

Abstract
Recent studies of postmortem brains from Alzheimer’s disease (AD) patients and transgenic AD mice suggest that oxidative damage, induced by amyloid beta, is associated with mitochondria early in AD progression. Amyloid beta and amyloid precursor protein are known to localize to mitochondrial membranes, block the transport of nuclear-encoded mitochondrial proteins to mitochondria, interact with mitochondrial proteins, disrupt the electron transport chain, increase reactive oxygen species production, cause mitochondrial damage and prevent neurons from functioning normally. Further, accumulation of amyloid beta at synaptic terminals may contribute to synaptic damage and cognitive decline in patients with AD. This article describes the latest research in identifying factors that may affect AD progression, particularly synaptic damage and cognitive decline in AD patients.
Keywords: Adenosine triphosphate, Aging, Alzheimer’s disease, Amyloid beta, Amyloid precursor protein, Cognitive decline, Electron transport chain, Mitochondrial therapeutics, Oxidative stress, Reactive oxygen species, Synaptic damage, Synaptic mitochondria
Introduction
Oxidative stress and synaptic damage are known to implicate in Alzheimer’s disease (AD) pathogenesis [1–6], and may play a critical role in cognitive decline in elderly individuals and AD patients. Synaptic damage causing cognitive impairment is generally accepted in the AD field. However, the precise cellular changes at synaptic terminals that ultimately cause cognitive decline are not fully understood. Based on recent biochemical, cellular, and molecular biology data from cell and mouse models of AD, and on studies of postmortem brains from persons with AD, we propose that mitochondrial failure and amyloid beta (Aβ) accumulation at synapses may cause synaptic damage, impair neurotransmission, and cause cognitive decline in elderly individuals and AD patients.
AD is a progressive, neurodegenerative disorder, characterized by an age-dependent loss of memory and an impairment of multiple cognitive functions. The major pathological features of AD are extra-cellular Aβ plaques and intracellular neurofibrillary tangles. AD is also associated with loss of neurons, synapses, and synaptic function, mitochondrial abnormalities, and inflammatory responses [7–13]. Neuron loss may contribute to 20–30% of brain-weight loss reported in AD patients [13–14]. The loss of synapses, synaptic damage, and mitochondrial oxidative damage have been reported as early events in AD progression [1–4,15,16].
Gene mutations in amyloid precursor protein (APP), presenilin1 (PS1), and presenilin 2 (PS2) are responsible for a small proportion (2% of total) of early-onset familial AD [9]. Apolipoprotein E gene allele 4 (ApoE4) genotype [17] and a genetic variant in the sortilin-related receptor 1 gene [18] are risk factors for developing late-onset AD. Recent studies have revealed that diet and environmental exposure are critical factors in the development of late-onset AD. In addition, aging is considered ‘the number one risk factor’ for the development of late-onset and familial AD. Estimates are that by the year 2050, 50% of people 85 years and older will be afflicted with AD [9]. With such a large population poised to be afflicted, AD is a major health concern for society. Early detection and therapeutic interventions are urgently needed. Here we discuss the possible role of Aβ accumulation in the mitochondria of the brain, mitochondrial dysfunction, and synaptic damage in neuronal processes leading to cognitive decline in aging and AD.
Role of amyloid beta in Alzheimer’s disease
Aβ is a major component of neuritic plaques or amyloid deposits found in AD brain [19]. Aβ is generated by abnormal processing of APP in AD neurons. The APP processing occurs in 2 pathways -amyloidogenic and non-amyloidgenic: in the amyloidogenic pathway, APP undergoes sequential proteolysis of β- and γ-secretases, resulting Aβ, and in the non-amyloidogenic pathway, cleavage occurs by the α-secretase, within the Aβ domain and prevents the formation of full Aβ (Fig. 1). In early-onset AD, genetic mutations in APP, PS1, and PS2 genes activate β- and γ-secretases, and cleave Aβ [9,20]. In late-onset AD, oxidative stress has been proposed to activate β-secretase and to facilitate the secretion of Aβ [21]. A time-course analyses of Aβ in transgenic AD mice lines revealed that Aβ production and deposits increase in an age-dependent manner [1,22]. Postmortem brain studies of the aged humans and patients with AD found an age-dependent increase of Aβ levels in elderly individuals with mild cognitive impairment and in AD patients [23]. These findings suggest that aging plays a role in the production and depositing of Aβ in the brains of AD patients and in transgenic AD mice.
Figure 1. APP processing in non-demented healthy individuals and AD patients.

APP processing occurs via 2 pathways. Beta secretase based amyloidogenic and α-secretase based non-amyloidogenic: In non-amyloidogenic pathway, cleavage occurs by α-secretase within the Aβ domain and generates the large soluble N-terminal fragment (sAPPα) and a non-amyloidogenic C-terminal fragment of 83 aminoacid residues (C83). Further cleavage of this C-terminal fragment by γ-secretase generates the non-amyloidogenic peptide (P3) and APP intracellular domain (ACID). These products are nontoxic. The non-amyloidogenic α-secretase pathway occurs in over 90% of humans, and these individuals generally do not develop dementia. In amyloidogenic pathway, cleavage occurs by β-secretase at the beginning of the Aβ domain and generates a soluble N-terminus fragment (sAPPβ , and amyloidogenic C-terminal fragment of 99 residues (C99). This C-terminal fragment, further cleaved by γ-secretase and generates Aβ. Cleavage by multiple γ-secretases can generate Aβ1-40 and Aβ1-42 fragments. Amyloid beta can accumulate in cellular compartments such as mitochondria and lysosomes and impair cellular functions. Amyloidogenic pathway occurs in about 10% of total humans and these individuals may develop dementia and AD. Age-dependent decrease of Aβ degrading enzymes such as neprilysin and insulin degrading enzyme may not clear Aβ in neurons that contribute to Aβ accumulation in the brain.
