

Abstract
The pufferfish Fugu rubripes has a compact 400-Mb genome that is ∼7.5 times smaller than the human genome but contains a similar number of genes. Focusing on the distal short arm of the human X chromosome, we have studied the evolutionary conservation of gene orders in Fugu and man. Sequencing of 68 kb of Fugu genomic DNA identified nine genes in the following order: (SCML2)-STK9, XLRS1, PPEF-1, KELCH2, KELCH1, PHKA2, AP19, and U2AF1-RS2. Apart from an evolutionary inversion separating AP19 and U2AF1-RS2 from PHKA2, gene orders are identical in Fugu and man, and all nine human homologs map to the Xp22 band. All Fugu genes were found to be smaller than their human counterparts, but gene structures were mostly identical. These data suggest that genomic sequencing in Fugu is a powerful and economical strategy to predict gene orders in the human genome and to elucidate the structure of human genes.
[Sequence data for this article were deposited with the EMBL/GenBank data libraries under accession nos. AJ011381 and AF094327.]
Isolation of human genes and their structural characterization is hampered by the fact that >95% of the DNA is noncoding and that intronic sequences can be very large. The Japanese pufferfish Fugu rubripes (Fugu) has a small genome of ∼400 Mb, which is 7.5 times smaller than the human genome. Therefore, Fugu has been proposed as a model organism for rapid analysis of vertebrate genes (Brenner et al. 1993). The structure of genes appears to be conserved, because most splice sites reside in identical positions to those found in man (Baxendale et al. 1995; Elgar et al. 1995; Macrae and Brenner 1995; Mason et al. 1995). In regions of the Fugu genome that exhibit conservation of gene order, sequencing would reduce the effort required for isolation of candidate genes in human disease loci and for studying genome organization. However, reports showing conserved linkage over larger distances are scarce. The few available examples include the report of Trower et al. (1996), who have demonstrated that three genes that are linked to FOS in the familial Alzheimer disease locus on human chromosome 14 have homologs in the Fugu genome adjacent to cFOS. Conserved linkage of the genes encoding the platelet-derived growth factor receptor and the macrophage colony-stimulating factor receptor (How et al. 1996), of complement C9 and DOC-2 (Yeo et al. 1997), and of MAP-2, MYL-1, and CPSIII (Schofield et al. 1997) have been described. In contrast, the gene order of the Surfeit genes that map to three separate loci in the Fugu genome is largely different from that found in mammals in which these genes are clustered (Armes et al. 1997; Gilley et al. 1997). These findings gave rise to a controversial discussion on the potential of Fugu as a model organism to accelerate the prediction and discovery of genes by applying comparative sequencing or positional cloning strategies (Aparicio and Brenner 1997; Elgar et al. 1997; Gilley et al. 1997). Important questions concerning the extent and average length of conserved linkage groups and the distribution of conserved segments still wait to be answered. Very recently, Miles et al. (1998) found extensive conservation of synteny between a 1.5-Mb region of human chromosome 11 and <100 kb of the Fugu WAGR region. Here we report on another highly conserved segment in the Fugu genome that encompasses nine different genes from the human Xp22.2–p22.1 region.
RESULTS
Isolation of Fugu Cosmids Homologous to Human Xp22.2–Xp22.1
Initially, three partially overlapping human liver cDNA probes encoding PHKA2 were used to screen a Fugu cosmid library (kindly provided by the Resource Center of the German Human Genome Project). One cosmid, ICRFc66A2095Q1.2 (A2095), was entirely sequenced because it also showed positive hybridization signals with human PPEF-1 cDNA. Rescreening of this and another Fugu cosmid library [kindly provided by Greg Elgar, British Resource Center of the Human Genome Project (HGMP-RC)] with end fragments of A2095 resulted in the isolation of overlapping cosmids ICRFc66L2390Q1.2 and F089J4. Together, these three cosmids encompass 80 kb of contiguous Fugu DNA, of which 68 kb has been sequenced.
