

Abstract
Lung dysfunction is an important part of the pathology of the neurodegenerative disorder, Niemann-Pick C1 (NPC1). We have studied the pulmonary disease in the Npc1NIH/NIH mouse model. On histology, we find large numbers of alveolar foamy macrophages but no alveolar proteinosis. Lung weight as percent of body weight was markedly increased; using the flexiVent small animal ventilator (SCIREQ, Inc.), we find inspiratory capacity, elastance and hysterisivity to be increased while resistance was not changed. Cholesterol measurements show a doubling of lung cholesterol levels. Collagen is also increased. Treatment of Npc1−/− mice with hydroxypropyl-β-cyclodextrin (HPBCD), despite efficacious effects in brain and liver, results in little difference from age-matched controls (using a CNS-expressed transgene to extend the life expectancy of the Npc1−/− mice) for these variables.
Keywords: Niemann-Pick C1, intracellular cholesterol transport, foamy macrophages, small animal ventilator, hydroxypropyl-beta-cyclodextrins
Introduction
It has long been known that pulmonary disease, along with liver and spleen storage, are important aspects of NPC. In the period before Niemann-Pick A and B were distinguished from NPC, a variety of pulmonic x-ray descriptions were provided including micronodularity and diffuse changes similar to those seen in tuberculosis [1]. Even when the 3 types were distinguished, similarities of lung disease among NPCA, B, and C, with finding of interstitial lung pathology and foamy cells obtained by bronchoalveolar lavage, were confirmed [2]. Pulmonary disease in a case of the longer surviving NPCD (a milder mutant of NPC1) was well described and included decreased diffusion capacity for CO [3]. As NPC1 and NPC2 were distinguished, it became apparent that NPC2 has much earlier onset and showed more severe lung disease [4–5] which included pulmonary alveolar proteinosis [6]. The lung disease has been studied in the Npc1NIH/NIH mouse model by electron microscopy [7]. Lung endothelial cells and type I pneumocytes were heavily laden with stored material. Foamy macrophages were abundant in the alveolar spaces [7].
Nearly a decade ago, an efficacious effect of hydroxypropyl-beta-cyclodextrins (HPBCD) on slowing the neurodegeneration in the mouse model of Niemann-Pick C1 was shown [8]. HPBCDs are substituted rings of 7 glucoses and this same publication showed they do not cross the blood brain barrier (BBB). Perhaps because it seems counter-intuitive to treat a CNS disorder with a drug that does not cross the BBB, and perhaps because the effects are rather small when the drug is only started at the time of weaning, the treatment was not pursued. Interest was re-ignited when HPBCDs were used as a carrier for neurosteroids and injections were started at 7 days, when the BBB is still open in mice [9]. It was soon found that HPBCD alone was equally efficacious as HPBCD with the neurosteroid [10].
We have studied the pulmonary disease in the Npc1NIH/NIH model by histology, pulmonary function tests, and biochemical assays. We have also studied the effects of HPBCD therapy and find that it does not ameliorate the lung disease.
Materials and Methods
Mice
The Npc1NIH/NIH (Npc1−/−) mutant is essentially a “natural” knockout: an active mouse retroposon inserted 1,100 bp and deleted 800 bp of the Npc1 gene [11]. This also created a frameshift. Thus, only a small amount of the apparently truncated Npc1 mRNA [12] and no protein is detectable in homozygous recessive mice. These mice are maintained on the BALB/cJ background and derived from heterozygous matings. Tail tips were routinely obtained at 14 days and DNA was typed by PCR according to Loftus, et al [11]. Only 17%, instead of the expected 25% Npc1 −/− are weaned and untreated mice survive (16) 73 +/− 2 days.
We also used these mice carrying the glial fibrillary acidic protein promoter-promoted Npc1 cDNA. These mice express Npc1 only in fibrillary astrocytes and have significantly prolonged survivals [13]. The transgene is not expected to have any significant influence on lung function, although it does influence the intestine because of the presence of glia in enteric ganglia [14]. Since there was likely to be progression of lung disease with age, we used these mice to provide age-matched controls for the cyclodextrin-treated mice. These are also on the BALB/cJ genetic background (N6 or greater).