In the last decade, a large body of research has been devoted to understanding Aβ toxicity, particularly intracellular Aβ. It is now generally accepted that extracellular Aβ deposits are the by-products of AD pathology. Recent studies of AD patients and transgenic AD mice found intracellular Aβ present in AD-affected brain regions [22–24] and that Aβ1-42 participates in fibrillogenesis and the formation of Aβ plaques. In AD patients, intracellular Aβ has been found to precede extracellular Aβ deposits [23]. In addition, several studies of transgenic AD mice reported that intracellular Aβ accumulates early in AD progression [22,25–27].
Aβ is generated in neurons, wherever APP and β- and γ-secreteses are present, in particular, in several intracellular sites, including Golgi apparatus, endoplasmic reticulum, endosomal-lysosomal systems, and multivesicular bodies [13]. Several lines of evidence suggest that as AD progresses, Aβ accumulates in cellular compartments, interferes with normal cell function [13], and promotes cellular changes. These findings suggest that Aβ plays a major role in AD development and progression.
Cognitive decline and synaptic damage in elderly individuals and in patients with Alzheimer’s disease
Cognitive changes
Synaptic damage is a critical factor that may contribute to cognitive decline in aging persons and persons with AD. Researchers have studied cognitive changes known to occur with age in humans [28], including changes in learning and memory, and the speed of mental processing [28–30]. They found that long-term memory changes relatively little with age, but short-term memory begins declining around the sixth decade. In addition, it takes elderly persons longer to remember events and people than it takes persons 30 years and younger. Language has also been known to change with age [30]. Syntax and word usage of 80-year-old persons are preserved, but naming and verbal fluency decline [30]. It is now generally accepted that age-related cognitive impairment and language changes are due to cellular changes in neurons, particularly at synapses [5], but the extent and nature of these age-related changes are not known.
Synaptic damage
In non-diseased synapses, the terminals transmit signals between cells in order to process information [31]. During aging, the number of synapses and their transmission of signals dramatically decrease [32,33]. These decreases have been documented in different brain regions of elderly persons, supporting the hypothesis that such synaptic changes are ubiquitous features of the aging brain [31].
In a study of synaptic loss, brain samples were taken at autopsy from the cerebellum (unaffected in AD) and hippocampus (affected in AD) of adult and elderly persons without AD, and of patients with AD. The synapse-to-neuron ratio was found to vary according to the brain regions from which the samples were taken and the individual’s health [34]. No significant differences in the synapse-to-neuron ratio were found in samples taken from the cerebellum of adult and elderly persons without AD, and of elderly AD patients. However, in samples from the hippocampus, the synapse-to-neuron ratio decreased more than 50% in the adult and elderly persons without AD, compared to the ratio in the elderly AD patients. In several studies investigating the extent that synaptic loss correlates with cognitive decline in AD [6,35], researchers found a 25-30% decrease in synapses in the cortex and a 15-35% decrease in synapses per cortical neuron, suggesting that synaptic loss in AD patients may correlate more with cognitive decline than with the number of Aβ plaques, neurofibrillary tangles, neuronal loss, or the extent of cortical gliosis. Further, recent studies of synaptic proteins revealed decreased levels of presynaptic (synaptophysin) and postsynaptic proteins (synaptopodin and PSD95) in AD patients compared to age-matched control subjects, suggesting that presynaptic and postsynaptic proteins are critically involved in AD progression [15,31,36,37] and that the loss of synapses and the loss of synaptic proteins are confined to brain regions known to be affected in AD [15,31,36,37].
Evidence of Aβ in synaptic damage
Several recent in vitro and in vivo studies revealed that soluble oligomeric Aβ is responsible for a decease in the long-term potentiation and disruption of synaptic plasticity in AD neurons [38–40]. Further, recent electrophysiology data also suggest that a trimeric form of Aβ inhibits long-term potentiation in rodents [40]. These lines of evidence suggest that synaptic changes caused by soluble Aβ may contribute to the loss of synapses and of synaptic proteins, and may be responsible for cognitive decline in AD patients. However, the precise mechanisms involved in such synaptic changes– in terms of neurotransmission, mitochondrial ATP, and cognitive decline – are still unclear. Further, we still need to know the types of changes occurring at synaptic terminals in elderly persons without AD and in AD patients.
Role of mitochondria in aging and Alzheimer’s disease
Mitochondrial DNA changes in aging and AD
It is well -documented that mtDNA changes are responsible for aging phenotypes [41-43]. For example, many tissues from aged individuals have a lower respiratory function compared to those from younger individuals. mtDNA defects (point mutations and deletions) are highly prevalent in aged cells, and there is evidence that 8-hydroxy-2-deoxyguanosine (damaged DNA) is more prevalent in aged tissues [41]. Further, mice carrying a mitochondrial DNA mutation (in DNA polymerase-γ gene) showed features of aging and reduced life span, suggesting that mtDNA changes are critical for aging phenotypes [42,43].
Mitochondrial DNA defects have not only been found in elderly persons without AD but also in AD patients [44,45], and have been associated with decreased cytochrome oxidase activity in non-AD aging and aging AD brains [44,45]. Recent cytoplasmic hybrid studies indicate that in AD, reduced cytochrome oxidase activity is associated with increased mtDNA defects [46]. However, the effects of specific mtDNA changes on cytochrome oxidase activity have not yet been studied. Overall, findings from these studies suggest that somatic mtDNA mutations are involved in aging phenotypes and may contribute to AD development.
