

Abstract
Helicobacter pylori (H. pylori) is a gram negative bacterium that can cause diseases such as peptic ulcers and gastric cancer. IL-17A, a proinflammatory cytokine that can induce the production of CXC chemokines for neutrophil recruitment, has recently been shown to be elevated in both H. pylori-infected patients and mice. Furthermore, studies in mouse models of vaccination have reported levels significantly increased over infected, unimmunized mice and blocking of IL-17A during the challenge phase in immunized mice reduces protective immunity. Because many aspects of immunity had redundant or compensatory mechanisms, we investigated whether mice could be protectively immunized when IL-17A function is absent during the entire immune response using IL-17A and IL-17A receptor knockout (KO) mice immunized against H. pylori. Gastric biopsies were harvested from naïve, unimmunized/challenged, and immunized/challenged wild type (WT) and KO mice and analyzed for inflammation, neutrophil, and bacterial levels. Groups of IL-17A KO mice were also treated with anti-IFNγ or control antibodies. Surprisingly, all groups of immunized KO mice reduced their bacterial loads comparably to WT mice. The gastric neutrophil counts did not vary significantly between IL-17A KO and WT mice, whereas IL-17RA KO mice had on average a four-fold decrease compared to WT. Additionally, we performed an immunization study with CXCR2 KO mice and observed significant gastric neutrophils and reduction in bacterial load. These data suggest that there are compensatory mechanisms for protection against H. pylori and for neutrophil recruitment in the absence of an IL-17A-CXC chemokine pathway.
INTRODUCTION
Helicobacter pylori is a gram negative bacterium that colonizes the stomachs of approximately half the world’s population.1 Infection may progress to pathologic states, such as peptic ulcer disease or gastric adenocarcinoma, resulting in significant morbidity and mortality worldwide.2, 3 Infection can be successfully treated with antibiotics and proton pump inhibitors, but antibiotic resistance is an increasing concern,4 and current treatments are not practical in endemic areas due to costs and because eradication by these methods does not provide resistance to reinfection.
A vaccine against H. pylori is therefore desirable, but clinical trials have been disappointing and vaccination in most animal models fails to completely protect against challenge. It may be important for vaccine strategies to preferentially elicit specific aspects of the immune system in order to improve efficacy. This could allow for the development of new vaccines and drugs focusing on these important targets. One such target may be TH-17 cells. Mice deficient in the IL-12 family p40 subunit (common to both IL-12 and IL-23) fail to be protected,5, 6 but IFNγ knockout (KO) mice have achieved immunity in some but not all studies.5–8 This has lead to speculation of the potential significance of the IL-23 and proinflammatory cytokine IL-17 pathway. Since IL-17 can induce CXC chemokines for the rapid recruitment of neutrophils, an IL-23 and TH-17-mediated neutrophil activation pathway may play a role in clearance of the bacteria.
IL-17 has been suggested to play a role in Helicobacter infection and immunity. It is present in gastric biopsies of H. pylori-infected patients and mice, leading to speculation that IL-17 contributes to the pathology associated with the bacteria.9–11 Murine models of protection demonstrate an association of protection with severe inflammation,12–15 and therefore it is possible that the proinflammatory effects of IL-17, if enhanced relative to the activities generated during chronic infection, may be important for the eradication of the bacteria. Indeed, we previously observed a robust IL-17 recall response by CD4+ cells from immunized mice and significantly more IL-17 mRNA levels in the stomachs of immunized/challenged (I/C) mice compared to unimmunized/challenged mice (U/C).16
It has recently demonstrated that antibody-mediated in vivo neutralization of IL-17A following challenge of immunized mice results in reduced protective immunity against both H. felis and H. pylori in mice, thus demonstrating a potentially vital role for IL-17 in the vaccine induced protective immune response and suggesting that immunization strategies designed to optimize the TH-17 response might result in improved vaccine efficacy.17, 18 Neutralizing antibody was not administered during the immunization phase in either of these studies, but the ability to abrogate immunity by neutralizing IL-17 during the effector phase indicates that activation of TH-17 during immunization is an important effector mechanism. However, it is possible that limiting the TH-17 response during immunization might result in compensatory mechanisms capable of promoting protective inflammation. We now demonstrate using both IL-17A and IL-17 receptor A (IL-17RA) gene-targeted KO mice that reduced IL-17A activity can be overcome. We found that vaccinated mice significantly reduced their bacterial load despite the absence of IL-17A or IL-17RA. Interestingly, we also found that both IL-17A KO mice and CXCR2 KO immune mice each had equivalent levels of neutrophils compared to their corresponding wild type controls, reaffirming the possible importance of neutrophils in the eradication of H. pylori from the gastric mucosa.16
MATERIALS AND METHODS
Bacteria
H. pylori Sydney strain 1 (HpSS1)19 was grown on Columbia blood agar plates plus antibiotics (7% horse blood (Cleveland Scientific, Bath, OH), 20 µg/ml trimethoprim, 16 µg/ml cefsulodin, 6 µg/ml vancomycin, and 2.5 µg/ml amphotericin B (Sigma, St. Louis, MO) at 37°C for 4–5 days under microaerophilic conditions as previously described.16 Bacteria were transferred to Brucella broth containing 10% FBS and antibiotics and grown at 37°C and 5% C02. H. felis, originally isolated from the gastric biopsy of a cat by our laboratory20 were grown on agar plates as described for H. pylori with Polymixin B substituting for cefsulodin. For culture of bacteria from harvested stomach biopsies, plates also contained 20 µg/ml bacitracin (Sigma).
