as does the purified P element transposase (Tang et al., 2007).
One clear example of a dedicated protein multimerization domain was identified in the IS3 family where the vast majority of the almost 500 members of this family exhibit a leucine-rich region predicted to form a coiled coil structure or leucine zipper (LZ) (Rousseau et al., 2002). The functionality of this region was experimentally tested for one family member, IS911 (Haren et al., 1998, 2000). Site-specific mutagenesis of key LZ residues based on analysis of the jun/fos system demonstrated that this region is required for formation of transposition intermediates as judged both by genetic and physical tests in vivo and by a standard in vitro transposition assay. The inability of these mutants to multimerize was confirmed directly by their inability to undergo co-immunoprecipitation with a tagged wild-type transposase derivative (Haren et al., 1998). These studies also revealed a second region of the protein required for correct multimerization. This region has no particular motifs to suggest how it might function directly in multimerization. Similar leucine-rich regions have been identified in other transposases including the regulatory KP element involved in regulation in P element transposition (Andrews and Gloor, 1995; Lee et al., 1996); IS1111 (Hoover et al., 1992) and other members of the IS110 family; the IS66 family (Gourbeyre et al., personal communication); and Tc1 (Ivics et al., 1996).
An unusual example of a multimerization domain appears in the Hermes transposase and RAG1 where an entirely α-helical domain forms a tightly intertwined dimer. This is the same domain that appears to function in site-specific DNA binding (Hickman et al., 2005; Yin et al., 2009). A region of the hAT transposases that had been previously implicated in dimerization (Michel et al., 2003), and which has infiltrated the Conserved Domains Database (Marchler-Bauer et al., 2009) as pfam 05699, does not appear to be involved in dimerization; rather, as the structure of Hermes revealed, this region meanders through the protein holding adjacent domains together (Hickman et al., 2005), a role that provides an alternative explanation for the original observation that disruption of this region leads to loss of multimerization.
In some cases, the formation of multimers may be the product of protein–protein interactions between several domains, so there may be no single dedicated multimerization domain. For instance, Mos1 uses one of its two N-terminal HTH DNA binding domains and a motif between strands β1 and β2 of the catalytic RNase H-like domain to dimerize (Richardson et al., 2009) which also facilitates DNA binding. On the other hand, DNA binding can also contribute to oligomerization, since transposon ends are often bound in “trans” such that a site-specific DNA binding domain of one protomer binds the transposon end that is processed by the catalytic domain of another protomer; in this way, the DNA acts as the “glue” holding (or contributing to holding) the oligomer together. This seems to be the case so far in all three-dimensionally resolved transposase–DNA complexes. For retroviral integrases, the catalytic domain itself is a dimerization domain (Dyda et al., 1994); however, in the formation of the functional tetramer, it is very likely that other protein–protein and protein–DNA interactions are also involved.
While distinct from transposase multimerization, transposases are sometimes intimately involved through protein–protein interactions with other proteins encoded by the transposon. Well-known examples are bacteriophage Mu and the Tn7 transposon where target immunity, the ability to avoid integrating into a DNA molecule already containing the transposon, is mediated by a transposon-encoded ATPase. This ATPase is a part of the assembled transpososome and interacts directly with the transposase (Baker et al., 1993; Wu and Chaconas, 1994; Craig, 2002; Ronning et al., 2004). V(D)J recombination also relies on a hetero-oligomer between RAG1 and RAG2 for function (Bailin et al., 1999).
Sequence expansion
Over the past few years, the number of prokaryotic and eukaryotic transposons which have been identified has grown tremendously. This is due largely to the extraordinary growth in sequence data deposited in the public databases. For example, there are at present about 1000 completed and publicly deposited prokaryotic genome sequences listed in the GOLD database (Liolios et al., 2008), the majority of which are poorly annotated for ISs and other mobile genetic elements. Attempts to collate sequences of transposons in a usable form have fallen to a very small number of researchers who have managed to obtain the resources to maintain databases. These include ISfinder (http://www-is.biotoul.fr) which provides a list of ISs identified in eubacteria and archaea (Siguier et al., 2006), and Repbase Update (http://www.girinst.org/repbase/index.html) which maintains sequences of large families and subfamilies of repetitive elements from eukaryotes (Jurka et al., 2005). A third database, ACLAME (http://aclame.ulb.ac.be/) collates a more general collection of prokaryotic mobile genetic elements (Leplae et al., 2004).
The ISfinder database (which is freely consultable and does not require registration) includes over 3000 bacterial ISs. When ISfinder was initiated, ISs were identified from experimental data (e.g. inactivation of a particular gene by insertion) and, while some ISs deposited are still identified in this way, the majority are now defined by homology searches between transposase enzymes and other characteristics of the IS such as genetic organization and the terminal inverted repeats characteristic of transposons with RNase H-like transposases. Thus, in general there is no demonstration that a given IS is active although this can be inferred from those which are present in multiple identical copies in one or several genomes, or by comparison with other closely related members of the family to which they belong. The same vexing problem with deducing whether an element is active also exists among the collated eukaryotic DNA transposon sequences.
Of prokarytic ISs, 25 families have been defined in ISfinder (Siguier et al., 2006) and 20 superfamilies of eukaryotic DNA transposons are currently listed on Repbase (Jurka et al., 2005). Recent reviews that provide useful summaries of the properties of prokaryote and archaea ISs and eukaryotic DNA transposons on a family-by-family basis include those by Mahillon and Chandler (1998) (and its update; see Chandler and Mahillon, 2002), Filée et al. (2007), Feschotte and Pritham (2007), and Wicker et al. (2007). We note in passing that the designation of “families” (for ISs) as opposed to “superfamilies” (among eukaryotes) appears entirely a matter of convention, rather than a statement regarding “superiority”.
To understand how these families and superfamilies are related to each other – evolutionarily, mechanistically, and structurally – would ideally involve integrating genetic, biochemical, and structural data with results obtained from computational biology, or bioinformatic, approaches. Unfortunately at present, despite the vast amounts of sequence data that are publicly available, only a few transposons and their transposases have been studied experimentally. Nevertheless, the results obtained from classical biochemical and structural approaches serve as the filter through which we are forced to make sense of the sequence data.
Do we now know the folds of the catalytic cores of all DNA transposases?
Of necessity, in the absence of any three-dimensional structural data for most transposition systems, transposase sequence alignments have been the main tool for analyzing these proteins in an effort to determine how they might be related to each other and to transposases of known structure. Using sequence alignments to identify the catalytic DDE/D residues and other conserved amino acids, among the prokaryotic ISs, 17 families have been annotated in ISfinder (Siguier et al., 2006) as containing a transposase with a DDE/D catalytic domain. Among the remaining eight families, the IS607 family possesses a serine transposase, and the IS91 and IS200/IS605 families are members of the HUH superfamily of nucleases, consistent with their known transposition mechanisms which use 5′ phosphotyrosine intermediates. This leaves five families currently unannotated.
Among the 20 superfamilies of eukaryotic transposases identified in Repbase (Jurka et al., 2005), six have been described only in Repbase (Mirage, Rehavkus, Nobosib, Kolobok, ISL2EU, and Chapaev – but see Panchin and Moroz, 2008), making it difficult to objectively assess their significance. Of these six superfamilies, some are annotated as encoding a transposase with a DDE catalytic core (Kolobok, ISL2EU, and Chapaev) whereas others are annotated as possessing a novel “cut and paste” transposase (for example, Rehavkus-1_DY). We eagerly await further description of these transposon super-families in order to evaluate the relevance or validity of these assignments.
Of the remaining 14 identified superfamilies, the Helitrons, Cryptons, and the Mavericks can be considered sui generis. The Helitron transposases (Kapitonov and Jurka, 2001) have Rep domains, suggesting that they are related to the prokaryotic IS91 family, and thus any active members are expected to transpose using a mechanism related to rolling-circle replication (Koonin and Ilyina, 1993; Mendiola et al., 1994). The few identified Cryptons (Goodwin et al., 2003) encode a protein similar to tyrosine recombinases, and therefore they have been proposed to constitute a rather unusual class of LTR retrotransposons (Poulter and Goodwin, 2005). At present it is unclear whether or not these elements are transposons or indeed if they are mobile. The Mavericks/Polintons are unusually large elements, generally encoding between four and nine proteins which include a protein-primed DNA polymerase, a retroviral-like integrase, a cysteine protease, and usually an ATPase, suggesting perhaps a novel – and potentially complex – transposition mechanism (Feschotte and Pritham, 2005; Kapitonov and Jurka, 2006; Pritham et al., 2007). Nevertheless, their integrase ORF clearly indicates the presence of an RNase H-like catalytic domain. Although unrelated, a similarly complex organization occurs in the IS66 (Han et al., 2001) and Tn7 (Parks and Peters, 2009) families.