Biochemical changes
Compared to other organs, the brain is vulnerable to oxidative stress due to its high lipid content, its relatively high oxygen metabolism, and its low levels of anti-oxidant defenses [21]. Mitochondrial oxidative stress occurs early in AD progression, before the onset of Aβ pathology [1,2,16,47]. Oxidative stress was reported in the mitochondria of the brain, platelets, and fibroblasts from AD patients [48,49]. Oxidative stress was also found in the brains of AD patients [50–54] and in those of AD transgenic mice [3,55–58].
Mitochondrial abnormalities have been found both in neurons and astrocytes [47,54,59], suggesting that both neurons and astrocytes may be damaged by free radicals in the AD brain. Superoxide radicals (O2 −) may be produced in mitochondrial ETC complexes I and III (see Fig. 3) [21,50] and in components of the tricarboxylic acid cycle, including α-ketoglutarate dehydrogenase [11,21]. In addition, O2 - may be generated in the outer mitochondrial membrane. H2O2 and O2 −, released from the mitochondrial matrix and from the inner and outer mitochondrial membranes, may be carried to the cytoplasm and, ultimately, may lead to the oxidation of cytoplasmic proteins [21]. Decreased levels of 3 mitochondrial enzymes – pyruvate dehdrogenase complex, alpha ketoglutarate dehydrogenase complex, and cytochrome oxidase – have also been reported in AD brain [49].
Figure 3. Mitochondrial hypothesis and AD.
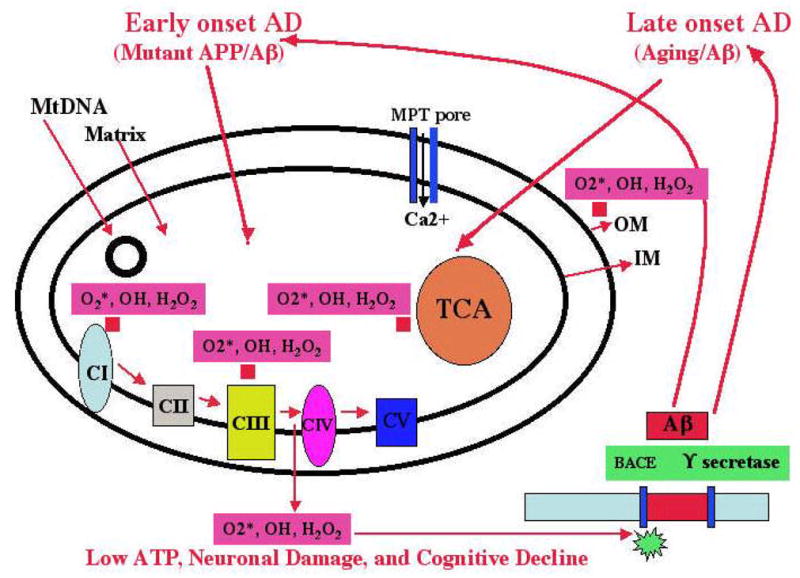
In early-onset AD, mutant APP and soluble Aβ are hypothesized to localize to synaptic mitochondria, leading to the generation of free radicals (O2*, H2O2, OH). The free radicals, in turn, may decrease cytochrome oxidase activity and inhibit cellular ATP. In late-onset AD, the free radicals that are generated due to aging may activate BACE and facilitate the cleavage of the Aβ. The cleaved Aβ may enter mitochondria and induce free radicals (O2*, H2O2, OH), leading to the disruption of the ETC, a decrease in cytochrome oxidase activity, and the inhibition of ATP. This feedback loop may ultimately lead to neuronal damage, to the degeneration of neurons, and to cognitive decline in AD patients.
Cell body mitochondria in AD neurons
To determine the role of mitochondrial abnormalities in AD, Hirai et al. [54] studied post-mortem brain specimens from AD patients and healthy persons, using in situ hybridization, electron microscopy, and immunohistochemistry techniques. They found increased oxidative damage in AD patients, a striking and significant increase in mtDNA in their pyramidal neurons, and cytochrome oxidase in their neuronal cytoplasm. They found that the increase in oxidative damage was not due to an increase in morphologically normal mitochondria, but rather due to an accumulation of products from degraded mitochondrial cell bodies, suggesting that such degradations may be important in AD progression.
Synaptic mitochondria in AD neurons
Synaptic mitochondria are synthesized in the cell body of neurons and are then transported down the axon or dendrite to serve cellular energy demands [60,61], a phenomenon termed mitochondrial trafficking [62]. If mitochondria localized in the cell body are damaged or are otherwise degraded, such as by aging or Aβ, these defective mitochondria may be transported to synaptic terminals via natural mitochondrial trafficking, where they produce low levels of ATP due to their degradation. Supporting the hypothesis that Aβ causes synaptic mitochondrial dysfunction in AD, Mungarro-Manchaca [63] found that the Aβ peptide potentiates mitochondrial dysfunction in the presence of ryanodine, and induces morphological changes, including mitochondrial swelling. Further, Gillardon et al. [64] found Aβ oligomers in synaptosomal mitochondrial fractions and decreased energy metabolism in AD transgenic mice. The findings from these two studies suggest that in AD, the Aβ peptide may be responsible for synaptic mitochondrial damage, low ATP production, and, ultimately, the degeneration of synapses [63,64]. However, further research is needed to understand the mechanisms of synaptic mitochondrial damage, changes in ATP production, and the relationship between ATP production and cognitive function in AD patients.