Mice
Wild type BALB/c mice (The Jackson Laboratory, Bar Harbor, ME), IL-17A KO mice (generous gift of Dr. Robert Fairchild, Cleveland Clinic, Cleveland, OH and permission of Dr. Yoichiro Iwakura of the Institute of Medical Science, University of Tokyo, Japan), and CXCR2 KO mice (generous gift of Dr. Eric Pearlman, Case Western Reserve University (CWRU), Cleveland, OH) on the BALB/c background were housed under pathogen-free conditions in microisolator cages at CWRU’s Animal Resource Center (ARC). Mouse protocols were approved by the Institutional Animal Care and Use Committee. C57BL/6 mice (The Jackson Laboratory) and IL-17RA-deficient mice backcrossed onto the C57BL/6 background were maintained in a conventional animal care facility at the University of Virginia (Charlottesville, VA). All procedures were approved by the Animal Care and Use Committee at the University of Virginia. The genotype of the WT and IL-17A KO mice were confirmed by PCR using the method and primers described by Nakae et al.21
For infection, live HpSS1 or H. felis (approximately 107 CFU) was administered directly by oral gavage on two consecutive days. For immunization, mice received 100 µg HpSS1 or H. felis lysate antigen plus 5 µg cholera toxin adjuvant (Sigma) in 20 µl PBS intranasally once a week for 4 weeks. Lysate antigens were prepared by sonication and filtration as previously described.16
IL-17A KO study
Groups consisted of 16 week old naïve, unimmunized/challenged (U/C), and immunized/challenged (I/C) wild type (WT) and IL-17A KO mice with 5–8 mice/group. In one of two studies using IL-17A KO mice, two additional groups of I/C KO mice received either anti-IFN-γ antibody XMG1.2 (BioXCell, West Lebanon, NH, #BE0055) or control IgG (Jackson ImmunoResearch, West Grove, PA, #012000003) according to the following treatment: 1 mg IgG in PBS administered i.p. 1 day prior to and 1 week following the first live HpSS1 challenge. We have previously shown that treating mice with this dose of monoclonal antibody XMG1.2 one day before and 10 days after infection with H. felis, significantly reduced gastric inflammation in these mice 21 days post-challenge.22 Groups of naïve mice were female, and all other groups consisted of male mice. All mice were harvested at 2 weeks post-challenge, a time point previously demonstrated to be sufficient to achieve significant reductions in bacterial load and which would avoid the need for protracted treatment with injected antibodies.16, 23 Stomach biopsies were taken for bacterial load quantification, inflammation grading, and RNA isolation. Bacterial load was determined by colony forming units (CFU) on homogenates of pre-weighed biopsies as described below. The bacterial load for H. felis was also determined by quantitative PCR as described below. Another biopsy was cut along the outer curvature of the stomach, fixed in 10% buffered formalin, and evaluated for gastritis and neutrophil counts using H&E-stained sections by a pathologist blinded to sample identities. The most inflamed area of each section was given a score of 0–5 after being evaluated in blind fashion for the extent, depth, and makeup of cellular infiltrate as well as changes in tissue architecture, as previously described in detail for H. pylori infection.15 Neutrophil scores refer to the number of neutrophils per high power field in that most inflamed area. A third stomach biopsy was stored in RNAlater (Ambion, Austin, TX) at −80°C, and RNA was isolated with a RiboPure kit according to the manufacturer’s protocol (Ambion). RNA was used for reverse transcription quantitative PCR for TNFα and IL-22.
IL-17RA KO study
Female mice approximately 20 weeks old at harvest were divided into mock-immunized, mock-immunized/challenged, and I/C groups and harvested at 2 weeks post-challenge. Mock-immunized mice received intranasal PBS. Stomach biopsies were processed as described above with the following exceptions: biopsies for histology were fixed in Bouin’s solution and sections were stained for myeloperoxidase (MPO) in addition to H&E. The bacterial load was determined by Q-PCR.
CXCR2 KO study
Male and female WT and CXCR2 KO littermates were divided into groups of naïve, U/C, and I/C with 5–8 mice/group. Mice were approximately 16 weeks old at the time of harvest. Stomach biopsies were harvested at 2 weeks post-challenge and processed as described for the IL-17A KO study, except that bacterial load was determined by Q-PCR.
Antibody titer determination
Serum IgG titers for reactivity to H. felis lysate antigens were measured by endpoint titer determination. Ninety-six well Maxisorp plates (Nalge NUNC International, Rochester, NY) were coated with 100 µl of 10 µg/ml of H. felis sonicate antigen/well in 0.05 M carbonate-bicarbonate buffer, pH 9.6 and incubated in a humidified chamber overnight at 4°C. Plates were emptied and washed three times between all subsequent steps with PBS containing 0.1% bovine serum albumin (BSA) (USB, Cleveland, Ohio). Wells were blocked for 2 h at room temperature with 200 µl PBS containing 1% bovine BSA, incubated with 100 µl/well serum samples in half log dilutions in PBS with 1% BSA for 90 min, and then with 100 µl/well alkaline phosphatase-conjugated goat anti-mouse IgG (Southern Biotechnology, Birmingham, Ala.) diluted 1/1,500 in PBS containing 1% BSA for 60 minutes. Assays were developed using 1 mg/ml p-nitrophenyl phosphate [Sigma] substrate in 50 mM glycine-1 mM MgCl2 buffer [pH 9.6]. Absorbance was recorded after 60 min at 405 nm with a BioTek EL-312 microplate reader (BioTek Instruments, Winooski, VT.). Endpoint titers were determined by interpolating the serum dilution to the nearest quarter log yielding an O.D. of at 0.5. above conjugate (no experimental serum sample added) control.
Bacterial load determination
A 2 mm wide longitudinal strip of the greater curvature of each stomach was placed into pre-weighed 1.5 ml microcentrifuge tubes containing 200µl urease test broth, weighed again, and then homogenized with disposable pellet pestles (Kontes, Vineland, NJ). Homogenate was diluted serially in 10-fold dilutions in sterile PBS to 1:1000 and 10µl of each dilution was plated. Colonies were counted after 5 to 7 days of incubation, and representative colonies were tested for urease, oxidase and catalase activities to confirm their identity as H. pylori. The gas generating envelope system used for maintaining microaerobic cultures was discontinued by the manufacturer. Therefore, bacterial load determinations for all remaining studies were performed using quantitative PCR. For bacterial load determination, total DNA was extracted from frozen gastric tissue using DNeasy (Qiagen) but with an additional step following Proteinase K digestion in which the samples were incubated at 95°C for 10 min. to help release bacterial DNA. PCR amplification was performed on an Eppendorf Realplex real time thermocycler (Westbury, NY) using primers for ureC as previously reported24 and a standard curve consisting of purified chromosomal DNA from H. pylori SS1. H. felis was quantified using primers specific for 16S rRNA.25 For each sample the PCR reaction was performed in duplicate with the SYBR Green supermix (Fermentas, Glen Burnie, MD).