Sequence Comparison Identified Nine Genes in Fugu with Conserved Order of a Five-Gene Cluster and a Two-Gene Cluster
Nine Fugu genes were identified following exon prediction and database searches (Fig. 1; see Materials and Methods). The gene order is as follows SCML2 (related to the Drosophila Sex comb on midleg repressor protein), STK9 (Serine–threonine kinase 9), XLRS1 (X-linked Retinoschisis), PPEF-1 (Protein phosphatase with EF calcium-binding domain), KELCH2 and KELCH1 (related to the Drosophila Kelch protein), PHKA2 (Phosphorylase kinase 2-subunit), AP19 (Golgi adaptor AP-1 19-kD adaptin), and U2AF1-RS2 (U2 small nuclear ribonucleoprotein auxiliary factor 35-kD subunit-related protein 2). We subsequently compared our results with the gene order in human Xp22 (Kitagawa et al. 1995; Montini et al. 1997; Sauer et al. 1997; Sherman et al. 1997). Very recently, sequencing of overlapping PAC clones dJ757P12 and dJ958B3, which contain the human SCML2 gene, and of dJ245G19 and dJ436M11, which cover the STK9–PPEF-1 region, have been completed at the Sanger Centre, Cambridge, UK (http://www.sanger.ac.uk/), facilitating comprehensive comparison. Human PHKA2 had been mapped to Xp22.2–p22.1 (Davidson et al. 1992) and our YAC and PAC hybridization results confirm its location proximal to PPEF-1 (not shown), identical to the position in Fugu. The human KELCH homologs are presently unknown and their isolation is in progress. To map human AP19, we performed FISH and Southern blot hybridizations of YAC clones containing human PHKA2. FISH assigned AP19 to Xp22, whereas YAC hybridizations were negative. While this work was in progress, the sequence of a BAC clone was published (AC004106) showing linkage of both human AP19 and U2AF1-RS2 genes and two STS markers, located distal to SCML2. Therefore, we extended Southern hybridizations of AP19 to YAC clones CEPHy904D10984 and CEPHy904D11161 and positive signals confirmed its location telomeric to SCML2 (Fig. 1).
Figure 1.

Scale representation of the Fugu genomic region encompassing 80 kb compared with the equivalent genomic region of human Xp22 (not drawn to scale). In total, 68 kb of Fugu genomic DNA have been sequenced, of which 62 kb are contiguous. (Broken horizontal line) The region not completely sequenced in both Fugu and humans, which, in Fugu, spans ∼18 kb. (Dotted line) A distance of ∼1 Mb in human DNA. (Cross-lines) The borders of conserved gene order. The complete coding region of each Fugu gene is represented by aligned solid boxes and the transcriptional direction is indicated by the arrow above each gene. The results of the exon prediction programs FGENES, GENSCAN, and GRAIL 2 are shown (bottom), with each predicted exon shown by a solid box.
For a more detailed structural analysis of the Fugu genes, amino acid sequences were deduced for each putative coding region and were aligned to the sequences of homologous proteins from various species, including human, mouse, rabbit, Drosophila, Dictyostelium, yeast, and Caenorhabditis. Sizes of all Fugu genes, protein sequences, and their conservation in other species are given in Table 1.
Table 1.
Structural Analysis of Fugu Genes
| Fugu genes | Length
|
Homologs | Amino acid
|
SwissProt/GenBank accession no. | ||
|---|---|---|---|---|---|---|
| kb | amino acid | similarity (%) | identity (%) | |||
| SCML2 | 2.0 | 250 | Drosophila SCM | 70 | 63 | Q24191 |
| human SCML2 | 74 | 65 | Z93023, AL031007 | |||
| STK9 | 12.5 | 1104 | human STK9 | 67 | 62 | Y15057 |
| yeast SLT2 | 46 | 34 | Q00772 | |||
| Dictyostelium CDK7 | 49 | 38 | P54685 | |||
| XLRS1 | 4.1 | 280 | human XLRS1 | 78 | 71 | AF014459 |
| mouse XLRS1 | 78 | 71 | AJ011381 | |||
| PPEF-1 | 5.5 | 683 | Drosophila rdgC | 54 | 42 | P40421 |
| Caenorhabditis PPEF | 56 | 46 | AF023454 | |||
| human PPEF-1 | 64 | 54 | AF023455 | |||
| human PPEF-2 | 63 | 54 | AF023456 | |||
| mouse PPEF-2 | 64 | 55 | AF023458 | |||
| KELCH2 | 2.1 | 531 | human putative protein Q14145 | 39 | 31 | AC004021 |
| KELCH1 | 2.1 | 518 | human putative protein Q14145 | 45 | 33 | AC004021 |
| PHKA2 | 10 | 1229 | human PHKA1 | 74 | 67 | P46020 |
| mouse PHKA1 | 73 | 67 | P18826 | |||
| rabbit PHKA1 | 73 | 67 | P18688 | |||
| human PHKA2 | 77 | 72 | P46019 | |||
| rabbit PHKA2 | 78 | 72 | P46018 | |||
| AP19 | 1.2 | 157 | human AP19 | 98 | 96 | AC004106 |
| mouse AP19 | 91 | 86 | Q00382 | |||
| yeast AP19 | 67 | 54 | P35181 | |||
| U2AF1–RS2 | 1.8 | 605 | human U2AF1–RS1 | 65 | 54 | D49676 |
| mouse U2AF1–RS1 | 69 | 58 | D85430 | |||
| human U2AF1–RS2 | 65 | 54 | D49677 | |||
| mouse U2AF1–RS2 | 68 | 58 | D45205 | |||
The nine putative Fugu genes are listed together with sets of homologs found by database search. The approximate length of each gene is given in kb. The percent similarity and identity were calculated by the GCG program BESTFIT.