We initially used 3 Npc1+/+ or Npc1+/− as wild-type, age-matched controls, ages 44 to 47 days, for the untreated Npc1−/− mice, and 5, ages 94 to 112 days, as age-matched controls initially intended for treated mice. There were no statistical differences in any of the two wild-type age group measurements so they were combined as one group. We studied 8 untreated Npc1−/− mice (ages 72 to78 days) which were compared to the above controls. Five treated (ages 133 to 154 days) Npc1−/− mice were studied. As mentioned above, to provide age-matched Npc1−/− controls for the treated Npc1−/− mice, we studied 7 Npc1−/− GFAP tg mice (ages 106 to 116 days). Since not every study was successful on every mouse (e.g. some died on the flexiVent), actual numbers for each determination are provided in the figures of results. Treated mice that were not sacrificed for studies live to 150+ days.
Hydroxypropyl-beta-cyclodextrin treatment
(2-hydroxypropyl)-beta-cyclodextrin from Sigma, batch 094K1435, degree of substitution not provided, was dissolved in normal saline at 200 mg/ml. 0.02 c.c./g were injected subcutaneously, to achieve a dose of 4g/kg. This was injected weekly starting at 7 days post-natal. All pups of a litter were injected at days 7 and 14, genotyping was performed as described above, and injections were continued on the Npc1−/− mice. Control mice for pulmonary studies were not injected. Some Npc1+/+ received weekly injections of HPBCD for lung cholesterol and collagen determinations.
Pulmonary Function Tests
Mice were anaesthetized with 0.017–0.020 ml of 2.5% Avertin per g body weight and the trachea was dissected free and cannulated with a 20 or 21 gauge cannula (depending on the size of the trachea) which was kept in place with a single tie suture. The animals were then connected to a small animal ventilator (flexiVent, SCIREQ, Inc., Montreal, Quebec, Canada) and ventilated with a tidal volume of 10ml/kg, inspiratory/expiratory ratio of 66.67%, respiratory rate of 150/min and maximum pressure of 30 cm H20. Positive end-expiratory pressures (PEEP) were maintained by submerging the expiratory limb from the ventilator into a water trap (3 cm H20 pressure).
The flexiVent apparatus ventilates the mouse and then generates pressure-volume curves over an 8 sec. interval before continuing the respirations. The machine is calibrated for gas compressibility and resistive and accelerative losses in all the connections and calculates inspiratory capacity, elastance, hysterisivity, and resistance on the basis of the entered mouse weight.
Cholesterol determinations
Cholesterol was determined with the InfinityTM reagent (ThermoFisherScientific, Inc., Middletown, VA) which measures total cholesterol. Briefly, tissue was homogenized in 20mmol. Tris, 2 mmol EDTA, 150 mmol NaCl with 1% Triton X-100, pH 8.0. An aliquot was taken for protein determination by the bicinchoninic acid technique (BCA Protein Assay, Pierce, Rockford, IL). Standards were dissolved in the same buffer. The homogenate was stirred for 5 min at RT, centrifuged at 18,000g for 5 min., up to 10 ul of supernatant incubated at 37 C for 5–10 min. with 0.3 ml.s of InfinityTM reagent, and absorbance read at 500 nm.
Collagen determinations
Hydroxyproline was determined in lung samples after collagenase digestion of the sample. Briefly, tissue was homogenized in a citrate:acetate buffer (26 mmol citrate, 166 mmol acetate, pH 5.8) and an aliquot taken for protein determination by the bicinchonic acid technique. Collagenase (Clostridiopeptidase A, EC 3.4.24.3, Sigma) was added to 0.1% and the samples were incubated at 37 C with agitation for 16 hours. After digestion, the samples were centrifuged at 18,000g for 3 min. Hydroxyproline standards were diluted in the same homogenization buffer. Supernatants were incubated with one-half vol. of resolving buffer (freshly prepared 62mmol Chloramine T in 26% propanol and half-strength homogenization buffer) for 20 min at RT and then with another one-half vol. of colorimetric reagent (freshly prepared 116 mmol p-aminobenzaldehyde in 70% propanol and 18% percholoric acid) for 5 min. at 60 C. Absorption was measured at 562 nm.
Statistics
Paired t-tests were performed between Npc1−/− and controls and between GFAP tg Npc1−/− and HPBCD-treated Npc1−/− mice. Testing was done for differences between group variances and, if they differed, the t-test that did not assume equal variances was used.