RT-Quantitative-PCR
For relative quantification of cytokines, total RNA was extracted using Trizol (Invitrogen) and then RNA (1 µg) was converted to cDNA using a reverse transcription kit (Qiagen). PCR amplification was performed using a two step cycle of 95°C for 15 sec. followed by 60°C for one min for 40 cycles in an Eppendorf realplex2 Mastercylcer (Hamburg, Germany). Primer sequences were as follows; TNF forward CCCAAAGGGATGAGAAGT and TNF reverse ACAGGCTTGTCACTCGAA; IL-22 forward ATACATCGTCAACCGCACCTTT and IL-22 reverse AGCCGGACATCTGTGTTGTTAT; GAPDH forward TGTAGACCATGTAGTTGAGGTCA and GAPDH reverse AGGTCGGTGTGAACGGATTTG. Gastric tissue RNA from a naïve mouse of each group was chosen as a calibrator using relative analysis real-time PCR. Fold differences in the expression of genes in the tissue were calculated as 2(CtGene-CtGAPDH)-(CtGene-CtGAPDH).
Statistics
Statistics were determined using ANOVA. Differences between groups were considered significant at an interval level of p<0.05.
RESULTS
Vaccinated WT and IL-17A KO mice respond comparably to H. pylori challenge
We investigated whether an IL-17A deficiency can be overcome to achieve protective immunity using naïve, U/C, and I/C WT and IL-17A KO mice. We also included groups of I/C IL-17A KO mice receiving anti-IFN-γ or control antibody treatments to examine a possible compensatory mechanism. WT I/C mice and all three groups of IL-17A KO I/C mice exhibited comparable levels of gastritis that was significantly increased over their U/C counterparts (Fig 1a; P < 0.004). The levels of gastritis in U/C WT and IL-17A KO mice were also not significantly different from each other, although unimmunized mice generally exhibit very little inflammation at 2 weeks post-infection. Consistent with elevated inflammation and neutrophil counts, all I/C IL-17A KO and WT mice were able to significantly reduce the bacterial load compared to their corresponding U/C mice (Fig. 1b; P = 0.02). There were no significant differences in H. pylori levels among these three I/C IL-17A KO groups regardless of antibody treatments (P > 0.05). We did observe, however, that U/C IL-17A KO mice had significantly lower bacterial colonization than U/C WT mice (P < 0.001).
Figure 1. Protective immunity against H. pylori in IL-17A KO mice.
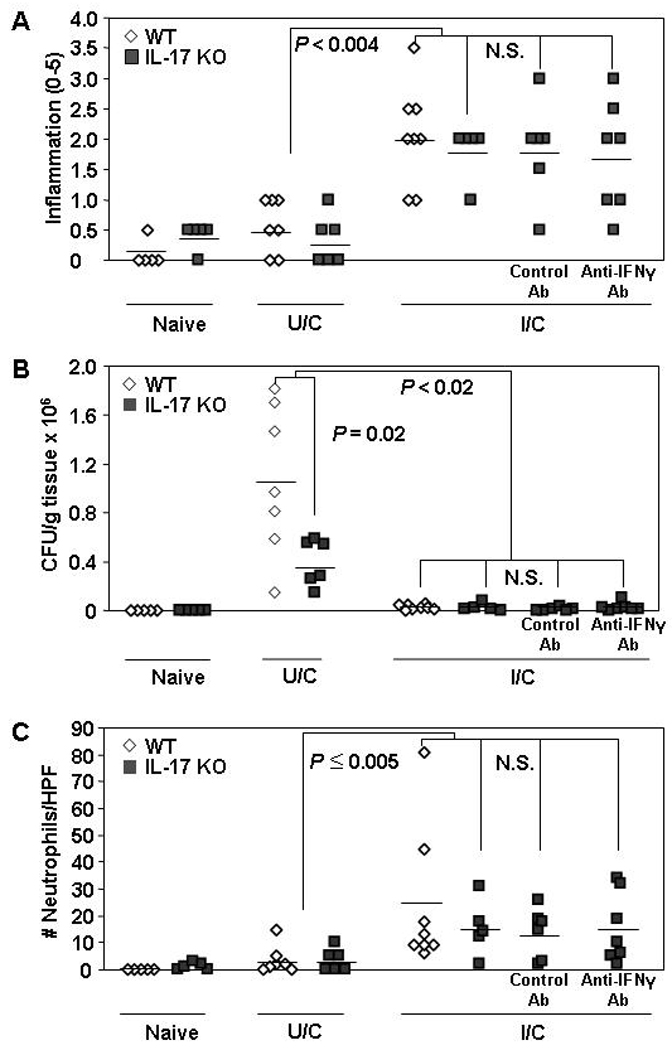
WT and IL-17A KO mice were intranasally immunized once a week for four weeks with 100 µg of H. pylori sonicate plus 5 µg of cholera toxin. On days 7 and 8 post-immunization, mice were challenged by orogastric gavage with 1–2 × 107 cfu live H. pylori. Two additional groups of I/C mice were inoculated i.p. with 1 mg of either control rat IgG or anti-IFNγ monoclonal Ab 1 day prior to and 1 week following the first bacterial challenge. Naive mice were untreated. Gastric biopsies were harvested from all mice two weeks post-challenge and evaluated for gastritis (A), H. pylori load (B) and gastric neutrophil numbers (C). Similar results were obtained in two experiments. n = 5 – 8 mice/group. N.S. = not significant.
All I/C mice, regardless of genotype or antibody treatment had similar gastric neutrophil levels (Fig 1c). Similarly, neutrophil counts between WT and IL-17A KO U/C mice were also comparable but only WT I/C mice had neutrophil counts significantly elevated over their respective U/C mice (p < 0.005). Additionally, since cytokines such as TNFα and IL-22 can also promote granulocyte activity we quantified these cytokines to determine if they might be upregulated in the absence of IL-17A. The gastric mRNA expression levels for both of these cytokines, however, were comparable between WT and IL-17A KO mice (data not shown).