Fugu SCML2, STK9, and XLRS1
By making use of the computer program NIX (http://www.hgmp.mrc.ac.uk/NIX/), detailed sequence analysis at one end of our contig revealed the presence of a gene encoding a homolog of the Drosophila Scm gene, which is not the ortholog of the recently identified human SCML1 gene (van de Vosse et al. 1998) and was therefore designated SCML2 (Fig. 1). The human SCML2 homolog spans base pairs 69966–107467 of PAC clone dJ958B3 and base pairs 47167–31846 of overlapping clone dJ757P12. BLAST search identified two human ESTs, one of which represents the full-length cDNA. Its database sequence contains exons 1–4 and part of exon 5 (accesssion no. AA227320). The other EST starts in exon 7 and extends into the 3′ UTR (accession no. AA618466). Fugu and human SCML2 splice sites are conserved and both genes contain a total of six coding exons. Human SCML2 harbors one additional 5′ exon that is noncoding and which, by sequence comparison, could not be identified in Fugu. Predicted SCML2 proteins consist of 250 amino acids in Fugu and 268 amino acids in humans, which are 65 % identical (Table 1).
The same Fugu cosmid contains the STK9 and XLRS1 genes. Comparison of the predicted Fugu STK9 protein revealed homology to several serine/threonine kinases of the CMGC group (Hanks and Hunter 1995; Fig. 2). The human homolog has been described recently (Montini et al. 1998) and consists of 20 exons coding for a protein of 1030 amino acids. Fugu STK9 is composed of 18 exons encoding 1104 amino acids. Gene structures of Fugu and human STK9 are similar with 8 exons having identical length. Amino acid comparison revealed 62% identity spanning amino acids 33–819 of the human protein. Multiple alignment of the Fugu STK9 predicted protein showed high conservation throughout the kinase domain, which, in Fugu, consists of the amino-terminal 283 amino acids. All invariant residues found in 60 kinase domains representative of members of the eukaryotic protein kinase superfamily (Hanks and Hunter 1998) are also invariant in Fugu STK9. In addition, the Fugu protein harbors a polyglutamine stretch that also is present in homologous serine/threonine kinases of Dictyostelium and yeast but is absent in the human protein (Fig. 2).
Figure 2.