Results
Npc1NIH/NIH compared to wild-type sibs
Histological staining of age-matched wild-type sibling mice shows normal pulmonary tissue (Fig 1A and E) while the affected mice show foamy macrophages but no alveolar proteinosis (Fig 1B and F). Lung weight as percent of body weight was increased (Fig 3A). When studied on the small animal ventilator, differences in pressure-volume loops are apparent (representative loops in Fig 2A).The calculations of the pulmonary function output from these loops showed increases in inspiratory capacity, elastance and hysterisivity (Fig 3) but no changes in resistance (not shown). Cholesterol and collagen were also increased (Fig 4).
Figure 1.
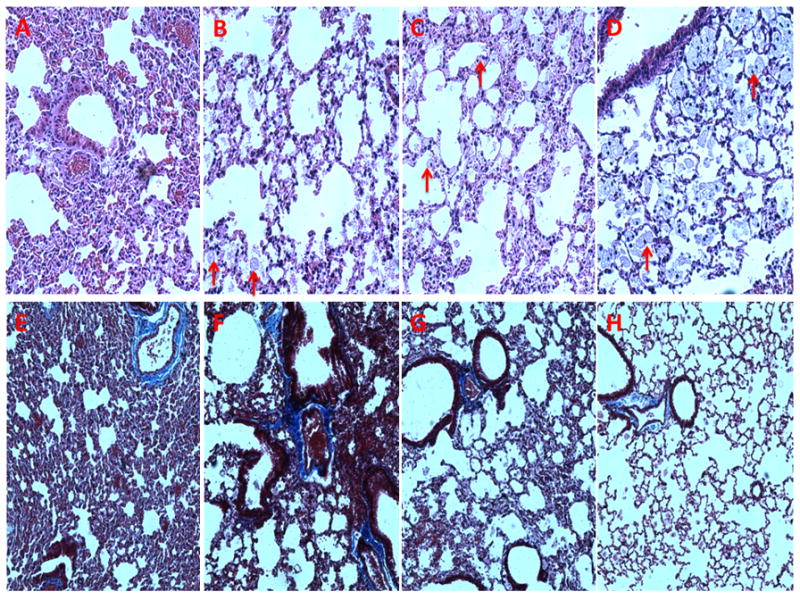
Histological examination of lungs from control (16 weeks), Npc1−/− (11 weeks), GFAP transgenic (16 weeks), and HPBCD-treated Npc1−/− (18 weeks) mice. A. Hematoxylin and eosin (H&E) stained control lung. B. H&E stained Npc1−/− lung. C. H&E stained Npc1−/−, GFAP tg lung. D. H&E stained HPBCD-treated Npc1−/− lung. E. Trichrome stained control lung. F. Trichrome stained Npc1−/− lung. G. Trichrome stained Npc1−/− GFAP tg lung. H. Trichrome stained HPBCD-treated Npc1−/− lung. Foamy macrophages indicated by arrows.
Figure 3.
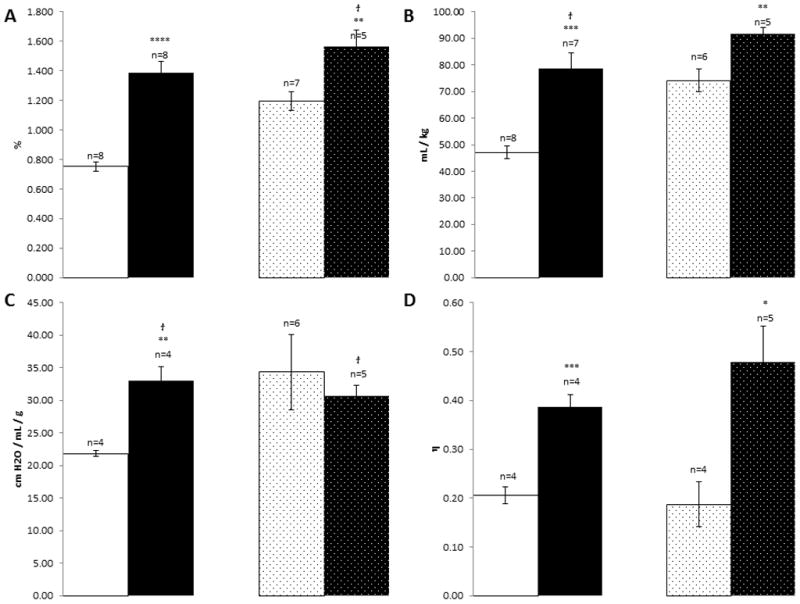
Pulmonary function values for control mice (white), Npc1−/− (black), age-matched Npc1−/−, GFAP transgenic mice (dotted white), and Npc1−/− treated with HPBCDs (dotted black). A. Lung weight as percent of body weight. B. Inspiratory capacity over body weight. C. Elastance. D. Hysterisivity. *p≤0.05; **p≤0.01; ***p≤0.005; ****p≤0.0005. group variances differed so the t-test that did not assume equal variances was used.