Protein sequence alignments have been used in an attempt to place the remaining 11 eukaryotic super-families of DNA transposons within the known transposon classification groups. In this way, six superfamilies have been reported to have transposases that are homologous to bacterial IS transposases (summarized in Feschotte and Pritham, 2007; and Table 1): the transposases of the Tc1/mariners are related to those of IS630 (Doak et al., 1994) as are the Zator transposases (Bao et al., 2009); Mutator/MuDr transposases are related to those of IS256 (Eisen et al., 1994; Hua-Van and Capy, 2008); piggyBac to the IS4/5 family (Sarkar et al., 2003); Harbinger/PIF to IS5 (Kapitonov and Jurka, 1999; Zhang et al., 2001); Merlin to IS1016 (Feschotte, 2004); leaving only the En/Spm (CACTA), hAT, P element, Transib, and Sola superfamilies without presently identified prokaryotic equivalents. The crystal structures of the catalytic domain of Hermes, a representative hAT transposase, and of Mos1, a Tc1/mariner element, established that both possess an RNase H-like catalytic domain (Hickman et al., 2005; Richardson et al., 2006).
Table 1.
List of mobile elements whose transposases have been examined by secondary structure prediction programs.
| Family | Element (or protein) analyzed | Active or # copies in genome1 | From secondary structure, type of DDE/D motif2 | Relevant references3 |
|---|---|---|---|---|
| IS1 | IS1N | >40* | DD(24)E | * Nyman et al., 1981; Ohta et al., 2002, 2004; Siguier et al., 2009 |
| ISSto9 | 5 | DD(20)E | ||
| IS1595 | 1. ISPna2 | — | DD(36)N | Siguier et al., 2009 |
| 2. ISH4 | — | DD(36)E | Siguier et al., 2009 | |
| 3. IS1016C | — | DD(34)E | Siguier et al., 2009 | |
| 4. IS1595 | — | DD(35)N | Siguier et al., 2009 | |
| 5. ISSod11 | 13 | DD(34)H | Siguier et al., 2009 | |
| 6. ISNWi1 | — | DD(35)E | Siguier et al., 2009 | |
| 7. ISNha5 | — | DD(33)E | Siguier et al., 2009 | |
| Merlin: MERLIN1_SM | Consensus | DD(36)E | Feschotte, 2004 | |
| IS3 | IS911 | Active | DD(35)E | Polard and Chandler, 1995; Rousseau et al., 2002 |
| IS481 | IS481 | ~100* | DD(35)E | *Glare et al., 1990; Chandler and Mahillon, 2002 |
| IS4 | IS50R | Active | PDB ID: 1muh | Rezsöhazy et al., 1993; Davies et al., 2000 |
| DD(β-strand)E | ||||
| IS701 | IS701 | Active (15*) | DD(β-strand)E | *Mazel et al., 1991 |
| ISRso17 | 7 | |||
| ISH3 | ISC1359 | 5 | DD(β-strand)E | |
| ISC1439A | 13 | |||
| IS1634 | IS1634 | Active (~30*) | DD(β-strand)E | *Vilei et al., 1999 |
| ISMac5 | 7 | |||
| ISPlu4 | 7 | |||
| IS5 | IS903 | Active | DD(65)E | Derbyshire et al., 1987; Rezsöhazy et al., 1993; Tavakoli et al., 1997 |
| PIF/Harbinger: PIFa (Z. mays) | Active | DD(59)E | Zhang et al., 2001; Kapitonov and Jurka, 2004; Sinzelle et al., 2008 | |
| IS1182 | IS660 | 3 | DD(β-strand)E | Takami et al., 2001 |
| ISPsy6 | 14 | |||
| IS6 | IS6100 | Active | DD(34)E | Martin et al., 1990; Mahillon and Chandler, 1998 |
| IS21 | IS21 | Active | DD(45)E | Mahillon and Chandler, 1998; Berger and Haas, 2001 |
| IS30 | IS30 | Active | DD(33)E | Caspers et al., 1984; Mahillon and Chandler, 1998 |
| IS66 | IS679 | Active | DD(α-helical?)E | Han et al., 2001 |
| ISPsy5 | 33 | |||
| ISMac8 | 3 | |||
| IS110 | IS492 | Active | DEDD | Perkins-Balding et al., 1999; Buchner et al., 2005 |
| IS1111 | 20 | DEDD | ||
| IS256 | IS256 | Active | DD(α-helical)E | Mahillon and Chandler, 1998; Prudhomme et al., 2002 |
| MuDr/Foldback (Mutator) | Active | DD(α-helical)E | Eisen et al., 1994; Babu et al., 2006; Hua-Van and Capy, 2008 | |
| IS630 | ISY100 | Active | DD(34)E | Doak et al., 1994; Feng and Colloms, 2007 |
| Tc1/mariner: Mos1 (D. mauritiana) | Active | PDB ID: 2f7t | Plasterk et al., 1999; Richardson et al., 2006 | |
| DD(34)D | ||||
| Zator: Zator-1_HM | 36* | DD(43)E | *Bao et al., 2009 | |
| IS982 | ISPfu3 | 5 | DD(47)E | Mahillon and Chandler, 1998 |
| IS1380 | IS1380A | ~100* | DD(β-strand)E | *Takemura et al., 1991; Chandler and Mahillon, 2002 |
| piggyBac (T. ni) | Active | DD(β-strand)D | Cary et al., 1989; Sarkar et al., 2003; Mitra et al., 2008 | |
| ISAs1 | ISAzo3 | 7 | DD(β-strand)E/D? | |
| ISL3 | IS31831 | Active | DD(α-helical)E | Suzuki et al., 2006 |
| IS651 | 22 | |||
| Tn3 | Tn3 (E. coli) | Active | DD(α-helical?)E | Grindley, 2002 |
| hAT | Hermes (M. domestica) | Active | PDB ID: 2bw3 | Warren et al., 1994; Rubin et al., 2001; Hickman et al., 2005 |
| DD(α-helical)E insertion | ||||
| CACTA | CACTA1 (A. thaliana) | Active | DD(α-helical?)E/D? | Miura et al., 2001; DeMarco et al., 2006 |
| En/Spm ZM | ||||
| P | Drosophila | Active | ? | Rio, 2002 |
| Transib | Transib1_AG | Consensus | DD(α-helical)E | Kapitonov and Jurka, 2005; Chen and Li, 2008 |
| RAG1 (M. musculus) | Active | DD(α-helical)E | Kim et al., 1999; Landree et al., 1999; Lu et al., 2006 | |
| Sola | Sola3-3_HM | Multiple copies* | DD(40)E | *Bao et al., 2009 |
Information on the number of copies within the host genome was obtained from ISfinder or the reference indicated by the asterisk.
Where indicated, the secondary structure predicts an insertion domain between β5 and α4 with predominantly either β-strands or α-helices.
Relevant references include reviews or papers that report the results of secondary structure prediction, report sequence alignments or consensus sequences, identify the DDE/D catalytic residues, or demonstrate that the element is active. The association of certain eukaryotic superfamilies to specific IS families is as per Feschotte and Pritham (2005) and references therein.
How reliable are sequence alignments in predicting structure?
In the absence of structural and biochemical data for the vast majority of IS families and eukaryotic superfamilies, the question arises of how reliably sequence analysis can place all these transposases within the structural universe. Is it reasonable that primary sequence alone has established that the vast majority of dsDNA transposases possess an RNase H-like catalytic domain? Furthermore, where sequence alignments remain ambiguous, how likely is it that all the remaining unannotated ISs and eukaryotic DNA transposases are similarly structurally related?
One concern when relying solely on sequence alignments to identify a catalytic DDE/D motif is that there are many possible reasons why acidic residues might be conserved in proteins and the identification of three “does not necessarily a catalytic site make”. Prior to the deluge of DNA transposon sequences, the limited number of known eukaryotic elements and the lack of information about whether they were active, occasionally proved treacherous in predicting a DDE motif. For example, early efforts to identify the DDE triad in the hAT elements (Rubin et al., 2001; Michel et al., 2002) were hampered by the presence of an unrecognized insertion domain, leading investigators to search for relatively closely spaced Ds and Es, rather than allowing for a very large spacing between the D residues and the final E. This led to a brief foray into the possibility of a “DSE” catalytic motif (Bigot et al., 1996) before this provocative proposal was discarded (Zhou et al., 2004).