Synaptic terminals are sites of high-energy demand. Synaptic transmission requires high levels of cellular ATP for neurotransmitter exocytosis and the potentiation of neurotransmitter release. In addition, synaptic terminals require mitochondria for sequestering and releasing Ca2+ for post-tetanic potentiation [65]. Therefore, increased transport of mitochondria to synaptic terminals is necessary to deliver mitochondria with short morphology. The half-life of a brain mitochondrion is about one month [66]. However, synaptic mitochondria may be "older" than cell-body mitochondria. It is these older synaptic mitochondria that may exhibit greater damage from oxidative stress than cell-body mitochondria [67]. The increase in oxidative damage exhibited by synaptic mitochondria may affect neurotransmission and synaptic damage and loss, and may ultimately be responsible for cognitive decline in AD patients. We need further research to determine whether synaptic mitochondria play a role in synaptic loss in AD neurons.
Amyloid beta and mitochondrial dysfunction in Alzheimer’s disease
Several lines of evidence suggest that APP and Aβ are factors contributing to mitochondrial dysfunction in AD.
Abnormal mitochondrial gene expression
Increasing evidence suggests that abnormal mitochondrial gene expression is initiated by mitochondrial dysfunction in AD. The Reddy laboratory investigated gene expression profiles in brain slices from mice at 3 stages of AD progression: long before (2 months of age), immediately before (5 months), and after (18 months) the appearance of Aβ plaques in the cerebral cortex of APP transgenic mice (Tg2576) and age-matched wild-type mice [16]. Our analysis revealed that the genes related to mitochondrial energy metabolism and apoptosis were up-regulated in 2-month-old Tg2576 mice and that the same genes were up-regulated at 5 and 18 months of age [16]. We also found decreased cytochrome oxidase, increased free radicals, and increased carbonyl proteins in 2-month-old APP mice compared to the age-matched wild-type mice [1], suggesting that mitochondrial energy metabolism may be impaired by mutant APP and/or Aβ, and that the up-regulation of mitochondrial genes may be a compensatory response
Using quantitative reverse transcriptase polymerase chain reaction (RT-PCR) techniques, we also studied mRNA expression of 11 mitochondrial-encoded genes in patients with early AD and with definite AD, and in Age-matched control subjects. This analysis revealed a down-regulation of mitochondrial genes in complex I of OXPHOS in both early and definite AD brain specimens. Complex I showed a down-regulation of mitochondrial genes, but complexes III and IV showed increased mRNA expressions in the brain specimens of both the early and definite AD patients, suggesting a great demand on energy production [47]. Several other studies reported decreased cytochrome oxidase in mitochondria from platelets, fibroblasts, and brains of AD patients. The connection between decreased cytochrome oxidase and increased mitochondrial mRNA has been interpreted as a compensatory response in AD [21] to compensate for the loss of cytochrome oxidase, mitochondrial-encoded genes may be activated in the surviving brain neurons of AD patients.
Aβ and mitochondrial dysfunction - in vitro evidence
Several in vitro studies have provided compelling evidence that Aβ may cause mitochondrial dysfunction [8,68]. Sirk et al. [69] reported that the Aβ (25–35) peptide blocks the entry of nuclear-encoded proteins into mitochondria when Aβ are exposed to PC12 cells and that this blockage causes decreased mitochondrial membrane potential, increased ROS production, oxygen glucose deprivation, and altered mitochondrial morphology [69]. In another study, Aβ was found to increase ROS production, to activate mitochondrial fission proteins Drp1 and Fis1, and to cause mitochondrial fragmentation [70]. Recently, Casley et al. [71] reported that when Aβ peptides (25–35) were exposed to rat brain mitochondria, states 3 and 4 of respiration reduced, and activities of cytochrome oxidase and TCA cycle enzymes decreased [71]. Moreira et al. reported that in the presence Ca2+, Aβ peptides induce the permeability transition pore to open, which may play a key role in apoptosis [72,74]. These and several other in vitro studies [summarized in refs #8,68] suggest that Aβ plays a role in inducing mitochondrial dysfunction in AD.
APP/Aβ association with mitochondria
The 4 kDa Aβ peptide is a product of APP, cleaved via the sequential proteolysis of aspartyl β-secretase and presenilin-dependent γ-secretase [7]. APP is synthesized in the cell bodies of neurons and is anterogradely transported via axons to nerve terminals in the brain [74–76]. The localization of APP in nerve terminals suggests that they are the major source of Aβ in the Aβ plaques found in AD brain specimens. It is now clear that Aβ originates in neurons and may be released from synapses to outside the cell [77] (see Fig.2). In APP processing, monomeric Aβ forms oligomers in synaptic terminals. Oligomeric Aβ, with their sharp morphology, is hypothesized to enter cell organelles, such as mitochondria, by penetrating the membrane [78]. Oligomeric Aβ may preferentially enter synaptic mitochondria because Aβ is enriched at synaptic terminals before leaving the cell via the synapses (see Fig.2).
Figure 2. Hypothesized changes at the synaptic mitochondria in AD neurons.

In AD neurons, Aβ (1–40 and 1–42) are secreted via sequential cleavage of Aβ precursor protein (APP) by beta secretase (BACE) and gamma secretase. In late-onset AD, mitochondrially produced free radicals (O2*, H2O2, OH) activate beta secretase and facilitate the cleavage of APP. The cleaved Aβ may further enter mitochondria primarily localized at synapses, induce free radicals, cause oxidative damage, and inhibit cellular ATP. Increased levels of BACE activity have been observed in the postmortem brain specimens from late-onset AD patients. This increased BACE activity may be primarily due to decreased energy metabolism – in other words, it may be due to decreased ATP, increased Ca2+ influx, and increased free radicals. Reduced cellular ATP production, in turn, may cause the impairment of neurotransmission (glutamate and NMDA receptors). These events may occur primarily in learning and memory regions of the brain, and ultimately may cause cognitive decline in AD patients.