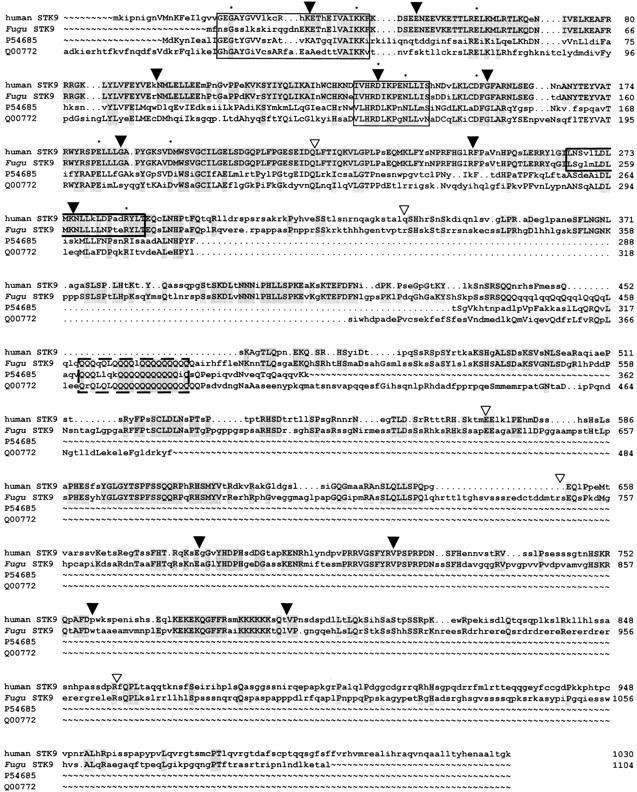
Multiple alignment of the human, Fugu, Dictyostelium (P54685), and yeast (Q00772) STK9 proteins. The alignment was performed with the GCG programs PILEUP and LINEUP. Amino acids identical in at least two proteins are shown in uppercase letters, shading indicates residues identical in Fugu and human and in all proteins, respectively. (▾) Intron/exon boundaries identical in Fugu and human STK9; (▿) splice sites only present in Fugu. The protein kinase ATP-binding region is presented by the first box. The second box indicates the predicted serine-threonine protein kinase active site. The predicted leucine zipper is boxed in bold and the polyglutamine stretch in broken lines. (Asterisks) Amino acid residues invariant in 60 members of the eukaryotic protein kinase superfamily (Hanks and Hunter 1995).
The structures of the Fugu and human XLRS1 genes are identical, except for longer 5′ exons in Fugu that contain 172 and 142 bp, respectively, compared with 52, 26, and 106 bp in human XLRS1. The Fugu protein contains 280 amino acids and is 71% identical to the 56 amino acids shorter mouse and human homologs. Sequence identity is mainly confined to the highly conserved discoidin domain at the carboxyl end. The amino-terminal ends of the mammalian proteins contain a secretory leader peptide sequence of 23 amino acids . By use of the program SIGNALP (Nielsen et al. 1997) one cleavage site of the signal peptide in Fugu has been predicted also between amino acids 23 and 24 (SQQ↓EK) (not shown).
Fugu PPEF-1
Fugu PPEF-1 belongs to a conserved branch of the phosphoprotein phosphatase (PPP) family of serine/threonine PPPs. In contrast to the 16 exons of the human PPEF-1 gene (Montini et al. 1997; GenBank accession no. Z94056, partly contained in PAC clone), we discovered 17 putative exons in Fugu. Interestingly, 12 of 17 Fugu exons are identical in position and length. The deduced protein is 54% identical to human PPEF-1 and PPEF-2 proteins (Table 1). Multiple alignment revealed a high degree of conservation throughout the protein, except for a segment that is apparently inserted into the catalytic core and is highly variable in both length and sequence (Fig. 3). In addition, protein phosphatases of the PPEF subtype possess two functional domains. In Fugu the catalytic domain encompasses amino acids 130–473. The second domain is present at the carboxyl end of the protein and encompasses amino acids 595–668. This domain contains two potential Ca2+-binding sites, as defined by the highly conserved EF hand motif. Taken together, these data strongly suggest that the PPEF-1-encoded protein is a functional phosphatase (Fig. 3). To assess the degree to which Fugu PPEF-1 is related to other members of the PPEF subfamily, a phylogenetic tree was generated from aligned conserved regions on the basis of nucleotide identity (see Materials and Methods). The unrooted tree shows that Fugu PPEF-1 and human PPEF-1 cluster in the same branch, whereas human and mouse PPEF-2 cluster in another branch (Fig. 4). Less-conserved regions that contained gaps when aligned, like exon 10–11 and the 5′ and 3′ ends of Fugu PPEF-1, were excluded. We generated 100 trees with the maximal likelihood method and bootstrapped them. Bootstrap values are indicated for each branch.
Figure 3.