In other earlier work, efforts to force unexpectedly placed – or simply missing – conserved acidic residues into a DDE motif led to contortions such as the initial “putative noncanonical DDE catalytic site” of the Transib transposases (Kapitonov and Jurka, 2003) and the “working alignment of DD(35)E family members” which aligned TnsA of the Tn7 transposon with retroviral integrases and the MuA transposase (Sarnovsky et al., 1996). To the credit of the authors of these papers, the clear indications that something was not quite right led quickly to an amended alignment in the first case (Kapitonov and Jurka, 2005) and was the impetus in the second case to experimentally determine the three-dimensional structure of TnsA which was unexpectedly found to resemble type II restriction enzymes rather than RNase H, and therefore does not possess a DDE active site (Hickman et al., 2000).
Today, there are many readily available bioinformatic tools that have the potential to bridge the gap between transposase amino acid sequences and protein structure prediction. One level of analysis that can be straightforwardly applied is to determine if the predicted secondary structure is consistent with an RNase H-like fold. To ask this question is, in part, to revert to the longstanding problem of protein folding: how well can we predict or calculate the three-dimensional structure of a protein knowing its primary sequence (reviewed in Dill et al., 2007)? Homology modeling is generally not a useful approach to predicting structure if the level of sequence identity is below 20–25% (Cavasotto and Phatak, 2009), and it is clear that most transposases do not have significant sequence identity to those with known structures. At present, the most reliable secondary structure prediction programs are generally considered to be ~80% accurate, and the reliability of protein threading (the prediction of protein structure by a combination of amino acid sequence homology and predicted secondary structure) also continues to improve (Zhang, 2008; Kryshtafovych et al., 2009; Qu et al., 2009).
Below, we present an overview of the capacity of these bioinformatic tools to provide information on the structural relationship between different transposases with an identical chemistry but with diverse but related molecular mechanisms. We integrate previously reported results with those using web-based servers that implement secondary structure prediction methods including PSIPRED (Jones, 1999; Bryson et al., 2005) and the Jnet algorithm (Cole et al., 2008), in additional to the pGenTHREADER and pDomTHREADER methods for fold recognition (Lobley et al., 2009). This is applied to representative members of all the IS families either annotated as DDE proteins or uncommented upon in ISfinder (Siguier et al., 2006), and nine of the 11 described eukaryotic superfamilies not yet directly demonstrated through structure determination to possess an RNase H-like catalytic core. The representative members were chosen because they are either known to be active or exist in multiple copies (suggesting that they are active); they are shown in Table 1. The analysis was not performed for the IS4 family, hAT transposases, or the Tc1/mariner superfamilies as RNase H-like catalytic cores have been demonstrated directly by three-dimensional structure determination; for these, PDB IDs are shown in Table 1. Similarly, the analysis of secondary structure has already been reported for the Mutator/MULE (“Mutator-like elements”) superfamily (Babu et al., 2006; Hua-Van and Capy, 2008); nevertheless, all of these elements are included in Table 1. The assumption is made throughout that, since identifiable sequence homology allows different IS families or eukaryotic superfamilies to be defined, the result for one representative member is highly likely to hold true for all family or superfamily members.
Prokaryotic IS transposases
The predicted secondary structure of representative members of the 17 IS families annotated in ISFinder as DDE/D transposases and of the five currently unannotated families reveals that essentially all have predicted folds consistent with an RNase H-like fold.
The first criterion considered was a conserved order of secondary structure elements with the approximate pattern β1-β2-β3-α1-β4-α2/3-β5-α4-α5. Prediction is, of course, not perfect and not all of these elements are always predicted with high confidence: sometimes they appear unreasonably short, are not predicted at all, or are occasionally interrupted by short bursts of predicted random coil. Nevertheless, the broad outline of the fold can be discerned by visual inspection. This was confirmed by protein threading (Lobley et al., 2009) where, for 19 of the 22 IS families, representative members yielded a match – with a varying degree of certainty – to one or more of the known transposase or retroviral integrase structures in the ranked results (Table 2).
Table 2.
Protein threading results for transposases of representative members of IS families and eukaryotic superfamilies. These results are accessible by obtaining the amino acid sequence of the IS transposase from ISfinder (http://www-is.biotoul.fr) and submitting it to the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/). For the eukaryotic transposases, the sequences were obtained either from RepBase or GenBank. “Rank on List” indicates where in the overall list of threading results the relevant DDE/D protein appeared.
| IS/transposon | PDB ID for best relevant threading result | Rank on list | P value (probability of false positive) |
|---|---|---|---|
| IS1 (IS1N) | 1exq | #3 | 1 × 10−3 |
| IS1595 (IS1595) | 1cxq | #1 | 1 × 10−3 |
| IS3 (IS911) | 1cxq | #1 | 6 × 10−7 |
| IS481 (IS481) | 1cxq | #1 | 7 × 10−7 |
| IS4 | Not necessary (see 1mus) | NA | NA |
| IS701 (IS701) | 1mus | #1 | 2 × 10−7 |
| ISH3 (ISC1439A) | 1mus | #1 | 2 × 10−4 |
| IS1634 (IS1634) | 1mus | #1 | 2 × 10−6 |
| IS5 (IS903) | 1cxq | #2 | 0.002 |
| IS1182 (IS660) | 1mus | #3 | 0.003 |
| IS6 (IS6100) | 1cxq | #1 | 1 × 10−4 |
| IS21 (IS21) | 1exq | #1 | 1 × 10−4 |
| IS30 (IS30) | 1cxq | #1 | 1 × 10−5 |
| IS66 (IS679) | None | NA | NA |
| IS110 (IS492) | 1hjr | #1 | 3 × 10−4 |
| IS256 (IS256) | 1cxq | #17 | 0.009 |
| IS630 (ISY100) | 1cxq | #3 | 9 × 10−4 |
| IS982 (ISPfu3) | 1exq | #2 | 0.003 |
| IS1380 (IS1380A) | 1mus | #1 | 3 × 10−7 |
| ISAs1 (ISAzo3) | 1mus | #1 | 6 × 10−5 |
| ISL3 (IS31831) | 2bw3 | #12 | 0.007 |
| Tn3 (Tn3; V00613) | None | NA | NA |
| En/Spm/CACTA (AAC97237) | None | NA | NA |
| hAT | Not necessary (see 2bw3) | NA | NA |
| Harbinger/PIF (AF412282) | 1bco | #26 | 0.011 |
| Tc1/mariner | Not necessary (see 2f7t) | NA | NA |
| Merlin (MERLIN1_SM_1) | 1cxq | #1 | 9 × 10−4 |
| MuDR/Mutator | Babu et al., 2006; Hua-Van and Capy, 2008 | NA | NA |
| P element (AAT96009) | None | NA | NA |
| piggyBac (ABC88680) | 1mus | #1 | 2 × 10−4 |
| Transib (Transib1_AGp) | None | NA | NA |
| Sola (Sola3-3_HM) | 1cxq | #2 | 0.002 |
| Zator (Zator-1_HM) | None | NA | NA |
NA: not applicable, either because the structure of a representative member has been determined, there were no relevant threading results, or the results have been previously reported. The PDB IDs correspond to the following proteins: 1exq (HIV-1 integrase); 1cxq (ASV integrase); 1mus (Tn5 transposase); 1hjr (RuvC); 2bw3 (Hermes); and 1bco (MuA).
The second criterion required for an RNase H-like fold, dictated by the determined structures, is that the DD of the DDE/D motif must fall on or very close to predicted β1 and β4, and the E/D must be on or close to a predicted downstream α-helix. Also supportive was the location of a conserved K/R at a characteristic downstream distance from the catalytic E.
One notable variant in these predictions is whether or not a transposase carries an insertion domain between the second and third conserved acidic residue. On this basis, the IS families can be grouped into those without a predicted insertion domain (IS1, IS3, IS5, IS6, IS21, IS30, IS110, IS481, IS630, IS982, IS1595), those with a largely α-helical insertion domain (IS256, ISL3, and possibly IS66 and Tn3), and those with a mostly β-strand insertion domain (IS4, IS701, ISH3, IS1634, IS1182, IS1380, ISAs1).
The three IS families that do not yield a straightforward prediction of an RNase H-like catalytic domain are IS110, IS66, and Tn3. For IS110, the predicted fold of the catalytic domain corresponds to that of RuvC, another member of the RNase H superfamily (Ariyoshi et al., 1994), a resemblance has been noted previously (Buchner et al., 2005). Although RuvC has the same topological organization as the RNase H-like transposases, the arrangement of acidic residues, DEDD, is different with the third and fourth aspartic acid residues appearing one helical turn apart in a C-terminal helix that differs in orientation from either of the C-terminal helices of the RNase H-like fold (reviewed in Yang and Steitz, 1995). For both IS66 and Tn3, the secondary structure prediction results suggest RNase H-like folds with α-helical insertion domains, but this was not supported by the results of protein threading.