In support of the hypothesis that APP and Aβ enter mitochondria, several studies have found APP and its derivatives (monomeric and oligomeric forms of Aβ) in mitochondrial membranes [Table 1, Ref# 1,2,3,55,56,63,64,79,80,81,82]. Lustbader et al. [55] found Aβ normally interacting with mitochondrial matrix protein, Aβ-binding alcohol dehydrogenase (ABAD), leading to mitochondrial dysfunction. Caspersen et al. [3] found Aβ in mitochondria from postmortem brain specimens of AD patients, and an accumulation of Aβ in the brain mitochondria from APP transgenic mice. Recently, our laboratory found Aβ monomers and oligomers in isolated mitochondria from the cerebral cortex of APP transgenic mice [1] and in isolated mitochondria from N2a cells expressing APP. Our digitonin fractionation analysis of isolated mitochondria from APP transgenic mice revealed Aβ in outer and inner mitochondrial membranes and matrix, and that mitochondrial Aβ decreases cytochrome oxidase activity and increases free radical production and carbonyl proteins [1].
Table 1.
Amyloid precursor protein and Aβ associated with mitochondria and mitochondrial dysfunction in AD
| Source of cells/neurons in AD patients | Methods determining APP/ Aβ association with mitochondria | APP/ Aβ associated with mitochondria and mitochondrial dysfunction | Study and Ref. |
|---|---|---|---|
| AD patients | Immuno-electron microscopy and immunohistochemistry | APP is associated with the outer mitochondrial membrane. | Yamaguchi et al. 1992 [79] |
| AD patients and APP mice (Tg mAPP-J mouse line) | Percoll gradient, immunoblotting, immuno-electron microscopy, and immuno-fluorescence | Aβ binds to the mitochondrial matrix protein ‘abeta binding alcohol dehydrogenase’ and causes mitochondrial dysfunction in AD. | Lustbader et al. 2004 [55] Takuma et al. 2005 [80] Caspersen et al. 2005 [3] |
| Human lymphocytes treated with Aβ1–42; APP mice (Tg2576 mouse line) | Immunoblotting | Isolated mitochondria from lymphocytes treated with Aβ1- 42, inhibits cytochrome oxidase activity in the presence of Cu2+. Aβ is found in isolated brain mitochondria, from Tg2576 mice. | Crouch et al. 2005 [81] |
| APP mice (Tg2576 mouse line) and APP cell- lines | Percoll gradient, digitonin fractionation, immunoblotting, and electron microscopy | Aβ monomers and oligomers are found in mitochondrial membranes in AD neurons from Tg2576 mice and in N2a cells expressing mutant human APP. Soluble Aβ correlates with increased H2O2 production. Decreased cytochrome oxidase and increased carbonyl proteins are found in Tg2576 mice. | Manczak et al. 2006 [1] |
| AD patients | Immunoblotting and immuno-electron microscopy | Non-glycosylated, full-length APP and C-terminal truncated APP, including Aβ, are found in import channels of mitochondria of AD brains. AD brains with ApoE4 genotype have higher levels of APP in the affected brain mitochondria than in nonaffected brain mitochondria. | Devi et al. 2006 [2] |
| Cell culture (incubation of rat brain mitochondria with Aβ 25– 35) | Immunoblotting and immuno-electron microscopy | Synaptosomal mitochondria containing Aβ show morphological changes, including mitochondrial swelling and depletion of synaptic vesicles. Synaptosomal Aβ diminishes mitochondrial redox functions. | Mungarro- Menchaca et al. 2002 [63] |
| APP mice (Tg2576 mouse line) | Synaptosomal fractionation and immunoblotting | Aβ oligomers are detected in synaptosmal fractions from Tg2576 mice, and reduced glucose metabolism is found. | Gillardon et al. 2007 [64] |
| APP mice (Tg2576 mouse line) | Immunoblotting, immuno-fluorescence, and immuno-electron microscopy | APP is targeted to cortical mitochondria in a transmembrane arrested form of Tg2576 mice. | Anandatheerthav arada et al 2003 [56] |
| Mouse neuroblastoma cells expressing (N2a) ApoE4 fragments | Immunoblotting and immuno-fluorescence | ApoE4 protein (1–272 amino acid residues) interact with mitochondria and cause mitochondrial toxicity. | Chang et al. 2005 [82] |
Aβ, amyloid beta; AD, Alzheimer’s disease; ApoE4, apolipoprotein allele 4; APP, amyloid precursor protein; N2a cells mouse neuroblastoma cells; and Tg, Transgenic;
Two recent studies found that APP directly interacts with mitochondrial proteins [1,83]. Schmidt et al. (77) found that mitochondrial ATP synthase subunit alpha is a binding partner of the extracellular domain of APP and Aβ. Transfection of APP-deficient neuroblastoma cells with APP resulted in increased surface localization of the ATP synthase alpha subunit, and in extracellular APP/Aβ inhibiting the extracellular generation of ATP. In another study, Devi et al. [2] found that nonglycosylated full-length and C-terminal truncated APP accumulates in the protein import channels of mitochondria of human AD brains, but not in age-matched controls. The accumulation of APP across mitochondrial import channels inhibited the entry of nuclear-encoded cytochrome c oxidase subunits IV and Vb proteins, and was associated with decreased cytochrome oxidase and increased free radical production [2]. Based on recent findings of APP/Aβ and mitochondria [1,2,3,55,56,63,64,79,80,81,82]. we propose that mutant APP/Aβ enter mitochondria and interact with mitochondrial proteins, disrupt the ETC, increase ROS production, and inhibit the generation of cellular ATP (Fig. 3).