Vaccination protects IL-17A KO mice from H. felis challenge
The use of IL-17A-neutralizing antibodies to demonstrate that IL-17A plays an important role in protective immunity to Helicobacter was predominantly studied using the mouse model of H. felis infection17. To test whether differences observed between those IL-17A neutralization studies and our results using IL-17A KO mice were due to differences between the H. pylori and H. felis infection models, we used IL-17A KO mice to test against H. felis challenge. Similar to the H. pylori challenge model we observed a significant increase in gastritis in both WT and IL-17A KO I/C mice compared to their respective U/C controls (Fig. 2a; P < 0.0001). Protection of IL-17A KO mice was also similar to the H. pylori model as both WT and IL-17A KO immunized mice achieved significant reductions in bacterial load (Fig. 2b; P < 0.0001). There were no significant differences in H. felis bacterial loads between WT and IL-17A KO IL-17A KO I/C mice although IL-17A KO U/C mice had significantly greater number of bacteria than WT U/C mice (P = 0.003).
Figure 2. Protective immunity against H. felis in IL-17A KO mice.
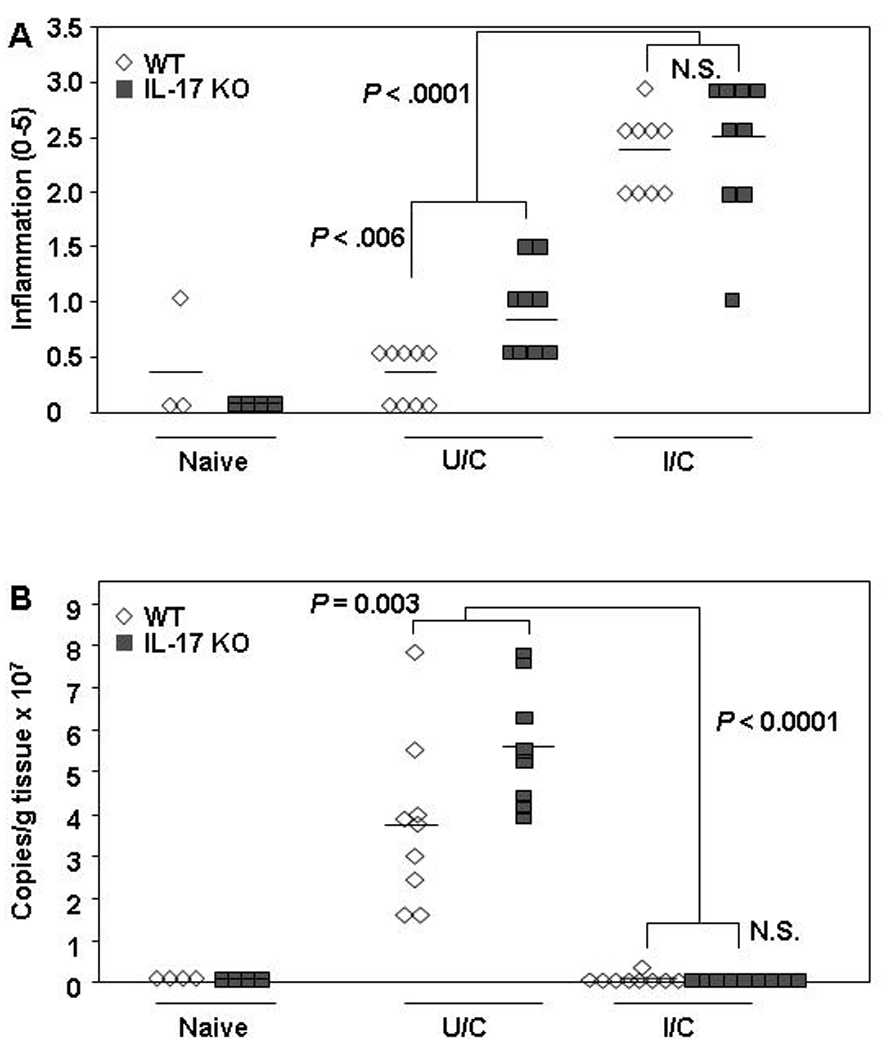
WT and IL-17A KO mice were intranasally immunized once a week for four weeks with 100 µg of H. felis sonicate plus 5 µg of cholera toxin. On days 7 and 8 post-immunization, both mice were challenged by orogastric gavage with 1–2 × 107 cfu live H. felis. Naive mice were untreated. Gastric biopsies were harvested from all mice two weeks post-challenge and evaluated for gastritis (A), and H. felis numbers (B). n = 4 – 9 mice/group.
We and others have demonstrated that antibodies are not required for vaccine-induced reductions in bacterial loads of H. pylori or H. felis.26–28 However, it is possible that anti-Helicobacter antibodies participate in bacterial clearance when present and they can also be used to monitor the immunogenicity of the vaccine. Therefore, we measured the serum antibody titers against H. felis antigens using blood isolated form the mice at sacrifice. Comparable titers of 1:103.9 were achieved between WT and IL-17A KO I/C mice (data not shown). Titers for U/C were reduced relative to I/C mice with WT and IL-17A KO achieving titers of 1:101.9 and 1:102.3 respectively. The titers for I/C WT and IL-17A KO mice were significantly greater than their respective U/C counterparts (P < 0.0001).
Vaccination of IL-17AR KO mice also reduces H. pylori load
In light of our IL-17A KO mouse results, we also used IL-17RA KO mice in our H. pylori model. IL-17RA KO I/C mice had significantly elevated gastritis over WT I/C mice (P < 0.001), although both these groups had increased inflammation versus the respective groups of U/C mice (Fig. 3a; P < 0.02). Similar to the previous experiment using IL-17A KO mice, the WT and IL-17RA KO I/C mice significantly reduced their bacterial load compared to their U/C counterparts (Fig. 3b; P < 0.01). Unlike IL-17A KO mice, there was no significant difference in the baseline colonization levels as WT and IL-17RA KO U/C mice had comparable bacterial loads. Interestingly, the IL-17RA KO I/C mice had, on average, a four-fold decrease in gastric neutrophils compared to WT I/C mice (P = 0.003) despite the increase in general inflammation (Fig 3c). The gastric neutrophil levels in IL-17RA KO I/C mice were not significantly different than IL-17RA KO U/C mice.
Figure 3. Protective immunity against H. pylori in IL-17AR KO mice.