A curious observation has recently come to light regarding the IS1595 family whose members can be separated into seven distinct groups (Siguier et al., 2009). Of the seven identified groups (shown in Table 1), only four have the expected DDE triad; members of the other three appear to have either an Asn or His as the conserved third catalytic residue, and two of the three have yet another conserved E further towards the C terminus. For two of the three subfamilies with apparent DDN or DDH motifs, predicted secondary structure places the conserved downstream E on α5, rather than α4. As is evident from Figure 2, it seems unlikely that a third catalytic residue located on α5 would be able to form a two metal ion binding site with residues from β1 and β4. It would be extremely revealing to determine the three-dimensional structures of these variant transposases to establish whether they differ from canonical DDE enzymes by assembling an active site using a DDN or DDH motif (discussed in Nowotny, 2009), or if the structure of the catalytic domain changes relative to others to allow a predicted fifth α-helix carrying the canonical E residue to move into the proximity of β1 and β4. In this context, it is worth noting that the human SETMAR protein, a fusion between a SET domain with protein methyltransferase activity and a mariner transposase (Robertson and Zumpano, 1997), has measurable in vitro activity for many of the steps of transposition despite an active site that has a DD(34) N motif (Cordaux et al., 2006; Liu et al., 2007; Miskey et al., 2007).
Eukaryotic DNA transposases
The type of secondary structure analysis outlined above for the prokaryotic IS families has been reported for representative members of several eukaryotic superfamilies including the hATs (Zhou et al., 2004) and piggyBac (Mitra et al., 2008), and for the Mutator/MULE superfamily (Babu et al., 2006; Hua-Van and Capy, 2008). For Hermes and piggyBac, where transposition has been recapitulated in vitro with purified transposases, the secondary structure prediction demonstrated that conserved acidic residues that are crucial for activity are located precisely where expected, a result confirmed crystallographically for Hermes (Hickman et al., 2005). Analysis of the Mutator transposases (Hua-Van and Capy, 2008), which was more extensive than simple protein secondary structure prediction and protein threading by incorporation of aspects of 3D structure prediction, could serve as a paradigm for further studies on the entire range of eukaryotic transposon superfamiles.
The task of deciding whether a given DNA transposase has a predicted secondary structure consistent with an RNase H-like fold is far less straightforward for eukaryotic elements than for the bacterial ISs. Although in many cases, appropriately placed β-strands relative to a few helices suggest an RNase H-like fold, as shown in Table 2, the results are compelling only for members of the Harbinger/PIF, Merlin, Mutator, piggyBac, and Sola superfamilies.
The predicted secondary structure of piggyBac from Trichoplusia ni is completely consistent with a fold resembling that of the Tn5 transposase (onto which it can be threaded; Table 2) with several β-strands comprising an insertion domain between β5 and α4. The presence of an insertion domain is in accord with the observed large spacing between the second and third identified catalytically essential acidic residues of the DDD motif, D346 and D447 (Mitra et al., 2008). Although the catalytic mechanism of piggyBac proceeds through hairpin intermediates on the transposon ends as does the Tn5 system, there is as yet no evidence for two Trp residues partaking in a “push-and-pull” mechanism like Tn5 nor is there a conserved YREK motif. This may well be related to the possibility that a substantially larger loop at the tip of the hairpin is formed on the piggyBac ends, requiring less distortion to form than does that of the short loop generated during Tn5 transposition and therefore requiring less assistance from the transposase.
For the CACTA transposases, a primary sequence alignment revealed regions of conservation that suggested the possible location of the two aspartate residues of a DDE/D motif (DeMarco et al., 2006). These are indeed located on predicted β-strands that fall in a pattern that strongly suggests a classical RNase H-like fold. Although DeMarco et al. (2006) also suggested a possible candidate for the third acidic residue of a DDE/D motif located between the first two Asp residues, it seems far more likely that the CACTA transposases possess an α-helical insertion domain and that the final E is located much further towards the C-terminus.
It was proposed some time ago that the V(D)J recombination system, central to the adaptive immune system of jawed vertebrates, likely evolved from an ancient mobile element (Sakano et al., 1979; reviewed in Jones and Gellert, 2004). The obvious conceptual link between DNA transposition and V(D)J recombination was tightened by the observation that the RAG proteins can carry out transpositional strand transfer, leading to 5-bp target site duplications upon insertion (Hiom et al., 1998). An exciting recent development has been the identification of the Transib transposons as the likely ancestors (Kapitonov and Jurka, 2005). Not only is there significant amino acid sequence similarity between the RAG1 core domain and that of the Transibs, but the resemblance between Transib TIRs and V(D)J recombination signal sequences (RSS) is particularly persuasive.
The secondary structure predicted for the Transibs and RAG1 (Kim et al., 1999; Lu et al., 2006) concurs with previous conclusions reached from mutational analysis and sequence alignments (Kim et al., 1999; Landree et al., 1999; Kapitonov and Jurka, 2005) that these proteins likely have catalytic domains with an RNase H-like fold, although this could not be confirmed by protein threading (Table 2). Furthermore, members of the Transib superfamily are predicted to have an essentially-all α-helical insertion domain as predicted for RAG1 (Lu et al., 2006). The presence of an insertion domain is consistent with the reported large spacing (206–214 amino acids) among the Transibs between the second D and E residues proposed to serve as the catalytic residues (Kapitonov and Jurka, 2005).
One curious conserved sequence feature of the Transibs and RAG1 is a CxxC motif (RAG1 residues 727–730 in Mus musculus). In one alignment of the insertion domains (Kapitonov and Jurka, 2005), these would occur immediately following β5 of the RNase H-like fold. Curiously, a conserved and similarly placed CxxH sequence also occurs in the hAT transposases (Zhou et al., 2004); the Hermes structure shows that these residues indeed follow β5 and are buried amino acids, at least in the apo-structure (Hickman et al., 2005). A CxxC motif is found in essentially the same place in the predicted RNase H-like fold in CACTA transposases (DeMarco et al., 2006) and a CxxH motif in the MULE transposases, both of which are predicted to have large α-helical insertion domains (Babu et al., 2006; Hua-Van and Capy, 2008). (In the “rough alignment” of Lu et al. (2006), the RAG1 CxxC motif is aligned slightly differently, placing these residues between predicted strands β4 and β5 of the catalytic core.)
The RAG1 CxxC motif has been proposed to be part of a C2H2 zinc finger (Rodgers et al., 1996), and would represent an intriguing structural insertion-within-aninsertion. For the Transib and hAT transposases, nearby conserved histidine residues have not been identified, suggesting that their CxxC/H residues might not participate in assembly of a similar Zn2+-binding site.
For transposases of the newly identified Sola and Zator superfamilies (Bao et al., 2009), the predicted catalytic core secondary structures are consistent with the proposal that these are DDE/D transposases. Among the three groups of Sola transposases, the Sola3 group bears some limited sequence resemblance to piggyBac transposases (Bao et al., 2009). However, Sola3 transposases do not have a predicted insertion domain between the identified second and third conserved acidic residue, and the alignment presented in Bao et al. (2009) aligns the third D of the Sola3 DDD motif with a D in piggyBac that was not one identified as a catalytic residue by mutational analysis (Mitra et al., 2008). The curious parallel that Sola3 elements integrate specifically into TTAA target sites as does piggyBac (Cary et al., 1989) suggests that the two types of transposases have found different structural solutions to achieve the same outcome, at least as they relate to the catalytic domain.
Finally, the structural aspects of the P element superfamily transposases have been, and remain, a mystery and secondary structure analysis sheds no new light on the issue. The P element transposases have always stood slightly apart from other DNA transposases due to unusual features of transposition such as cuts at the end of the transferred strand and within the non-transferred strand that are staggered by 17 bases, and a requirement for GTP (reviewed in Rio, 2002). Acidic residues that have been determined to be catalytically important are spaced as D(83)D(2)E(13)D, which – regardless of how they are arranged – cannot conform to the expected location of catalytic residues on an RNase H fold.
It appears probable that the eukaryotic DNA transposase superfamilies, like those of the prokaryotic transposons, can be grouped into those without an insertion domain (Tc1/mariner, Merlin, Sola, Zator, Harbinger/PIF), those with a largely α-helical insertion domain (hAT, Mutator, Transib, and probably CACTA), and those with a mostly β-strand insertion domain (piggyBac); the protein fold of the catalytic domain of the P element transposases remains unknown.