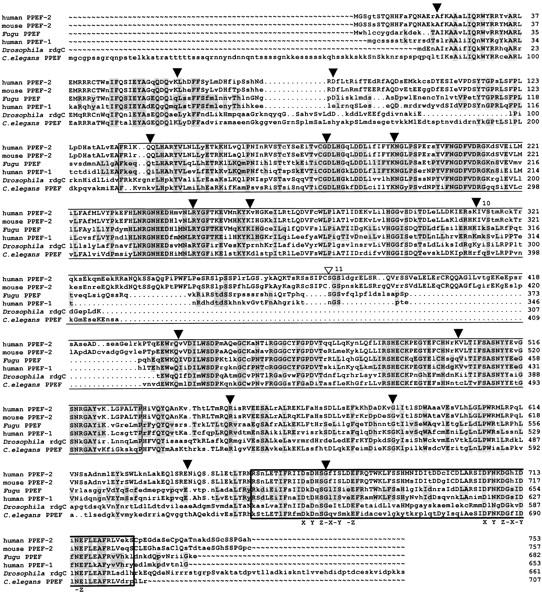
Alignment of Fugu and C. elegans PPEF, Drosophila RdgC, mouse PPEF-2, and human PPEF-1 and PPEF-2 proteins. The alignment was performed with the GCG programs PILEUP and PRETTY. Amino acids at least identical in two aligned sequences are shown in uppercase letters. Residues identical in Fugu and human PPEF-1 and in all proteins, respectively, are shaded. (▾) Identical intron/exon boundaries of Fugu PPEF-1 and human PPEF-1; (▿) Position of the putative additional splice site in Fugu. Fugu exons 10–11 are numbered. The catalytic domain and the EF hand motif of the PPEF proteins are boxed. The two positions of the Ca2+ chelating side chains are labeled X–Z.
Figure 4.
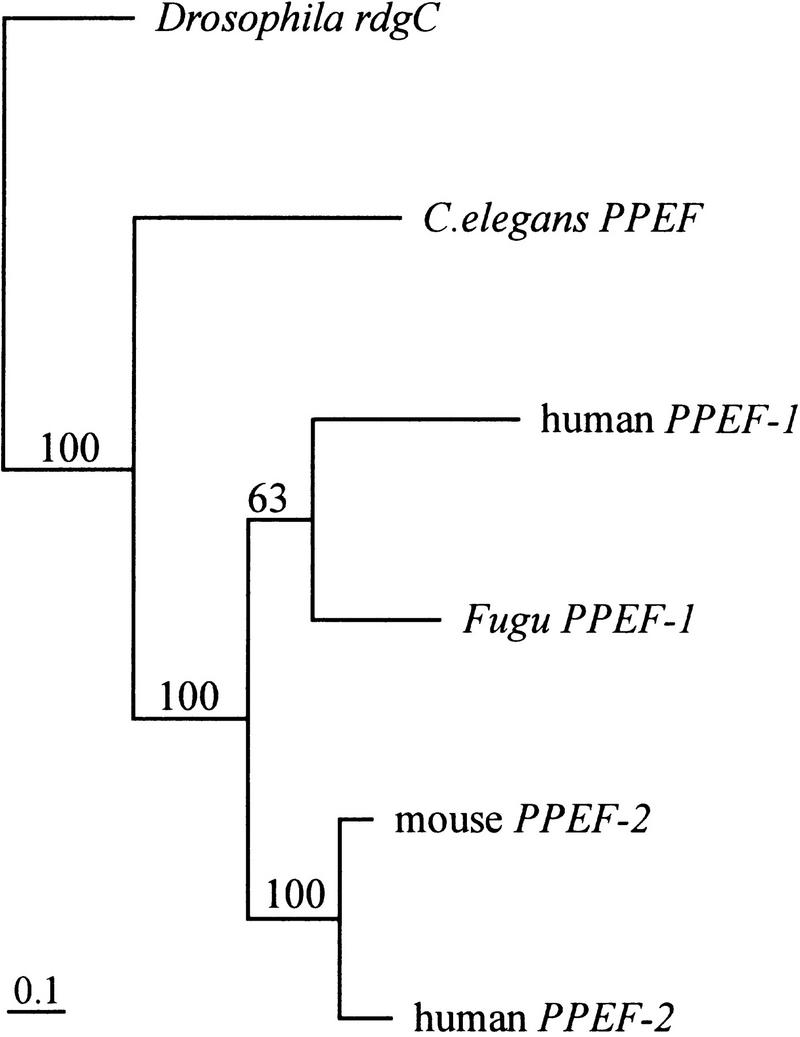
Phylogenetic tree of the PPEF subfamily. The unrooted tree was generated from aligned nucleotide sequences by the DNAML program of the PHYLIP software package. Regions of ambiguous alignment were excluded from the analysis. Scale bar, 0.1 expected substitutions per nucleotide site. Bootstrap values were generated from 100 bootstrap replicates of the alignment and are shown. Fugu PPEF-1 and human PPEF-1 cluster in the same branch; human and mouse PPEF-2 cluster in a second branch.