Synaptic Aβ and mitochondrial dysfunction
There is limited evidence to support the hypothesis that synaptic Aβ causes mitochondrial dysfunction and cognitive decline in AD. However, two recent studies found Aβ in the synaptosomal fractions of brain mitochondria and that Aβ diminishes the overall capacity of the ETC [63,64], suggesting that mitochondrial Aβ, localized at synapses, may be associated with synaptic damage in AD neurons. However, we do not know whether Aβ enters presynaptic mitochondria or postsynaptic mitochondria, or both, and we do not know how Aβ functionally damages synapses. Further, we do not know whether mitochondrial Aβ ultimately leads to cognitive decline in patients with AD. Further research may provide answers to these critical issues.
Mitochondrial Therapeutics of Alzheimer’s Disease
Oxidative stress and synaptic damage are early events in AD progression, and aging and Aβ are key players that may cause mitochondrial dysfunction and synaptic damage. Intracellular Aβ accumulates in cell organelles, including mitochondria, and causes cellular dysfunction. Strategies that decrease toxicity caused by age-related mitochondrial dysfunction, Aβ production, and synaptic damage may be useful in protecting AD neurons from this toxicity. Initial investigations of calorie-restricted diets and Aβ in AD transgenic mice lines found that calorie-restricted diets may decrease Aβ production by activating SIRT1 and non-amyloidogenic α-secretase [84,85]. An advantage of calorie-restricted diets is that toxic Aβ1-42 production decreases because increased α-secretase activity chops the intact Aβ into 2 small molecules: β-secretase site to α-secretase site (and soluble APP N-terminus to α-secretase site) and α-secretase site to γ-secretase site and γ site to end of C-terminal APP (see Fig. 1). These chopped APP molecules are nontoxic. By decreasing Aβ production in AD neurons, it is possible to minimize intracellular accumulation of Aβ in cell organelles and to decrease toxicity caused by the intracellular organelle Aβ, and sequential Aβ formations, including monomers, oligomers, protofribrils and fibrils and deposits. Calorie-restricted diets reduced defects in the ETC, decreased mitochondrial ROS, and decreased mitochondrial damage in neurons from the human brain [84,85].
Recent studies of AD transgenic mice and antioxidants have shown decreased Aβ pathology and ameliorated cognitive deficits in the mice [85]. However, in studies of elderly persons and AD patients using antioxidants, results were mixed. Some studies found a reduced risk of AD in elderly persons treated with natural antioxidants, such as vitamins C and E [86–89]. Other studies found no effect of antioxidants in elderly individuals and in AD patients [90]. One reason for these mixed findings is that the naturally occurring antioxidants might not cross the blood-brain barrier and so cannot reach mitochondria to neutralize free radicals. In the last decade, significant progress has been made in developing antioxidants that target mitochondria, including MitoQ, MitoVitE, MitoPBN, MitoPeroxidase, SS-tetra peptides, choline esters of glutathione, and N-acetyl-L-cysteine [91-94]. Initial studies of aging and ALS transgenic mice found that these antioxidants enter the mitochondria several hundred fold, rapidly neutralize free radicals, and decrease mitochondrial toxicity [91–94]. Therefore, such mitochondrially targeted antioxidants may be promising candidates to treat elderly persons and AD patients.
Conclusions and perspectives
There is increasing evidence to suggest that synaptic damage and mitochondrial dysfunction play a significant role in aging and AD development. Mitochondria are the major source of energy for the brain. The accumulation of mtDNA changes may increase ROS production and reduce mitochondrial ATP in an age-dependent manner. Recent studies of neurons from postmortem AD brain specimens and from transgenic AD mice brain specimens suggest that oxidative damage induces Aβ production. Aβ and APP are reported to localize to mitochondrial membranes, block the transport of nuclear-encoded mitochondrial proteins to mitochondria, interact with mitochondrial proteins, disrupt ETC activities, increase ROS production, cause mitochondrial damage, and prevent neurons from functioning normally. However, the mechanisms of Aβ and APP imported into mitochondrial membranes are unclear. Experiments that focus on the import of APP/Aβ into mitochondrial membranes may provide new insights in understanding the mechanisms of APP/Aβ to import to mitochondria. Further, future experiments that focus on mitochondrial functional association with APP/Aβ may be useful for developing mitochondrial drug targets.
Aβ accumulates at synaptic terminals and impairs synaptic function. Aβ also enters synaptic mitochondria and causes damage. Mitochondrial damage is expected to be greater in synaptic mitochondria (which are isolated) than in cell-body mitochondria (which are clustered). The damaged, synaptic mitochondria may not satisfy the high energy demands required at synapses, which may lead to impaired neurotransmission and, ultimately, to cognitive failure. However, further research is needed to understand the activities at synapses, particularly how Aβ interferes with functions of: (1) NMDA and glutamate receptors, (2) synaptic vesicles, and (3) Ca2+ influx. Future in vivo and in vitro experiments investigating synapses and Aβ, and electron microscopy of synapses in AD neurons may provide new insights into synaptic damage.