Figure 2.
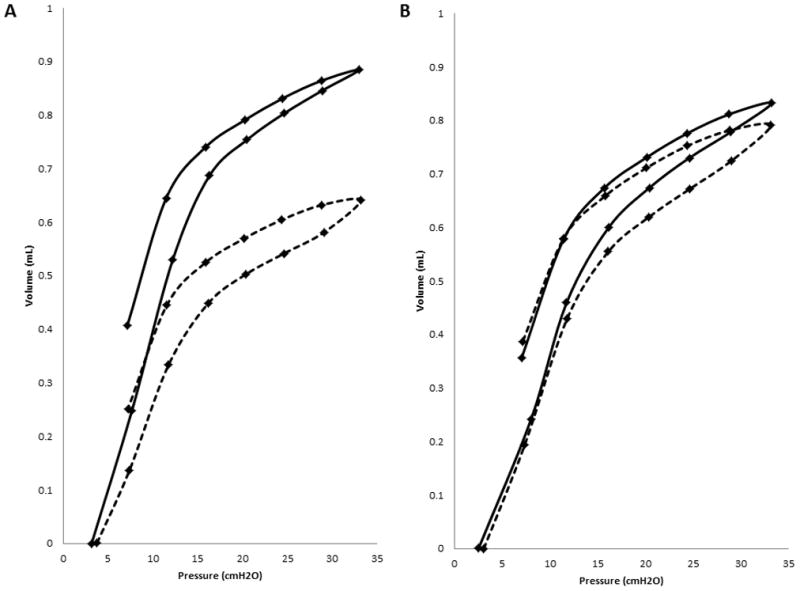
Representative pressure-volume loops from the flexiVent small animal ventilator (statistical analyses of the values derived from multiple loops are provided in Fig 3). A. One Npc1−/− (dotted) compared to a wild-type Npc1+/+ mouse (solid). B. Npc1−/− treated with HPBCDs (dotted) compared to age-matched Npc1−/−, GFAP transgenic mouse (solid).
Figure 4.
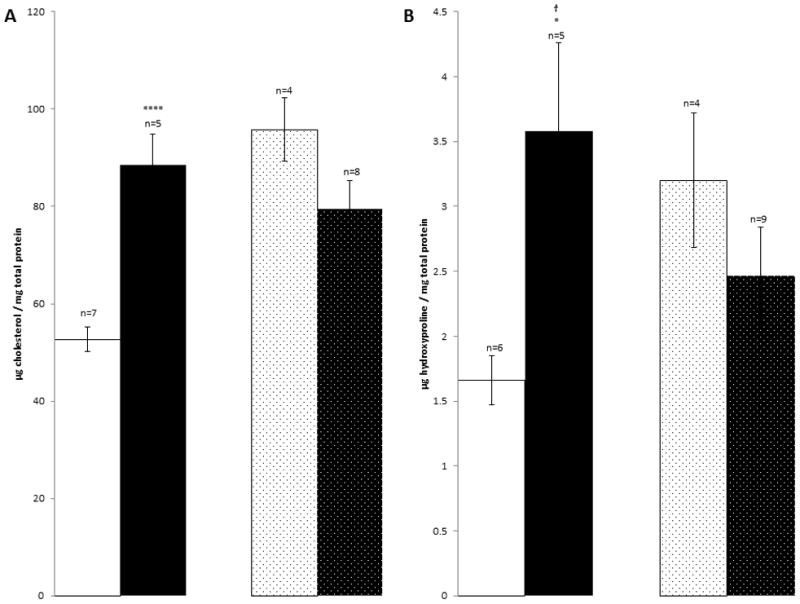
Biochemical determinations in lungs for control mice (white), Npc1−/− (black), age-matched Npc1−/−, GFAP transgenic mice (dotted white) and Npc1−/− treated with HPBCDs (dotted black). A. μg Cholesterol / mg protein. B. Collagen, determined as μg hydroxyproline / mg protein. *p≤0.05; ****p≤0.0005. group variances differed so the t-test that did not assume equal variances was used.