Conclusion
An exciting consequence of studies involving DNA transposons has been their development as tools for manipulating and modifying genomes. The application of prokaroytic transposons to genetically modify prokaryotic organisms is a well-developed technology. Startling advances have recently been made in higher organisms using eukaryotic transposons such as Sleeping Beauty, a resurrected salmonid Tc1/mariner element (Ivics et al., 1997; 2009); piggyBac from the moth Trichoplusia ni (Cary et al., 1989; Wu et al., 2006); Mos1, a Tc1/mariner element from Drosophila mauritiana (Medhora et al., 1991); and Tol2, a hAT transposon from the medaka fish (Koga et al., 1996; Kawakami, 2007). Just a few recent examples of the types of applications include using piggyBac to introduce transcription factors into mouse and human stem cells to convert them to a pluripotent state – and then to subsequently excise the exogenous genes (Woltjen et al., 2009); using Sleeping Beauty in a genomic screen to randomly disrupt genes in mice to identify genes and pathways responsible for cancer induction (Dupuy et al., 2005); and cleverly exploiting the tendency of Sleeping Beauty to hop locally to investigate regions (within 1.5 Mb of the donor site) that may have cis-regulatory effects on nearby genes (Kokubu et al., 2009).
It is astonishing to realize that, in spite of much effort in obtaining high resolution three-dimensional structures of prokaryotic DNA transposases likely to have RNase H-like folds, that only the Tn5 transposase and the catalytic subdomain of that of bacteriophage MuA have been solved. Similarly, among the eukaryotic transposases, there are only two structures that confirm that representative members of the Tc1/mariner family and the hAT transposases are members of this larger RNase H-like structural superfamily.
The notion that all the IS families – with the exceptions of IS607, IS91 and IS200/IS605 – are likely on the basis of secondary structure prediction to have RNase H-like folds is not a particularly surprising conclusion. It is, perhaps, precisely what was to be expected. However, it would be fascinating to determine if the predicted presence – and type – of insertion domain correlates with a particular mechanistic aspect of transposition. To date, though, for many of these IS families the necessary biological information (in vivo and in vitro) is missing.
Among the eukaryotic DNA transposases, secondary structure prediction appears to support the notion that essentially all of the nine superfamilies discussed above do possess catalytic domains with RNase H-like folds. Nevertheless, in the absence of biochemical information on the catalytic relevance of conserved acidic residues – or even data indicating which elements within some superfamilies are active – the evidence is more compelling for some than for others. The jury remains out on the P element. Is it truly the structural outlier, as was eventually established for TnsA of the Tn7 transposon, or does an RNase H-like fold lie somehow encrypted within its primary sequence?
As shown in Figure 1, the known pathways by which dsDNA transposons move from one location to another are wonderfully varied. So are the ever-expanding families and superfamilies of IS and transposon sequences. To conceptually link primary sequence to function will clearly require more experimental structural data; without this, the roles of identifiable transposase features such as insertion domains or multimerization domains or conserved sequence motifs remain a matter of speculation, conjecture, and imagination. In turn, the ability to determine high-resolution crystal structures of transposases and their complexes with DNA rests on the shoulders of in vitro and in vivo studies on these myriad transposition systems. In many cases, obtaining experimental results has been difficult due to the often inherently recalcitrant biochemical and biophysical properties of the transposases themselves. One hopes that the availability of vastly expanded sequence information will yield candidates whose experimental study will become possible. The challenges await!
Acknowledgments
We thank Patricia Siguier for supplying alignments of the IS1595 subfamilies and Julia Richardson for graciously providing information on the Mos1/DNA complex prior to publication. We also thank Bob Craigie and Andrea Regier Voth for helpful comments on the manuscript. At the NIH, this work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases. In France, this work was supported by intramural funding from the CNRS and by ANR grant MOBIGEN both to M.C.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Allingham JS, Wardle SJ, Haniford DB. Determinants for hairpin formation in Tn10 transposition. EMBO J. 2001;20:2931–2942. doi: 10.1093/emboj/20.11.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JD, Gloor GB. A role for the KP leucine zipper in regulating P element transposition in Drosophila melanogaster. Genet. 1995;141:587–594. doi: 10.1093/genetics/141.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyoshi M, Vassylyev DG, Iwasaki H, Nakamura H, Shinagawa H, Morikawa K. Atomic structure of the RuvC resolvase: A Holliday junction-specific endonuclease from E. coli. Cell. 1994;78:1063–1072. doi: 10.1016/0092-8674(94)90280-1. [DOI] [PubMed] [Google Scholar]
- Babu MM, Iyer LM, Balaji S, Aravind L. The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucl Acids Res. 2006;34:6505–6520. doi: 10.1093/nar/gkl888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailin T, Mo XM, Sadofsky MJ. A RAG1 and RAG2 tetramer complex is active in cleavage in V(D)J recombination. Mol Cell Biol. 1999;19:4664–4671. doi: 10.1128/mcb.19.7.4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Mizuuchi M, Savilahti H, Mizuuchi K. Division of labor among monomers within the Mu transposase tetramer. Cell. 1993;74:723–733. doi: 10.1016/0092-8674(93)90519-v. [DOI] [PubMed] [Google Scholar]
- Bao KK, Wang H, Miller JK, Erie DA, Skalka AM, Wong I. Functional oligomeric state of avian sarcoma virus integrase. J Biol Chem. 2003;278:1323–1327. doi: 10.1074/jbc.C200550200. [DOI] [PubMed] [Google Scholar]
- Bao W, Jurka MG, Kapitonov VV, Jurka J. New superfamilies of eukaryotic DNA transposons and their internal divisions. Mol Biol Evol. 2009;26:983–993. doi: 10.1093/molbev/msp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabas O, Ronning DR, Guynet C, Hickman AB, Ton-Hoang B, Chandler M, Dyda F. Mechanism of IS200/IS605 family transposases: Activation and transposon-directed target site selection. Cell. 2008;132:208–220. doi: 10.1016/j.cell.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B, Haas D. Transposase and cointegrase: specialized transposition proteins of the bacterial insertion sequence IS21 and related elements. Cell Mol Life Sci. 2001;58:403–419. doi: 10.1007/PL00000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigot Y, Auge-Gouillou C, Periquet G. Computer analyses reveal a hobo-like element in the nematode Caenorhabditis elegans, which presents a conserved transposase domain common with the Tc1-Mariner transposon family. Gene. 1996;174:265–271. doi: 10.1016/0378-1119(96)00092-3. [DOI] [PubMed] [Google Scholar]
- Bischerour J, Chalmers R. Base-flipping dynamics in a DNA hairpin processing reaction. Nucleic Acids Res. 2007;35:2584–2595. doi: 10.1093/nar/gkm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischerour J, Chalmers R. Base Flipping in Tn10 Transposition: An active flip and capture mechanism. PLoS ONE. 2009;4:e6201. doi: 10.1371/journal.pone.0006201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucl Acids Res. 2005;33:W36–W38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner JM, Robertson AE, Poynter DJ, Denniston SS, Karls AC. Piv site-specific invertase requires a DEDD motif analogous to the catalytic center of the RuvC Holliday junction resolvases. J Bacteriol. 2005;187:3431–3437. doi: 10.1128/JB.187.10.3431-3437.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary LC, Goebel M, Corsaro BG, Wang HG, Rosen E, Fraser MJ. Transposon mutagenesis of baculoviruses: Analysis of Trichoplusia ni Transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virol. 1989;172:156–169. doi: 10.1016/0042-6822(89)90117-7. [DOI] [PubMed] [Google Scholar]
- Caspers P, Dalrymple B, Iida S, Arber W. IS30, a new insertion sequence of Escherichia coli K12. Mol Gen Genet. 1984;196:68–73. doi: 10.1007/BF00334094. [DOI] [PubMed] [Google Scholar]
- Cavasotto CN, Phatak SS. Homology modeling in drug discover: current trends and applications. Drug Disc Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Chaconas G, Harshey RM. Transposition of phage Mu DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 384–402. [Google Scholar]
- Chandler M, Mahillon J. Insertion sequences revisited. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 305–366. [Google Scholar]