In AD, tremendous progress has been made in developing therapeutic strategies to decrease Aβ production and toxicity. However, further research is needed to develop molecules that chop or degrade intact Aβ in brain neurons affected by AD. Further research is also needed to test the efficacies of mitochondrially targeted antioxidants in AD mouse models.
Outstanding questions.
In AD, why and how are neurons, in particular those in the hippocampus and cortex, susceptible to oxidative damage over other neurons, such as Purkinje neurons of the cerebellum?
Recent studies have provided evidence that, in AD, Aβ enters the mitochondria. If so, do the Aβ enter presynaptic mitochondria or postsynaptic mitochondria, or both?
Based on findings from in vitro and in vivo studies, it has been proposed that Aβ plays a significant role in synaptic dysfunction and cognitive decline in AD patients. If so, what roles do mtDNA changes and mitochondrial dysfunction play at synaptic terminals?
Are synaptic mitochondria more susceptible to mitochondrial fragmentation in AD?
Do synaptic mitochondria cause greater oxidative damage and, ultimately, synaptic loss in AD.
Acknowledgments
Our sincere apologies to all those work could not be cited owing to space restrictions. This article is supported by grants from the KaloBios Pharmaceuticals, Inc., and NIH (AG028072 and AG02651).
Glossary
- Endocytosis
Endocytos is a process whereby cells absorb molecules such as proteins from the outside by engulfing it with their cell membrane. It is used by all cells of the body because most substances important to them are large polar molecules, and thus cannot pass through the hydrophobic plasma membrane. The function of endocytosis is the opposite of exocytosis
- Mitochondrial trafficking
Movement of mitochondria in the neuronal cell - from cell body (or soma) to axons, dendrites and synapses, and back to cell body
- Sirtuin
Sirtuin is a class of enzyme, specifically NAD-dependent histone deacetylases, found in both prokaryotes and eukaryotes. Sirtuins have been known to affect cellular metabolism through selective gene expression in eukaryotes. The name comes from silent information regulator two, the gene responsible for cellular regulation in yeast
- Synapse
The junction between the axon terminals of a neuron and the receiving cell is called a synapse
- Synaptic mitochondria
Mitochondria localized at synaptic terminals
References
- 1.Manczak M, et al. Mitochondria are a direct site of Abeta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 2.Devi L, et al. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caspersen C, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 4.Nunomura A, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 6.DeKosky ST, et al. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 8.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer's disease? Brain Res Brain Res Rev. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Reddy PH, McWeeney S. Mapping cellular transcriptosomes in autopsied Alzheimer's disease subjects and relevant animal models. Neurobiol Aging. 2006;27:1060–1077. doi: 10.1016/j.neurobiolaging.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Nunomura A, et al. Involvement of Oxidative Stress in Alzheimer Disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 11.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 12.Parihar MS, Brewer GJ. Mitoenegrtic failure in Alzheimer’s disease. Am J Physiol Cell Physiol. 2007;292:C8–23. doi: 10.1152/ajpcell.00232.2006. [DOI] [PubMed] [Google Scholar]
- 13.LaFerla FM, et al. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 14.Vina J, et al. Effect of Gender on Mitochondrial Toxicity of Alzheimer's Abeta Peptide. Antioxid Redox Signal. 2007;9:1677–1690. doi: 10.1089/ars.2007.1773. [DOI] [PubMed] [Google Scholar]
- 15.Reddy PH, et al. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–117. doi: 10.3233/jad-2005-7203. [DOI] [PubMed] [Google Scholar]
- 16.Reddy PH, et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 17.Saunders AM, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 18.Rogaeva E, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 20.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer's disease. J Neurochem. 2006;96:1–13. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- 22.Oddo S, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 23.Gouras GK, et al. Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy MP, et al. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Knobloch M, et al. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 26.Kuo YM, et al. The evolution of A beta peptide burden in the APP23 transgenic mice: implications for A beta deposition in Alzheimer disease. Mol Med. 2001:609–618. [PMC free article] [PubMed] [Google Scholar]
- 27.Oakley H, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flicker C, et al. Mild cognitive impairment in the elderly: predictors of dementia. Neurology. 1991;41:1006–1009. doi: 10.1212/wnl.41.7.1006. [DOI] [PubMed] [Google Scholar]
- 29.Rubin EH, et al. A prospective study of cognitive function and onset of dementia in cognitively healthy elders. Arch Neurol. 1998;55:395–401. doi: 10.1001/archneur.55.3.395. [DOI] [PubMed] [Google Scholar]
- 30.Kaye J, Quinn J. Clinical changes associated with normal aging. In: Ckark Christopher M, Trojanowski John Q., editors. Neurodegenerative dementias. McGraw-Hill publications; 2000. pp. 1–13. [Google Scholar]
- 31.Bertoni-Freddari C, et al. Synaptic structural dynamics and aging. Gerontology. 1996;1996:170–180. doi: 10.1159/000213789. [DOI] [PubMed] [Google Scholar]
- 32.Scheff SW, et al. Strain comparison of synaptic density in hippocampal CA1 of aged rats. Neurobiol Aging. 1985;6:29–34. doi: 10.1016/0197-4580(85)90068-5. [DOI] [PubMed] [Google Scholar]
- 33.Scheff SW, et al. Quantitation of synaptic density in the septal nuclei of young and aged Fischer 344 rats. Neurobiol Aging. 1991;12:3–12. doi: 10.1016/0197-4580(91)90032-f. [DOI] [PubMed] [Google Scholar]
- 34.Bertoni-Freddari C, et al. Morphological adaptive response of the synaptic junctional zones in the human dentate gyrus during aging and Alzheimer's disease. Brain Res. 1990;517:69–75. doi: 10.1016/0006-8993(90)91009-6. [DOI] [PubMed] [Google Scholar]
- 35.Terry RD, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 36.Gylys KH, et al. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almeida CG, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Cleary JP, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 39.Billings LM, et al. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 40.Townsend M, et al. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572(Pt 2):477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kujoth GC, et al. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res. 2006;66:7386–7389. doi: 10.1158/0008-5472.CAN-05-4670. [DOI] [PubMed] [Google Scholar]