HPBCD- treated Npc1NIH/NIH compared to Npc1NIH/NIH, GFAP Npc1 (Npc1−/−, GFAP transgenic) age-matched transgenics
Histological studies of HPBCD-treated Npc1−/− and similar-aged Npc1−/−, GFAP transgenic controls showed marked increases in the number of alveolar foamy macrophages in both groups (Fig 1C, D, G and H). There was not much difference in the pressure volume loops (Fig 2B) or the values for lung weight as percent of body weight and elastance but hysterisivity was normal in the Npc1−/− GFAP transgenic (Fig 3) while the treated, age-matched controls had a small increase in inspiratory capacity. Npc1+/+ mice treated with cyclodextrin showed lowered cholesterol ([3] 29.8±2.6, p≤0.0005) while collagen was not different ([3] 2.21±0.55, p=0.159). Cholesterol levels were not corrected in Npc1−/− mice, while there was a statistically insignificant trend for collagen to be decreased in them (Fig 4).
Discussion
Niemann-Pick disease type C (NPC) is an autosomal recessive, neurodegenerative disorder that usually presents in the first decade of life [15]. The classic presentation of NPC is a child of either sex developing coordination problems, dysarthria, and hepatosplenomegaly during early school-age years. The neurological progression of the disorder is relentless and is characterized by increasing severity of ataxia, developmental dystonia, and dementia, until death supervenes, usually during the second decade of life. Identification of the major gene responsible for the disorder, NPC1/Npc1, revealed one coding for a multipass transmembrane protein containing a sterol-sensing domain that shows homology to the genes for Patched, HMG CoA reductase, and SCAP (SREBP cleavage activating protein) [11,16] Analysis of NPC1 protein function suggest that it is involved in late endosomal lipid sorting/vesicular trafficking [17–19]. The fundamental role of NPC1 in intracellular lipid transport, and the severe visceral disease which sometimes causes death before the onset of neurodegeneration, established the idea that lipid accumulation is the primary defect in NPC and leads to secondary neurological impairment. Thus, the use of drugs to try and decrease cholesterol storage was an early goal for treatment of NPC1. However, classic cholesterol-lowering drugs did not have an effect on the neurodegeneration [20].
Beta-cyclodextrins have long been known to create inclusion complexes with cholesterol [21] and have been used to study cholesterol, and other steroid, metabolism in vitro [22–25] and in vivo [26–29], even in humans [30]. They appear to be non-toxic: even massive orally administered doses to pregnant rabbits resulted in no teratogenic or embryotoxic effects [31]. Much of the early literature is reviewed by Thompson [32] (337 references) and a critical discussion of beta-cyclodextrin modifications is provided by Blanchard and Proniuck [33].
HPBCDs are rapidly cleared by the kidney (90% in 4 hours); if pre-loaded with steroid, the steroid component is no longer present but has been transferred to serum proteins [34]. Uncomplexed HPBCD injected intracerebrally was rapidly cleared, cholesterol in such complexes was retained for at least 3 days. Uncomplexed HPBCDs given parentally to Npc1−/− mice result in significant clearance of cholesterol from liver [8, 10] but not from lung [10]. The sequestered cholesterol is secreted as bile acid [35]. The mechanism of cholesterol release from NPC1 cells involves endocytosis of the HPBCD [36] and corrects deficient transport of cholesterol from lysosomes to the endoplasmic reticulum [37]. Of interest, methyl-beta-cyclodextrin, which can lower brain cholesterol [38] and therefore, may be able to cross the BBB, was more effective in depleting cholesterol from NPC1 cells [36].