- Chen S, Li X. Molecular characterization of the first intact Transib transposon from Helicoverpa zea. Gene. 2008;408:51–63. doi: 10.1016/j.gene.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucl Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Udit S, Batzer MA, Feschotte C. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc Natl Acad Sci USA. 2006;103:8101–8106. doi: 10.1073/pnas.0601161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig NL. Tn7. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 423–456. [Google Scholar]
- Craigie R. HIV integrase, a brief overview from chemistry to therapeutics. J Biol Chem. 2001;276:23213–23216. doi: 10.1074/jbc.R100027200. [DOI] [PubMed] [Google Scholar]
- Curcio MJ, Derbyshire KM. The outs and ins of transposition: from Mu to Kangaroo. Nat Rev Mol Cell Biol. 2003;4:865–877. doi: 10.1038/nrm1241. [DOI] [PubMed] [Google Scholar]
- Davies DR, Braam LM, Reznikoff WS, Rayment I. The three-dimensional structure of a Tn5 transposase-related protein determined to 2.9-Å resolution. J Biol Chem. 1999;274:11904–11913. doi: 10.1074/jbc.274.17.11904. [DOI] [PubMed] [Google Scholar]
- Davies DR, Goryshin IY, Reznikoff WS, Rayment I. Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science. 2000;289:77–85. doi: 10.1126/science.289.5476.77. [DOI] [PubMed] [Google Scholar]
- DeMarco R, Venancio TM, Verjovski-Almeida S. SmTRC1, a novel Schistosoma mansoni DNA transposon, discloses new families of animal and fungi transposons belonging to the CACTA superfamily. BMC Evol Biol. 2006;6:89. doi: 10.1186/1471-2148-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire KM, Hwang L, Grindley ND. Genetic analysis of the interaction of the insertion sequence IS903 transposase with its terminal inverted repeats. Proc Natl Acad Sci USA. 1987;84:8049–8053. doi: 10.1073/pnas.84.22.8049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill KA, Ozkan SB, Weikl TR, Chodera JD, Voelz VA. The protein folding problem: when will it be solved? Curr Opin Struct Biol. 2007;17:342–346. doi: 10.1016/j.sbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Doak TG, Doerder FP, Jahn CL, Herrick G. A proposed superfamily of transposase genes: Transposon-like elements in ciliated protozoa and a common “D35E” motif. Proc Natl Acad Sci USA. 1994;91:942–946. doi: 10.1073/pnas.91.3.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon systems. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- Eisen JA, Benito M, Walbot V. Sequence similarity of putative transposases links the maize Mutator autonomous element and a group of bacterial insertion sequences. Nucl Acids Res. 1994;22:2634–2636. doi: 10.1093/nar/22.13.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayet O, Ramond P, Polard P, Prère MF, Chandler M. Functional similarities between retroviruses and the IS3 family of bacterial insertion sequences? Mol Microbiol. 1990;4:1771–1777. doi: 10.1111/j.1365-2958.1990.tb00555.x. [DOI] [PubMed] [Google Scholar]
- Feng X, Colloms SD. In vitro transposition of ISY100, a bacterial insertion sequence belonging to the Tc1/mariner family. Mol Microbiol. 2007;65:1432–1443. doi: 10.1111/j.1365-2958.2007.05842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C. Merlin, a new superfamily of DNA transposons identified in diverse animal genomes and related to bacterial IS1016 insertion sequences. Mol Biol Evol. 2004;21:1769–1780. doi: 10.1093/molbev/msh188. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. Non-mammalian c-integrases are encoded by giant transposable elements. Trends Genet. 2005;21:551–552. doi: 10.1016/j.tig.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filée J, Siguier P, Chandler M. Insertion sequence diversity in archaea. Microbiol Mol Biol Rev. 2007;71:121–157. doi: 10.1128/MMBR.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, Herschlag D, Brown PO. Stereospecificity of reactions catalyzed by HIV-1 integrase. J Biol Chem. 1999;274:33480–33487. doi: 10.1074/jbc.274.47.33480. [DOI] [PubMed] [Google Scholar]
- Glare EM, Paton JC, Premier RR, Lawrence AJ, Nisbet IT. Analysis of a repetitive DNA sequence from Bordetella pertussis and its application to the diagnosis of pertussis using the polymerase chain reaction. J Clin Microbiol. 1990;28:1982–1987. doi: 10.1128/jcm.28.9.1982-1987.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin TJD, Butler MI, Poulter RTM. Cryptons: a group of tyrosine-recombinase-encoding DNA transposons from pathogenic fungi. Microbiol. 2003;149:3099–3109. doi: 10.1099/mic.0.26529-0. [DOI] [PubMed] [Google Scholar]
- Grindley NDF. The movement of Tn3-like elements: Transposition and cointegrate resolution. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 272–302. [Google Scholar]
- Grindley NDF, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- Grundy GJ, Hesse JE, Gellert M. Requirements for DNA hairpin formation by RAG1/2. Proc Natl Acad Sci USA. 2007;104:3078–3083. doi: 10.1073/pnas.0611293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy GJ, Ramón-Maiques S, Dimitriadis EK, Kotova S, Biertümpfel C, Heymann JB, Steven AC, Gellert M, Yang W. Initial stagaes of V(D)J recombination: The organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol Cell. 2009;35:217–227. doi: 10.1016/j.molcel.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guynet C, Hickman AB, Barabas O, Dyda F, Chandler M, Ton-Hoang B. In vitro reconstitution of a single-stranded transposition mechanism of IS608. Mol Cell. 2008;29:302–312. doi: 10.1016/j.molcel.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Han CG, Shiga Y, Tobe T, Sasakawa C, Ohtsubo E. Structural and functional characterization of IS679 and IS66-family elements. J Bacteriol. 2001;183:4296–4304. doi: 10.1128/JB.183.14.4296-4304.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniford DB. Transpososome dynamics and regulation in Tn10 transposition. Crit Rev Biochem Mol Biol. 2006;41:407–424. doi: 10.1080/10409230600987415. [DOI] [PubMed] [Google Scholar]
- Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, Cherepanov P. Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009;5:e1000515. doi: 10.1371/journal.ppat.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haren L, Polard P, Ton-Hoang B, Chandler M. Multiple oligomerisation domains in the IS911 transposase: a leucine zipper motif is essential for activity. J Mol Biol. 1998;283:29–41. doi: 10.1006/jmbi.1998.2053. [DOI] [PubMed] [Google Scholar]
- Haren L, Normand C, Polard P, Alazard R, Chandler M. IS911 transposition is regulated by protein–protein interactions via a leucine zipper motif. J Mol Biol. 2000;296:757–768. doi: 10.1006/jmbi.1999.3485. [DOI] [PubMed] [Google Scholar]
- Hickman AB, Li Y, Mathew SV, May EW, Craig NL, Dyda F. Unexpected structural diversity in DNA recombination: the restriction endonuclease connection. Mol Cell. 2000;5:1025–1034. doi: 10.1016/s1097-2765(00)80267-1. [DOI] [PubMed] [Google Scholar]
- Hickman AB, Perez ZN, Zhou L, Musingarimi P, Ghirlando R, Hinshaw JE, Craig NL, Dyda F. Molecular architecture of a eukaryotic DNA transposase. Nat Struct Mol Biol. 2005;12:715–721. doi: 10.1038/nsmb970. [DOI] [PubMed] [Google Scholar]
- Higgins BP, Popkowski AC, Caruana PR, Karls AC. Site-specific insertion of IS492 in Pseudoalteromonas atlantica. J Bacteriol. 2009;191:6408–6414. doi: 10.1128/JB.00771-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- Hoover TA, Vodkin MH, Williams JC. A Coxiella burnetii repeated DNA element resembling a bacterial insertion sequence. J Bacteriol. 1992;174:5540–5548. doi: 10.1128/jb.174.17.5540-5548.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua-Van A, Capy P. Analysis of the DDE motif in the Mutator superfamily. J Mol Evol. 2008;67:670–681. doi: 10.1007/s00239-008-9178-1. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Izsvák Z, Minter A, Hackett PB. Identification of functional domains and evolution of Tc1-like transposable elements. Proc Natl Acad Sci USA. 1996;93:5008–5013. doi: 10.1073/pnas.93.10.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Li MA, Mátés L, Boeke JD, Nagy A, Bradley A, Izsvák Z. Transposon-mediated genome manipulation in vertebrates. Nature Meth. 2009;6:415–422. doi: 10.1038/nmeth.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009;276:2926–2946. doi: 10.1111/j.1742-4658.2009.07009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TM, Esposito D, Engelman A, Craigie R. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Jones JM, Gellert M. The taming of a transposon: V(D) J recombination and the immune system. Immunol Rev. 2004;200:233–248. doi: 10.1111/j.0105-2896.2004.00168.x. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Molecular paleontology of transposable elements from Arabidopsis thaliana. Genetica. 1999;107:27–37. [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA. 2001;98:8714–8719. doi: 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci USA. 2003;100:6569–6574. doi: 10.1073/pnas.0732024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Harbinger transposons and an ancient HARBI1 gene derived from a transposase. DNA Cell Biol. 2004;23:311–324. doi: 10.1089/104454904323090949. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci USA. 2006;103:4540–4545. doi: 10.1073/pnas.0600833103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. Tol2: a versatile gene transfer vector in vertebrates. Genome Biol. 2007;8(Suppl):57. doi: 10.1186/gb-2007-8-s1-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AK, Haniford DB, Mizuuchi K. Single active site catalysis of the successive phosphoryl transfer steps by DNA transposases: insights from phosphorothioate stereoselectivity. Cell. 2000;101:295–305. doi: 10.1016/s0092-8674(00)80839-9. [DOI] [PubMed] [Google Scholar]