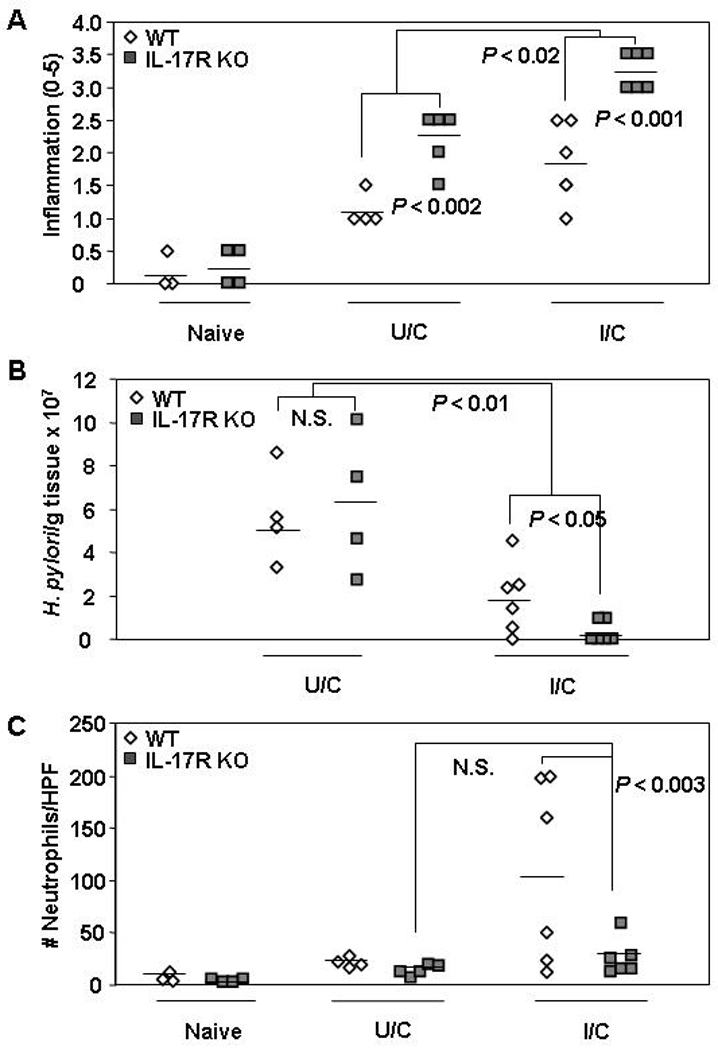
WT and IL-17AR KO mice received intranasal immunizations once a week for four weeks with 100 µg of H. pylori sonicate plus 5 µg of cholera toxin and received an orogastric challenge of 1–2 × 107 cfu H. pylori on days 7 and 8 following this treatment. Naive and unimmunized/challenged (U/C) mice received mock immunizations of intranasal PBS. U/C mice were infected at the same time as the I/C mice. Gastric biopsies were harvested from all mice two weeks post-challenge and evaluated for gastritis (A), H. pylori load (B) and gastric neutrophil numbers (C). n = 3 – 6 mice/group. N.S. = not significant.
Significant neutrophil recruitment occurs in mice lacking CXCR2
Since I/C IL-17A KO mice demonstrated considerable neutrophil infiltrate in the stomach, we tested whether factors such as the IL-17A-induced, neutrophil recruitment chemokines KC, MIP-2, and LIX are required for protective immunity. We performed immunization and challenge of CXCR2 mice which lack the common receptor for all three chemokines. General inflammation levels for U/C and I/C CXCR2 KO mice were not significantly different from their WT counterparts (Fig 4a). I/C CXCR2 KO mice did, however, have significantly elevated gastritis relative to the U/C CXCR2 KO mice (P = 0.02) and were also able to significantly reduce their bacterial load (Fig. 4b; P < 0.0005). The CXCR2 KO and WT mice responded similarly in terms of bacterial load, as there were no significant differences between the two I/C groups or between the two U/C groups. Despite the lack of CXCR2, U/C and I/C CXCR2 KO mice had neutrophil levels comparable to U/C and I/C WT mice, respectively (Fig 4c). There was also no statistical differences in gastric neutrophils between CXCR2 KO and WT I/C mice or between CXCR2 KO and WT U/C mice.
Figure 4. Protective immunity against H. pylori in CXCR2 KO mice.
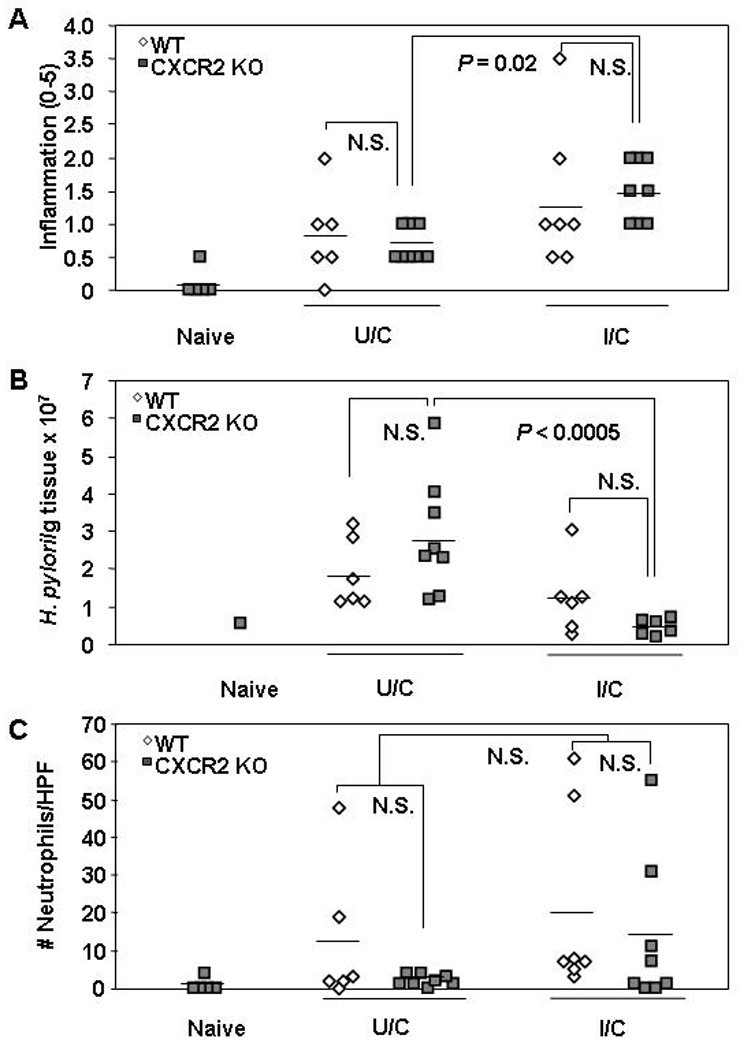
WT and CXCR2 KO mice received intranasal immunizations once a week for four weeks with 100 µg of H. pylori sonicate plus 5 µg of cholera toxin and received an orogastic challenge of 1–2 × 107 cfu H. pylori on days 7 and 8 following this treatment. U/C mice were also infected at that time. Gastric biopsies were harvested from all mice two weeks post-challenge and evaluated for gastritis (A), H. pylori load (B) and gastric neutrophil numbers (C). n = 5 – 8 mice/group. N.S. = not significant.