While Camargo, et al [8] found a modest effect with HPBCD injections at doses of 500mg/kg given 3 times a week starting at 28 days, Davidson, et al [39] found a doubling of life expectancy with every other day injections of 4,000mg/kg starting at 21 days. With this treatment, the authors found “a seemingly greater number of cholesterol positive cells present in the CD-treated lungs” , compared to age-matched untreated Npc1−/− mice. Using a similar treatment scheme, Ramirez, et al. [40] found a slight decrease in lung cholesterol levels but no decrease in CD68, CD11c, TNFα mRNA levels. Our histological results parallel those of Davidson, et al. [39] and Ramirez, et al. [40]: large numbers of foamy macrophages in the alveolar lining and alveolar spaces were found but no alveolar proteinosis was apparent. Trichrome staining suggested increased collagen which was confirmed by hydroxyproline assays.
We supplemented our histological results using quantitative measures of parameters of pulmonary function, cholesterol and collagen. Lung weight, inspiratory capacity, elastance and hysterisivity were increased in Npc1−/− as compared to Npc1+/+ mice. Cholesterol and collagen were also increased. Thus, this mouse model mostly replicates the pulmonary disease seen in Niemann-Pick C patients although the alveolar proteinosis seen in NPC2 [6] was not seen. We also monitored the effects of HPBCD treatment on the pulmonary disease. We used Npc1−/−, GFAP transgenics as age-matched controls. This GFAP-promoted Npc1 transgene expresses Npc1 in fibrilary astrocytes and would only affect lung neuronal activity by correcting astrocytes in their ganglia [41–42]. Interestingly, hysteresis (which may be influenced by muscle tone) was returned to normal in these mice but all other parameters were comparable to Npc1−/− mice at a younger age. HPBCD treatment did not alter these parameters although there was a trend towards decreased lung collagen.
While HPBCD’s decrease cholesterol in many tissues, including normal lung, they do not have this effect in diseased lung. Perhaps the large capillary bed of the lung results in transfer of cholesterol loaded into HPBCD from other organs to the capillary epithelia, and thus to the lung parenchyma. Possibly inhaled cyclodextrins would decrease the accumulation of cholesterol in the lung.
Acknowledgments
Support: This work was supported by NIH 5RO1 ED000343-5 (T. Trouard, P.I.), NIH 1R56 AI083403-01A1 (M. Daines, P.I.) and the Holsclaw Family Professorship of Human Genetics and Inherited Diseases (RPE).
We thank Dr. F. John Meaney for help with statistical analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hooft C, Delbeke MJ, Garmyn F, Vertruyen H. Lesions pulmonaire dans la maladie de Niemann-Pick. Ann De Pediatrie. 1963;39:385–93. [PubMed] [Google Scholar]
- 2.Guillemot N, Troadec C, Billete de Villemeur T, Clement A, Fauroux B. Lung disease in Niemann-Pick Disease. Ped Pulmon. 2007;42:1207–14. doi: 10.1002/ppul.20725. [DOI] [PubMed] [Google Scholar]
- 3.Minai OA, Sullivan EJ, Stoller JK. Pulmonary involvement in Niemann-Pick disease: Case report and literature review. Resp Med. 2000;94:1241–51. doi: 10.1053/rmed.2000.0942. [DOI] [PubMed] [Google Scholar]
- 4.Scholfer O, Mischo B, Puschel W, Harzer K, Vanier MT. Early-lethal pulmonary Niemann-Pick type C disease belonging to a second, rare genetic complementation group. Eur J Pediat. 1998;157:45–9. doi: 10.1007/s004310050764. [DOI] [PubMed] [Google Scholar]
- 5.Millat G, Chikh K, Naureckiene S, Sleat DE, Fensom AH, Higaki K, Elleder M, Lobel P, Vanier MT. Niemann-Pick Disease Type C: Spectrum of HE1 Mutations and Genotype/Phenotype Correlations in the NPC2 Group. Am J Hum Genet. 2001;69:1013–21. doi: 10.1086/324068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griese M, Brasch F, Aldana VR, Cabrera MM, Goelnitz U, Ikonen E, Karam BJ, Liebisch G, Linder MD, Lohse P, Meyer W, Schmitz G, Pamir A, Ripper J, Rolfs A, Schams A, Lezana FJ. Respiratory distress in Niemann-Pick C2 is caused by pulmonary alveolar proteinosis. Clin Genet. 2010;77:119–30. doi: 10.1111/j.1399-0004.2009.01325.x. [DOI] [PubMed] [Google Scholar]