- Kim DR, Dai Y, Mundy CL, Yang W, Oettinger MA. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 1999;13:3070–3080. doi: 10.1101/gad.13.23.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga A, Suzuki M, Inagaki H, Bessho Y, Hori H. Transposable element in fish. Nature. 1996;383:30. doi: 10.1038/383030a0. [DOI] [PubMed] [Google Scholar]
- Kokubu C, Horie K, Abe K, Ikeda R, Mizuno S, Uno Y, Ogiwara S, Ohtsuka M, Isotani A, Okabe M, Imai K, Takeda J. A transposon-based chromosomal engineering method to survey a large cis-regulatory landscape in mice. Nature Genet. 2009;41:946–952. doi: 10.1038/ng.397. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Ilyina TV. Computer-assisted dissection of rolling circle DNA-replication. Biosyst. 1993;30:241–268. doi: 10.1016/0303-2647(93)90074-m. [DOI] [PubMed] [Google Scholar]
- Kryshtafovych A, Fidelis K, Moult J. CASP8 results in context of previous experiments. Proteins: Struct Funct Bioinform. 2009;77 (Suppl 9):217–228. doi: 10.1002/prot.22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landree MA, Wibbenmeyer JA, Roth DB. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev. 1999;13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Mul YM, Rio DC. The Drosophila P-element KP repressor protein dimerizes and interacts with multiple sites on P-element DNA. Mol Cell Biol. 1996;16:5616–5622. doi: 10.1128/mcb.16.10.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Hebrant A, Wodak SJ, Toussaint A. ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 2004;32:D45–D49. doi: 10.1093/nar/gkh084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36:D475–D479. doi: 10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Bischerour J, Siddique A, Buisine N, Bigot Y, Chalmers R. The human SETMAR protein preserves most of the activities of the ancestral Hsmar1 transposase. Mol Cell Biol. 2007;27:1125–1132. doi: 10.1128/MCB.01899-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobley A, Sadowski MI, Jones DT. pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinform. 2009;25:1761–1767. doi: 10.1093/bioinformatics/btp302. [DOI] [PubMed] [Google Scholar]
- Lu CP, Sandoval H, Brandt VL, Rice PA, Roth DB. Amino acid residues in Rag1 crucial for DNA hairpin formation. Nat Struct Mol Biol. 2006;13:1010–1015. doi: 10.1038/nsmb1154. [DOI] [PubMed] [Google Scholar]
- Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Timm J, Rauzier J, Gomez-Lus R, Davies J, Gicquel B. Transposition of an antibiotic resistance element in mycobacteria. Nature. 1990;345:739–743. doi: 10.1038/345739a0. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH. CDD: specific functional annotation with the Conserved Domain Database. Nucl Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel D, Bernard C, Schwarz R, Castets AM, Houmard J, Tandeau de Marsac N. Characterization of two insertion sequences, IS701 and IS702, from the cyanobacterium Calothrix species PCC 7601. Mol Microbiol. 1991;5:2165–2170. doi: 10.1111/j.1365-2958.1991.tb02146.x. [DOI] [PubMed] [Google Scholar]
- Medhora M, Marayuma K, Hartl DL. Molecular and functional analysis of the mariner mutator element Mos1 in Drosophila. Genet. 1991;128:311–318. doi: 10.1093/genetics/128.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiola MV, Bernales I, de la Cruz F. Differential roles of the transposon termini in IS91 transposition. Proc Natl Acad Sci USA. 1994;91:1922–1926. doi: 10.1073/pnas.91.5.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Crucifix C, Granger F, Eiler S, Mouscadet JF, Korolev S, Agapkina J, Ziganshin R, Gottikh M, Nazabal A, Emiliani S, Benarous R, Moras D, Schultz P, Ruff M. Structural basis for HIV-1 DNA integration in the human genome, role of the LEDGF/P75 cofactor. EMBO J. 2009;28:980–991. doi: 10.1038/emboj.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel K, O’Brochta DA, Atkinson PW. Does the proposed DSE motif form the active center in the Hermes transposase? Gene. 2002;298:141–146. doi: 10.1016/s0378-1119(02)00981-2. [DOI] [PubMed] [Google Scholar]
- Michel K, O’Brochta DA, Atkinson PW. The C-terminus of the Hermes transposase contains a protein multimerization domain. Insect Biochem Mol Biol. 2003;33:959–970. doi: 10.1016/s0965-1748(03)00102-4. [DOI] [PubMed] [Google Scholar]
- Miskey C, Papp B, Mátés L, Sinzelle L, Keller H, Izsvák Z, Ivics Z. The ancient mariner sails again: Transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol. 2007;27:4589–4600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Fain-Thornton J, Craig NL. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008;27:1097–1109. doi: 10.1038/emboj.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;411:212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Transpositional recombination: Mechanistic insights from studies of Mu and other elements. Annu Rev Biochem. 1992;61:1011–1051. doi: 10.1146/annurev.bi.61.070192.005051. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cell. 1997;2:1–12. doi: 10.1046/j.1365-2443.1997.970297.x. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K, Adzuma K. Inversion of the phosphate chirality at the target sites of Mu DNA strand transfer: evidence for a one step transesterification mechanism. Cell. 1991;66:129–140. doi: 10.1016/0092-8674(91)90145-o. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K, Baker TA. Chemical mechanisms for mobilizing DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 12–23. [Google Scholar]
- Nowotny M. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 2009;10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: Substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Nyman K, Nakamura K, Ohtsubo H, Ohtsubo E. Distribution of the insertion sequence IS1 in Gram-negative bacteria. Nature. 1981;289:609–612. doi: 10.1038/289609a0. [DOI] [PubMed] [Google Scholar]
- Ohta S, Tsuchida K, Choi S, Sekine Y, Shiga Y, Ohtsubo E. Presence of a characteristic D-D-E motif in IS1 transposase. J Bacteriol. 2002;184:6146–6154. doi: 10.1128/JB.184.22.6146-6154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S, Yoshimura E, Ohtsubo E. Involvement of two domains with helix-turn-helix and zinc finger motifs in the binding of IS1 transposase to terminal inverted repeats. Mol Microbiol. 2004;53:193–202. doi: 10.1111/j.1365-2958.2004.04103.x. [DOI] [PubMed] [Google Scholar]
- Panchin Y, Moroz LL. Molluscan mobile elements similar to the vertebrate Recombination-Activating Genes. Biochem Biophys Res Commun. 2008;369:818–823. doi: 10.1016/j.bbrc.2008.02.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AR, Peters JE. Tn7 elements: Engendering diversity from chromosomes to episomes. Plasmid. 2009;61:1–14. doi: 10.1016/j.plasmid.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins-Balding D, Duval-Valentin G, Glasgow AC. Excision of IS492 requires flanking target sequences and results in circle formation in Pseudoalteromonas atlantica. J Bacteriol. 1999;181:4937–4948. doi: 10.1128/jb.181.16.4937-4948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RHA, Izsvák Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999;15:326–332. doi: 10.1016/s0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- Polard P, Chandler M. Bacterial transposases and retroviral integrases. Mol Microbiol. 1995;15:13–23. doi: 10.1111/j.1365-2958.1995.tb02217.x. [DOI] [PubMed] [Google Scholar]
- Poulter RTM, Goodwin TJD. DIRS-1 and the other tyrosine recombinase retrotransposons. Cytogenet Genome Res. 2005;110:575–588. doi: 10.1159/000084991. [DOI] [PubMed] [Google Scholar]
- Pritham EJ, Putliwala T, Feschotte C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene. 2007;390:3–17. doi: 10.1016/j.gene.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Prudhomme M, Turlan C, Claverys JP, Chandler M. Diversity of Tn4001 transposition products: the flanking IS256 elements can form tandem dimers and IS circles. J Bacteriol. 2002;184:433–443. doi: 10.1128/JB.184.2.433-443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Swanson R, Day R, Tsai J. A guide to template based structure prediction. Curr Prot Pept Sci. 2009;10:270–285. doi: 10.2174/138920309788452182. [DOI] [PubMed] [Google Scholar]