DISCUSSION
Our previous findings documented the prevalence of IL-17 in the gastric mucosa of immune mice compared to unimmunized controls and demonstrated the importance of neutrophil-based inflammation to protective immunity against H. pylori.16 These results implicated IL-17 as an important factor of the vaccine-induced protective immune response to H. pylori. Two laboratories have now confirmed the importance of IL-17 by administering IL-17-specific blocking antibodies during challenge of immunized mice using the H. felis and the H. pylori mouse models.17, 18 They demonstrated significantly reduced vaccine efficacy compared to mice treated with control antibody. An important aspect of their IL-17 neutralization however, was that antibody was applied subsequent to the immunization protocol and therefore, the host had likely committed to a specific adaptive immune pathway prior to antibody treatment. The investigators were able to successfully ameliorate immunity by blocking IL-17 during bacterial challenge, therefore nullifying the host recall response. Since there is often redundancy in the immune system, we tested whether complete elimination of IL-17A during both the immunization (immune induction) and challenge (immune recall) phase would result in the generation of immunity by some other mechanism. The present study now shows, using both IL-17A and IL-17RA KO mice, that in the absence of IL-17A or the capacity to respond to IL-17A, effective immunity is induced after vaccination against either H. pylori and H. felis. This immunity is capable of significantly reducing the bacterial load upon challenge.
We were unable to identify a compensatory mechanism in our IL-17A deficiency models. Challenge of immunized IL-17A KO mice that received anti-IFNγ antibody treatment resulted in H. pylori levels comparable to control antibody-treated and untreated I/C KO mice and I/C WT mice. This was surprising given that Otani et al recently showed that anti-IL-17 antibody treatment of mice chronically infected with H. pylori increased gastric IFNγ mRNA expression.29 It is also puzzling in light of studies demonstrating that immunized IL-12/23 p40 subunit KO mice fail to be protected against H. pylori5, 6 and the p40 knockout theoretically blocks both IL-12-induced Th1 responses and IL-23-induced Th-17 responses. It is possible that the timing of antibody administration was not optimal for blocking IFNγ and that additional or earlier applications might have produced a different result. However, we have shown that one mg doses of XMG1.2 monoclonal antibody at a somewhat less frequent dosing schedule (minus one and plus 10 days post challenge) than used in the current report had a highly significant (p < 0.005 to < 0.0005) impact on the gastritis observed in U/C and I/C mice infected with the related organism, H. felis.22 It is also possible there are other compensatory mechanisms capable of promoting protective immunity in vaccinated mice promoted by either IL-12 or IL-23. It may be that our anti-IFNγ treatments failed to sufficiently reduce the effects of this cytokine. Since prolonged use of an antibody decreases its efficacy over time, this question of redundancy might be further addressed with IL-17A/IFNγ double-KO mice.
Additional factors that might contribute to immunity include IL-22, an IL-10 cytokine family member secreted by Th-17 cells that can induce innate immune responses such as the secretion of host antimicrobial peptides.30, 31 Expansion of IL-22-producing cells and production of IL-22 are dependent on IL-23, but not IL-12.30 However, we did not observe increased levels of IL-22 mRNA in the stomachs of I/C IL-17A KO mice at the time of harvest. Another cytokine, TNFα plays a role in neutrophil recruitment and is produced by T cells, mast cells, and macrophages in response to H. pylori.15, 32, 33 We also quantified the expression of TNFα mRNA in the gastric mucosa of IL-17A KO mice at harvest to determine whether TNFα might compensate for the absence of IL-17A. Similar to IL-22 however, we did not observe any difference between I/C WT and I/C IL-17A KO mice, and levels were only marginally higher than nonimmunized controls. This does not exclude the possibility that increased TNFα levels were present earlier in the immune response and may have played a role in neutrophil recruitment.
Indeed, there are likely many overlapping mechanisms for neutrophil recruitment in this model. A key mechanism by which IL-17A results in neutrophil recruitment in mice is via the induction of the CXC chemokines KC, MIP-2, and LIX.34–36 Our data regarding gastric neutrophil levels in IL-17A KO mice suggest, not surprisingly, that there are mechanisms other than an IL-17A-CXC chemokine pathway of neutrophil recruitment in our H. pylori models. In support of this, we also showed that CXCR2 KO mice, which lack the receptor for KC, MIP-2, and LIX, are still able to recruit neutrophils to the stomach. Other mechanisms for attracting neutrophils include complement factors, acute phase cytokines, and membrane lipid derivatives, and any number of these factors may contribute. Thus, in the absence of CXCR2 activity, factors such as TNFα and others may compensate and promote neutrophil recruitment, but in the absence of TNFα, the CXC chemokines are sufficient. Elucidating the most common pathway of vaccine-induced neutrophil recruitment will most likely require the simultaneous elimination or blocking of several mechanisms at once.