- 7.Manabe T, Yamane T, Higashi Y, Pentchev PG, Suzuki K. Ultrastructural changes in the lung in Niemann-Pick type C mouse. Virchows Arch. 1995;427:77–83. doi: 10.1007/BF00203741. [DOI] [PubMed] [Google Scholar]
- 8.Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Hedienreich RA, Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sciences. 2001;70:131–42. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- 9.Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nature Med. 2004;10:704–11. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- 10.Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the Npc1−/− mouse. Proc Natl Acad Sci USA. 2009;106:2377–82. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–5. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- 12.Elrick MJ, Pacheco CD, Yu T, Dadgar N, Shakkottai VG, Ware C, Paulson HL, Lieberman AP. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet. 2010;19:837–47. doi: 10.1093/hmg/ddp552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Strnatka D, Donohue C, Hallows JL, Vincent I, Erickson RP. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1−/− mice. J Neurosci Res. 2008;86:2648–56. doi: 10.1002/jnr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapur R, Donohue C, Jelinek D, Erickson RP. Amelioration of Enteric Neuropathology in a Mouse Model of Niemann-Pick C by Npc1 Expression in Enteric Glia. J Neurosci Res. 2009;87:2994–3001. doi: 10.1002/jnr.22126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea ED, Neufeld EB, Blanchette-Mackie EJ, Pentchev PG. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The metabolic and molecular bases of inherited disease. 8. McGraw-Hill; New York: 2001. pp. 3611–34. [Google Scholar]
- 16.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Kirzman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O’Neill RR, vanDiggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–31. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 17.Garver WS, Heidenreich RA, Erickson RP, Thomas MA, Wilson JM. Localization of the murine Niemann-Pick C1 protein to two distinct intracellular compartments. J Lipid Res. 2000;41:673–87. [PubMed] [Google Scholar]
- 18.Higgins ME, Davies JP, Chen FW, Ioannou YA. Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab. 1999;68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- 19.Neufeld EB, Wastney M, Patel S, Suresh S, Cooney AM, Dwyer NK, Roff CF, Ohno K, Morris JA, Carstea ED, Incardona JP, Strauss JF, Vanier MT, Patterson MC, Brady RO, Pentchev PG, Blanchette-Mackie EJ. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J Biol Chem. 1999;274:9627–35. doi: 10.1074/jbc.274.14.9627. [DOI] [PubMed] [Google Scholar]
- 20.Erickson RP, Garver WS, Camargo F, Hossain GS, Heidenreich RA. Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J Inher Metab Dis. 2000;23:54–62. doi: 10.1023/a:1005650930330. [DOI] [PubMed] [Google Scholar]
- 21.De Caprio J, Yun J, Javitt NB. Bile acid and sterol solubilization in 2-hydroxypropyl-beta-cyclodextrin. J Lipid Res. 1992;33:441–3. [PubMed] [Google Scholar]
- 22.Kilsdonk DP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. Cellular Cholesterol Efflux Mediated by Cyclodextrins. J Biol Chem. 1995;270:17250–6. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 23.Neufeld EB, Cooney AM, Pitha J, Dawidowicz EA, Dwyer NK, Pentchev PG, Blanchette-Mackie EJ. Intracellular Trafficking of Cholesterol monitored with a Cyclodextrin. J Biol Chem. 1996;271:21604–13. doi: 10.1074/jbc.271.35.21604. [DOI] [PubMed] [Google Scholar]
- 24.Atger VM, de la Llera Moya M, Stoudt GW, Rodrigueza WV, Phillips MC, Rothblat GH. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J Clin Invest. 1997;99:773–80. doi: 10.1172/JCI119223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilangumaran S, Hoessli DC. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem J. 1998;335:433–40. doi: 10.1042/bj3350433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frijlink HW, Eissens AC, Hefting NR, Poelstra K, Lerk CF, Meijer DKF. The Effect of Parenterally Administered Cyclodextrins on Cholesterol levels in the Rat. Pharm Res. 1991;8:9–17. doi: 10.1023/a:1015861719134. [DOI] [PubMed] [Google Scholar]