- Ren G, Gao K, Bushman FD, Yeager M. Single-particle image reconstruction of a tetramer of HIV integrase bound to DNA. J Mol Biol. 2007;366:286–294. doi: 10.1016/j.jmb.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff WS. Transposon Tn5. Annu Rev Genet. 2008;42:269–286. doi: 10.1146/annurev.genet.42.110807.091656. [DOI] [PubMed] [Google Scholar]
- Rezsöhazy R, Hallet B, Delcour J, Mahillon J. The IS4 family of insertion sequences: evidence for a conserved transposase motif. Mol Microbiol. 1993;9:1283–1295. doi: 10.1111/j.1365-2958.1993.tb01258.x. [DOI] [PubMed] [Google Scholar]
- Rice PA. Visualizing Mu transposition: assembling the puzzle pieces. Genes Dev. 2005;19:773–775. doi: 10.1101/gad.1309305. [DOI] [PubMed] [Google Scholar]
- Rice P, Mizuuchi K. Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- Richardson JM, Dawson A, O’Hagan N, Taylor P, Finnegan DJ, Walkinshaw MD. Mechanism of Mos1 transposition: insights from structural analysis. EMBO J. 2006;25:1324–1334. doi: 10.1038/sj.emboj.7601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JM, Colloms SD, Finnegan DJ, Walkinshaw MD. Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell. 2009;138:1096–1108. doi: 10.1016/j.cell.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC. Drosophila P Elements. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 484–518. [Google Scholar]
- Robertson HM, Zumpano KL. Molecular evolution of an ancient mariner transposon, Hsmar1, in the human genome. Gene. 1997;205:203–217. doi: 10.1016/s0378-1119(97)00472-1. [DOI] [PubMed] [Google Scholar]
- Rodgers KK, Bu Z, Fleming KG, Schatz DG, Engelman DM, Coleman JE. A zinc-binding domain involved in the dimerization of RAG1. J Mol Biol. 1996;260:70–84. doi: 10.1006/jmbi.1996.0382. [DOI] [PubMed] [Google Scholar]
- Ronning DR, Li Y, Perez ZN, Ross PD, Hickman AB, Craig NL, Dyda F. The carboxy-terminal portion of TnsC activates the Tn7 transposase through a specific interaction with TnsA. EMBO J. 2004;23:2972–2981. doi: 10.1038/sj.emboj.7600311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau P, Normand C, Loot C, Turlan C, Alazard R, Duval-Valentin G, Chandler M. Transposition of IS. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Vol. 911. Washington, DC: ASM Press; 2002. pp. 367–383. [Google Scholar]
- Rubin E, Lithwick G, Levy AA. Structure and evolution of the hAT transposon superfamily. Genet. 2001;158:949–957. doi: 10.1093/genetics/158.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakano H, Huppi K, Heinrich G, Tonegawa S. Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature. 1979;280:288–294. doi: 10.1038/280288a0. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Sim C, Hong YS, Hogan JR, Fraser MJ, Robertson HM, Collins FH. Molecular evolutionary analysis of the wide-spread piggyBac transposon family and related “domesticated” sequences. Mol Gen Genomics. 2003;270:173–180. doi: 10.1007/s00438-003-0909-0. [DOI] [PubMed] [Google Scholar]
- Sarnovsky RJ, May EW, Craig NL. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Shapiro JA. Molecular model for the transposition and replication of bacteriophage Mu and other transposable elements. Proc Natl Acad Sci USA. 1979;76:1933–1937. doi: 10.1073/pnas.76.4.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Gagnevin L, Chandler M. The new IS1595 family, its relation to IS1 and the frontier between insertion sequences and transposons. Res Microbiol. 2009;160:232–241. doi: 10.1016/j.resmic.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Sinzelle L, Kapitonov VV, Grzela DP, Jursch T, Jurka J, Izsvák Z, Ivics Z. Transposition of a reconstructed Harbinger element in human cells and functional homology with two transposon-derived cellular genes. 2008. Proc Natl Acad Sci USA. 2008;105:4715–4720. doi: 10.1073/pnas.0707746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiniger-White M, Bhasin A, Lovell S, Rayment I, Reznikoff WS. Evidence for “unseen” transposase-DNA contacts. J Mol Biol. 2002;322:971–982. doi: 10.1016/s0022-2836(02)00877-x. [DOI] [PubMed] [Google Scholar]
- Steiniger-White M, Rayment I, Reznikoff WS. Structure/function insights into Tn5 transposition. Curr Opin Struct Biol. 2004;14:50–57. doi: 10.1016/j.sbi.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Okai N, Nonaka H, Tsuge Y, Inui M, Yukawa H. High-throughput transposon mutagenesis of Corynebacterium glutamicum and construction of a single-gene disruptant mutant library. Appl Environ Microbiol. 2006;72:3750–3755. doi: 10.1128/AEM.72.5.3750-3755.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami H, Han CG, Takaki Y, Ohtsubo E. Identification and distribution of new insertion sequences in the genome of alkaliphilic Bacillus halodurans C-125. J Bacteriol. 2001;183:4345–4356. doi: 10.1128/JB.183.14.4345-4356.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura H, Horinouchi S, Beppu T. Novel insertion sequence IS1380 from Acetobacter pasteurianus is involved in loss of ethanol-oxidizing ability. J Bacteriol. 1991;173:7070–7076. doi: 10.1128/jb.173.22.7070-7076.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Cecconi C, Bustamante C, Rio DC. Analysis of P element transposase protein–DNA interactions during the early stages of transposition. J Biol Chem. 2007;282:29002–29012. doi: 10.1074/jbc.M704106200. [DOI] [PubMed] [Google Scholar]
- Tavakoli NP, DeVost J, Derbyshire KM. Defining functional regions of the IS903 transposase. J Mol Biol. 1997;274:491–504. doi: 10.1006/jmbi.1997.1410. [DOI] [PubMed] [Google Scholar]
- Ton-Hoang B, Turlan C, Chandler M. Functional domains of the IS1 transposase: analysis in vivo and in vitro. Mol Microbiol. 2004;53:1529–1543. doi: 10.1111/j.1365-2958.2004.04223.x. [DOI] [PubMed] [Google Scholar]
- Turlan C, Chandler M. Playing second fiddle: second-strand processing and liberation of transposable elements from donor DNA. Trends Microbiol. 2000;8:268–274. doi: 10.1016/s0966-842x(00)01757-1. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Mizuuchi K, Gellert M. Similarities between initiation of V(D)J recombination and retroviral integration. Science. 1996;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- van Pouderoyen G, Ketting RF, Perrakis A, Plasterk RHA, Sixma TK. Crystal structure of the specific DNA-binding domain of Tc3 transposase of C.elegans in complex with transposon DNA. EMBO J. 1997;16:6044–6054. doi: 10.1093/emboj/16.19.6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilei EM, Nicolet J, Frey J. IS1634, a novel insertion element creating long, variable-length direct repeats which is specific for Mycoplasma mycoides subsp. mycoides small-colony type. J Bacteriol. 1999;181:1319–1323. doi: 10.1128/jb.181.4.1319-1323.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WD, Atkinson PW, O’Brochta DA. The Hermes transposable element from the house fly, Musca domestica, is a short inverted repeat-type element of the hobo, Ac, and Tam3 (hAT) element family. Genet Res. 1994;64:87–97. doi: 10.1017/s0016672300032699. [DOI] [PubMed] [Google Scholar]
- Watkins S, van Pouderoyen G, Sixma TK. Structural analysis of the bipartite DNA-binding domain of Tc3 transposase bound to transposon DNA. Nucleic Acids Res. 2004;32:4306–4312. doi: 10.1093/nar/gkh770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH. A unified classification system for eukaryotic transposable elements. Nature Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, Cowling R, Wang W, Liu P, Gertsenstein M, Kaji K, Sung H, Nagy A. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, Kaminski JM. piggyBac is a flexible and highly active transposon as compared to Sleeping Beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci USA. 2006;103:15008–15013. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Chaconas G. Characterization of a region in phage Mu transposase that is involved in interaction with the Mu B protein. J Biol Chem. 1994;269:28829–28833. [PubMed] [Google Scholar]
- Yang W, Steitz TA. Recombining the structures of HIV integrase, RuvC and RNase H. Struct. 1995;3:131–134. doi: 10.1016/s0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- Yin FF, Bailey S, Innis CA, Clubotaru M, Kamtekar S, Steitz TA, Schatz DG. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nature Struct Mol Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JF, Beniac DR, Chaconas G, Ottensmeyer FP. 3D reconstruction of the Mu transposase and the Type I transpososome: a structural framework for Mu DNA transposition. Genes Dev. 2005;19:840–852. doi: 10.1101/gad.1291405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Feschotte C, Zhang Q, Jiang N, Eggleston WB, Wessler SR. P instability factor: An active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. Proc Natl Acad Sci USA. 2001;98:12572–12577. doi: 10.1073/pnas.211442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Mitra R, Hickman AB, Dyda F, Craig NL. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature. 2004;432:995–1001. doi: 10.1038/nature03157. [DOI] [PubMed] [Google Scholar]