The impact on neutrophil recruitment to the gastric mucosa was not uniform in our study. Our results in IL-17A KO mice are consistent with a recent study by Shiomi et al who reported that H. pylori-infected C57BL/6 IL-17A KO mice had lower colonization than WT counterparts at one, three, and six months post-challenge.37 We also saw reduced bacterial colonization with our infected BALB/c IL-17A KO mice, although the endpoint of our study was two weeks. However, unlike Shiomi et al. who observed reduced neutrophil infiltration in infected IL-17A KO mice, our IL-17A KO mice did not show significant differences in neutrophil recruitment compared to WT mice. It is possible that harvest at two weeks post-infection was not sufficient to observe reliable differences in the ability of U/C KO and WT mice to recruit neutrophils, since there is generally little inflammation in U/C mice this early after challenge. It is also likely that, unlike the immune-suppressive environment of U/C mouse stomachs, there would be multiple compensating proinflammatory mechanisms to recruit neutrophils to the stomachs of I/C mice even in the absence of IL-17A. The relevance of the observed decreased colonization is unclear, particularly since we did not observe any differences in baseline bacterial colonization in our WT and IL-17RA KO U/C mice.
In contrast, we did observe decreased gastric neutrophil infiltration in our IL-17RA KO study. This suggests that IL-17A does contribute to neutrophil recruitment even though there are other mechanisms involved in that process. We are unsure as to why IL-17A and IL-17RA KO mice demonstrated varying results in terms of their inflammatory responses. One possible explanation, however, is the ability of isoforms other than IL-17A to signal through the IL-17RA. Our IL-17A KO mice are deficient in IL-17A but continue to produce IL-17F. Both IL-17A and IL-17F signal through IL-17RA and therefore mice deficient in IL-17RA might have even less IL-17A activity than the IL-17A KO mice. Alternatively, although the function of IL-17F has been shown to be dependent on IL-17RA, it also interacts with IL-17RC.38, 39 Furthermore, IL-17RA may heterodimerize with IL-17RC suggesting that the IL-17RA KO mice may have diminished function through IL-17RC as well.40 It is possible that IL-17F, as a proinflammatory cytokine, is also playing a role in the immune response against H. pylori. In any case, IL-17A likely has important functions despite not being solely required for bacterial load reduction in our vaccination model.
We also note that for unimmunized/challenged mice, IL-17A KO mice developed significantly greater inflammation than WT infected with H. felis, and IL-17RA KO mice developed significantly greater inflammation than WT mice infected with H. pylori. This observation was not uniform as there was no difference in the inflammation of unimmunized/challenged IL-17A KO mice infected with H. pylori. However, recent evidence indicates that TH-17 cells might have the potential to act as regulatory cells via the production of IL-10.41–44 Therefore, in the absence of such regulatory TH-17 cell activity inflammation might be expected to increase in response Helicobacter infection. We and others have demonstrated that in the absence of IL-10, H. pylori-associated gastritis in mice is significantly increased compared to WT mice.13, 45, 46
In summary, our data demonstrate that although IL-17A is prevalent in the gastric mucosa of immunized mice following challenge,16 and despite the previous demonstration that antibody-mediated neutralization during challenge of mice compromises the protective immune response,17 the complete absence of IL-17A or its receptor did not significantly impact the ability of the murine host to develop vaccine-induced protective immunity against H. pylori or H. felis. We further demonstrate that although neutrophil recruitment was negatively impacted in the IL-17RA KO model, no such compromise was observed in either IL-17A KO mice or immune CXCR2 KO mice. Taken together, these experiments indicate there are multiple mechanisms for activating vaccine based protective inflammatory responses against H. pylori and that in the absence of an IL-17A-CXC chemokine pathway of enhanced neutrophil recruitment the host can employ compensatory mechanisms of immunity.
ACKNOWEDGEMENTS
This work was supported by National Institutes of Health grants AI-055710 (T.G.B.), DK-46461 (S.J.C.), and AI-083694 (J.G.N.)
REFERENCES
- 1.Marshall BJ. The 1995 Albert Lasker Medical Research Award. Helicobacter pylori. The etiologic agent for peptic ulcer. J Am Med Assoc. 1995;274:1064–1066. doi: 10.1001/jama.274.13.1064. [DOI] [PubMed] [Google Scholar]
- 2.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 3.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 4.Becx MC, Janssen AJ, Clasener HA, de Koning RW. Metronidazole-resistant Helicobacter pylori. Lancet. 1990;335:539–540. doi: 10.1016/0140-6736(90)90772-w. [DOI] [PubMed] [Google Scholar]
- 5.Akhiani AA, Pappo J, Kabok Z, Schon K, Gao W, Franzen LE, Lycke N. Protection against Helicobacter pylori infection following immunization is IL-12-dependent and mediated by Th1 cells. J Immunol. 2002;169:6977–6984. doi: 10.4049/jimmunol.169.12.6977. [DOI] [PubMed] [Google Scholar]
- 6.Garhart CA, Heinzel FP, Czinn SJ, Nedrud JG. Vaccine-induced reduction of Helicobacter pylori colonization in mice is interleukin-12 dependent but gamma interferon and inducible nitric oxide synthase independent. Infect Immun. 2003;71:910–921. doi: 10.1128/IAI.71.2.910-921.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawai N, Kita M, Kodama T, Tanahashi T, Yamaoka Y, Tagawa Y, Iwakura Y, Imanishi J. Role of gamma interferon in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Infect Immun. 1999;67:279–285. doi: 10.1128/iai.67.1.279-285.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sayi A, Kohler E, Hitzler I, Arnold I, Schwendener R, Rehrauer H, Muller A. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J Immunol. 2009;182:7085–7101. doi: 10.4049/jimmunol.0803293. [DOI] [PubMed] [Google Scholar]
- 9.Algood HM, Gallo-Romero J, Wilson KT, Peek RM, Jr., Cover TL. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol Med Microbiol. 2007;51:577–586. doi: 10.1111/j.1574-695X.2007.00338.x. [DOI] [PubMed] [Google Scholar]
- 10.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 11.Khamri W, Walker MM, Clark P, Atherton JC, Thursz MR, Bamford KB, Lechler RI, Lombardi G. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun. 2010;78:845–853. doi: 10.1128/IAI.00524-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanchard TG, Yu F, Hsieh CL, Redline RW. Severe inflammation and reduced bacteria load in murine helicobacter infection caused by lack of phagocyte oxidase activity. J Infect Dis. 2003;187:1609–1615. doi: 10.1086/374780. [DOI] [PubMed] [Google Scholar]