- 27.Gerloczy A, Hoshino T, Pitha J. Safety of oral cyclodextrins: Effects of Hydroxypropyl Cyclodextrins, Cyclodextrin Sulfates and Cationic Cyclodextrins on Steroid Balance in Rats. J Pharm Sci. 1994;83:193–200. doi: 10.1002/jps.2600830215. [DOI] [PubMed] [Google Scholar]
- 28.Favier ML, Remesy C, Moundras C, Demigne C. Effect of Cyclodextrin on Plasma Lipids and Cholesterol Metabolism in the Rat. Metabolism. 1995;44:200–6. doi: 10.1016/0026-0495(95)90265-1. [DOI] [PubMed] [Google Scholar]
- 29.Ferezou J, Riottot M, Serougne C, Cohen-Solal C, Catala I, Alquier C, Parquet M, Juste C, Lafont H, Mathe D, Corring T, Luttton C. Hypocholesterolemic action of beta-cyclodextrin and its effects on cholesterol metabolism in pigs fed a cholesterol-enriched diet. J Lipid Res. 1997;38:86–100. [PubMed] [Google Scholar]
- 30.Carpenter TO, Gerloczy A, Pitha J. Safety of Parenteral Hydroxypropyl beta-cyclodextrin. J Pharm Sci. 1995;84:222–5. doi: 10.1002/jps.2600840220. [DOI] [PubMed] [Google Scholar]
- 31.Waalkens-Berendsen DH, Smits-van Prooije AD, Bar A. Embryotoxicity and Teratogenicity Study with gamma–cyclodextrin in Rabbits. Reg Toxicol and Pharm. 1998;27:172–7. doi: 10.1006/rtph.1998.1222. [DOI] [PubMed] [Google Scholar]
- 32.Thompson DO. Cyclodextrins-enabling excipients: their present and future use in pharmaceuticals. Criti Rev Ther Drug Carrier Syst. 1997;14:1–104. [PubMed] [Google Scholar]
- 33.Blanchard J, Proniuk S. Some Important Considerations in the Use of Cyclodextrins. Pharm Res. 1999;16:1796–8. doi: 10.1023/a:1011930821801. [DOI] [PubMed] [Google Scholar]
- 34.Pitha J, Gerloczy A, Olivi A. Parenteral Hydroxypropyl Cyclodextrins: Intravenous and Intracerebral Administration of Lipophils. J Pharm Sci. 1994;83:833–41. doi: 10.1002/jps.2600830615. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Ramirez M, Miller AN, Repa JJ, Turley SD, Dietschy JM. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J Lipid Res. 2010;51:933–44. doi: 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci USA. 2010;107:5477–82. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abi-Mosleh L, Infante RE, Radhakrishnan A, Goldstein JL, Brown MS. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc Natl Acad Sci USA. 2009;106:19316–21. doi: 10.1073/pnas.0910916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bar-On P, Rockenstein E, Adame A, Ho G, Hashimoto M, Masliah E. Effects of the cholesterol-lowering compound methyl-beta-cyclodextrin in models of alpha-synucleinopathy. J Neurochem. 2006;98:1032–45. doi: 10.1111/j.1471-4159.2006.04017.x. [DOI] [PubMed] [Google Scholar]
- 39.Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Walkley SU. Chronic Cyclodextrin Treatment of Murine Niemann-Pick C Disease ameliorates Neuronal Cholesterol and Glycosophingolipid Storage and Disease Pogression PLoS one. 2009;4(9):e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramirez CM, Liu B, Taylor AM, Repa JJ, Burns DK, Weinberg AG, Turley SD, Dietschy JM. Weekly Cyclodextrin Administration Normalizes Cholesterol Metabolism in Nearly Every Organ of the Niemann-Pick Type C1 Mouse and markedly Prolongs Life. Ped Res. 2010;68:309–15. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tollet J, Everet AW, Sparrow MP. Spatial and temporal distribution of nerves, ganglia, and smooth muscle during the early pseudoglandular stage of fetal mouse lung development. Dev Dyn. 2001;221:48–60. doi: 10.1002/dvdy.1124. [DOI] [PubMed] [Google Scholar]
- 42.Tollet J, Everet AW, Sparrow MP. Development of neural tissue and airway smooth muscle in fetal mouse lung explants: a role for glial-derived neurotrophic factor in lung innervation. Am J Respir Cell Mol Biol. 2002;26:420–9. doi: 10.1165/ajrcmb.26.4.4713. [DOI] [PubMed] [Google Scholar]