- 42.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 43.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 44.Lin MT, et al. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer's disease brain. Hum Mol Genet. 2002;11:133–1345. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 45.Coskun PE, et al. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swerdlow RH, et al. Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- 47.Manczak M, et al. Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromol Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 48.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 49.Reddy PH. Mitochondrial Dysfunction in Aging and Alzheimer’s Disease: Strategies to Protect Neurons. Antioxidants & Redox Signaling. 2007;9:1647–1658. doi: 10.1089/ars.2007.1754. [DOI] [PubMed] [Google Scholar]
- 50.Gibson GE, et al. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- 51.Parker WD, Jr, et al. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 52.Maurer I, et al. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 53.Smith MA, et al. Oxidative damage in Alzheimer's. Nature. 1996;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 54.Hirai K, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lustbader JW, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;16:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 56.Anandatheerthavarada HK, et al. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li F, Gouras GK, et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 58.Smith MA, et al. Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 59.Abramov AY, et al. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24:565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 61.Miller RJ. Neuronal Ca2+: getting it up and keeping it up. Trends Neurosci. 1992;15:317–319. doi: 10.1016/0166-2236(92)90045-a. [DOI] [PubMed] [Google Scholar]
- 62.Reynolds IJ, et al. Mitochondrial trafficking in neurons: a key variable in neurodegeneration? J Bioenerg Biomembr. 2004;36:283–286. doi: 10.1023/B:JOBB.0000041754.78313.c2. [DOI] [PubMed] [Google Scholar]
- 63.Mungarro-Menchaca, et al. beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J Neurosci Res. 2002;68:89–96. doi: 10.1002/jnr.10193. [DOI] [PubMed] [Google Scholar]
- 64.Gillardon F, et al. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007;7:605–616. doi: 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- 65.Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 66.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- 67.Hagen TM, et al. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci U S A. 1997;94:3064–3069. doi: 10.1073/pnas.94.7.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreira PI, et al. Alzheimer's Disease: A Lesson from Mitochondrial Dysfunction. Antioxid Redox Signal. 2007;9:1621–1630. doi: 10.1089/ars.2007.1703. [DOI] [PubMed] [Google Scholar]
- 69.Sirk D, et al. Chronic exposure to sub-lethal beta-amyloid inhibits the import of nuclear encoded proteins to mitochondria in differentiated PC12 cells. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04907.x. (in press) [DOI] [PubMed] [Google Scholar]
- 70.Barsoum MJ, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Casley CS, et al. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 72.Moreira PI, et al. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci Rep. 2001;21:789–800. doi: 10.1023/a:1015536808304. [DOI] [PubMed] [Google Scholar]
- 73.Moreira PI, et al. Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J Neurosci Res. 2002;69:257–267. doi: 10.1002/jnr.10282. [DOI] [PubMed] [Google Scholar]
- 74.Koo EH, et al. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buxbaum JD, et al. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lazarov O, et al. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schroeder BE, Koo EH. To think or not to think: synaptic activity and Abeta release. Neuron. 2005;48:873–875. doi: 10.1016/j.neuron.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 78.Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- 79.Yamaguchi H, et al. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol (Berl) 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- 80.Takuma K, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19(6):597–8. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 81.Crouch PJ, et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang S, et al. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmidt C. Amyloid precursor protein and amyloid beta-peptide bind to ATP synthase and regulate its activity at the surface of neural cells. Mol Psychiatry. 2007 doi: 10.1038/sj.mp.4002077. (in press) [DOI] [PubMed] [Google Scholar]
- 84.Anekonda TS, Reddy PH. Neuronal protection by sirtuins in Alzheimer's disease. J Neurochem. 2006;96:305–313. doi: 10.1111/j.1471-4159.2005.03492.x. [DOI] [PubMed] [Google Scholar]
- 85.Pasinetti GM, et al. Caloric intake and Alzheimer's disease. Experimental approaches and therapeutic implications. Interdiscip Top Gerontol. 2007;35:159–175. doi: 10.1159/000096561. [DOI] [PubMed] [Google Scholar]
- 86.Grundman M, et al. Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch Neurol. 2004;61:59–66. doi: 10.1001/archneur.61.1.59. [DOI] [PubMed] [Google Scholar]
- 87.Morris MC, et al. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–126. doi: 10.1097/00002093-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 88.Morris MC, et al. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA. 2002;287:3230–3237. doi: 10.1001/jama.287.24.3230. [DOI] [PubMed] [Google Scholar]
- 89.Morris MC, et al. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am J Clin Nutr. 2005;81:508–514. doi: 10.1093/ajcn.81.2.508. [DOI] [PubMed] [Google Scholar]
- 90.Luchsinger JA, et al. Antioxidant vitamin intake and risk of Alzheimer disease. Arch Neurol. 2003;60:203–208. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 91.Reddy PH. Mitochondrial oxidative damage in aging and Alzheimer's disease: implications for mitochondrially targeted antioxidant therapeutics. J Biomed Biotechnol. 2006:31372. doi: 10.1155/JBB/2006/31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sheu SS, et al. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim Biophys Acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 93.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–31. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Murphy MP, et al. A Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]