- 13.Berg DJ, Lynch NA, Lynch RG, Lauricella DM. Rapid development of severe hyperplastic gastritis with gastric epithelial dedifferentiation in Helicobacter felis-infected IL-10(−/−) mice. Am J Pathol. 1998;152:1377–1386. [PMC free article] [PubMed] [Google Scholar]
- 14.Ismail HF, Fick P, Zhang J, Lynch RG, Berg DJ. Depletion of neutrophils in IL-10(−/−) mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter. J Immunol. 2003;170:3782–3789. doi: 10.4049/jimmunol.170.7.3782. [DOI] [PubMed] [Google Scholar]
- 15.Garhart CA, Redline RW, Nedrud JG, Czinn SJ. Clearance of Helicobacter pylori infection and resolution of postimmunization gastritis in a kinetic study of prophylactically immunized mice. Infect Immun. 2002;70:3529–3538. doi: 10.1128/IAI.70.7.3529-3538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLyria ES, Redline RW, Blanchard TG. Vaccination of mice against H. pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterol. 2009;136:247–256. doi: 10.1053/j.gastro.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velin D, Favre L, Bernasconi E, Bachmann D, Pythoud C, Saiji E, Bouzourene H, Michetti P. Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterol. 2009;136:2237 e1–2246 e1. doi: 10.1053/j.gastro.2009.02.077. [DOI] [PubMed] [Google Scholar]
- 18.Flach CF, Ostberg AK, Nilsson AT, Malefyt Rde W, Raghavan S. Proinflammatory cytokine gene expression in the stomach correlates with vaccine-induced protection against Helicobacter pylori infection in mice: an important role for interleukin-17 during the effector phase. Infect Immun. 2011;79:879–886. doi: 10.1128/IAI.00756-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterol. 1997;112:1386–1397. doi: 10.1016/s0016-5085(97)70155-0. [DOI] [PubMed] [Google Scholar]
- 20.Czinn SJ, Cai A, Nedrud JG. Protection of germ-free mice from infection by Helicobacter felis after active oral or passive IgA immunization. Vaccine. 1993;11:637–642. doi: 10.1016/0264-410x(93)90309-l. [DOI] [PubMed] [Google Scholar]
- 21.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 22.Mohammadi M, Czinn S, Redline R, Nedrud J. Helicobacter-specific cell-mediated immune responses display a predominant Th1 phenotype and promote a delayed-type hypersensitivity response in the stomachs of mice. J Immunol. 1996;156:4729–4738. [PubMed] [Google Scholar]
- 23.Ding H, Nedrud JG, Wershil B, Redline RW, Blanchard TG, Czinn SJ. Partial protection against Helicobacter pylori in the absence of mast cells in mice. Infect Immun. 2009;77:5543–5550. doi: 10.1128/IAI.00532-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He Q, Wang JP, Osato M, Lachman LB. Real-time quantitative PCR for detection of Helicobacter pylori. J Clin Microbiol. 2002;40:3720–3728. doi: 10.1128/JCM.40.10.3720-3728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong L, Smith JG, Bramhill D, Abruzzo GK, Bonfiglio C, Cioffe C, Flattery AM, Gill CJ, Lynch L, Scott PM, Silver L, Thompson C, Kropp H, Bartizal K. A sensitive and specific PCR method to detect Helicobacter felis in a conventional mouse model. Clin Diagn Lab Immunol. 1996;3:73–78. doi: 10.1128/cdli.3.1.73-78.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanchard TG, Czinn SJ, Redline RW, Sigmund N, Harriman G, Nedrud JG. Antibody-independent protective mucosal immunity to gastric helicobacter infection in mice. Cell Immunol. 1999;191:74–80. doi: 10.1006/cimm.1998.1421. [DOI] [PubMed] [Google Scholar]
- 27.Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R, Lee CK, Kleanthous H, Monath TP. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J Exp Med. 1998;188:2277–2288. doi: 10.1084/jem.188.12.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutton P, Wilson J, Kosaka T, Wolowczuk I, Lee A. Therapeutic immunization against Helicobacter pylori infection in the absence of antibodies. Immunol Cell Biol. 2000;78:28–30. doi: 10.1046/j.1440-1711.2000.00881.x. [DOI] [PubMed] [Google Scholar]
- 29.Otani K, Watanabe T, Tanigawa T, Okazaki H, Yamagami H, Watanabe K, Tominaga K, Fujiwara Y, Oshitani N, Arakawa T. Anti-inflammatory effects of IL-17A on Helicobacter pylori-induced gastritis. Biochem Biophys Res Commun. 2009;382:252–258. doi: 10.1016/j.bbrc.2009.02.107. [DOI] [PubMed] [Google Scholar]
- 30.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Karttunen R. Blood lymphocyte proliferation, cytokine secretion and appearance of T cells with activation surface markers in cultures with Helicobacter pylori. Comparison of the responses of subjects with and without antibodies to H. pylori. Clin Exp Immunol. 1991;83:396–400. doi: 10.1111/j.1365-2249.1991.tb05650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, Hirayama T, Ra C. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J Immunol. 2002;168:2603–2607. doi: 10.4049/jimmunol.168.6.2603. [DOI] [PubMed] [Google Scholar]
- 34.Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76:135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- 35.Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, Jorres A. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 36.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiomi S, Toriie A, Imamura S, Konishi H, Mitsufuji S, Iwakura Y, Yamaoka Y, Ota H, Yamamoto T, Imanishi J, Kita M. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter. 2008;13:518–524. doi: 10.1111/j.1523-5378.2008.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman SJ, Pirhonen J, Kolls JK. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol. 2005;175:404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 41.Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CJ, Gran B, Ciric B, Rostami A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol. 2007;8:1372–1379. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- 42.Jankovic D, Trinchieri G. IL-10 or not IL-10: that is the question. Nat Immunol. 2007;8:1281–1283. doi: 10.1038/ni1207-1281. [DOI] [PubMed] [Google Scholar]
- 43.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 44.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O'Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 45.Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol. 2001;166:7456–7461. doi: 10.4049/jimmunol.166.12.7456. [DOI] [PubMed] [Google Scholar]
- 46.Matsumoto Y, Blanchard TG, Drakes ML, Basu M, Redline RW, Levine AD, Czinn SJ. Eradication of Helicobacter pylori and resolution of gastritis in the gastric mucosa of IL-10-deficient mice. Helicobacter. 2005;10:407–415. doi: 10.1111/j.1523-5378.2005.00349.x. [DOI] [PubMed] [Google Scholar]