Fugu KELCH1 and KELCH2
Linked to Fugu PPEF-1, we have identified two tandemly arranged genes. Nucleic acid comparison revealed 62% identity. Database searches showed that both genes encode proteins harboring the so-called kelch domain. The highest score with an expectation value of P ∼ 10−70, was assigned to the human putative protein Q14145 (Table 1). On the basis of the homology to Q14145 and the Drosophila Kelch protein, the Fugu genes were designated KELCH1 and KELCH2. The genes are in a head-to-tail orientation and are separated by ∼3.5 kb of genomic DNA. For both genes, exon prediction revealed four putative exons that form an identical structure. To confirm the predictions, we performed RT–PCR on total Fugu testis RNA with a combination of primer sets. Amplification with primer pairs spanning the predicted coding region resulted in the expected products, whereas no transcripts were detectable with a primer set that spans the presumptive intergenic region (not shown). The putative proteins share 48% identity (Fig. 5A). In both proteins the first repeated segment of the Kelch domain contains 48 amino acids, repeat 2–5 contains 47 amino acids , and repeat 6 contains 49 amino acids (Fig. 5B). This is in contrast to the variable length of the repeated segments in other Kelch-containing proteins. Comparison with the consensus sequence of Drosophila Kelch repeats revealed that conservation of the Kelch domain is confined to those amino acids that form the consensus sequence in Fugu (not shown). The first 12 residues at the amino-terminal end of both Fugu Kelch proteins are part of the BTB domain, which encompasses ∼24 residues in other homologs.
Figure 5.


(A) Alignment of Fugu KELCH1 and KELCH2 proteins. The alignment was performed with the GCG programs PILEUP and PRETTY. Residues identical at each position are shown in uppercase letters and shaded. Intron/exon boundaries are shown by closed arrowheads on either site of the alignment. Each repeated segment is boxed and the beginning of each repeat is indicated by its respective number. (B) Alignment of the six repeated segments of Fugu KELCH1 and KELCH2 proteins. The alignments were performed with the GCG programs PILEUP and PRETTY. Amino acid residues at least identical in three repeats are shaded and shown in the consensus sequence. Dashes were introduced at less conserved positions.
Fugu PHKA2
The Fugu PHKA2 gene exhibits significant homology to the liver (PHKA2) and muscle (PHKA1) isoforms of the α regulatory subunits of the phosphorylase kinase (Phk) of rabbit, mouse, and humans (Fig. 6). The gene structure for the human homolog has been determined for 11 exons (Hendrickx et al. 1995), corresponding to exons 20–28 in Fugu. Comparison revealed exons with identical length and position. Interestingly, human PHKA2 contains two exons that would reside between exon 27 and 28 in Fugu, but in this region splice sites are apparently not conserved. Mammalian PHKA muscle and liver isoforms and Fugu PHKA2 have large stretches of highly conserved amino acid sequences in common, which also include the regions of the hypothetical 5′ and 3′ calmodulin binding sites and several phosphorylation sites (Fig. 6).
Figure 6.


Alignment of Fugu, rabbit, and human PHKA2 and mouse, rabbit, and human PHKA1 proteins. The alignment was performed with the GCG programs PILEUP and PRETTY. Residues identical at each position are shown in uppercase letters and are shaded. (▾) Intron/exon boundaries of Fugu PHKA2. Fugu exons 18–29 are numbered. (▿) Known positions of splice sites in the human PHKA2 gene. The PHKA muscle and liver isoforms have two subunit-specific domains (boxed) with comparatively low amino acid sequence similarities of <50%. These domains encompass amino acids 612–788 and amino acids 1027–1055 in Fugu with a negatively charged cluster spanning amino acids 620–645. The hypothetical 5′ and 3′ calmodulin binding sites of all Phk α subunits are highly conserved (boxed in bold) and encompass amino acids 828–855 and amino acids 1056–1087 in Fugu. Three autophosphorylation sites, serine 987, serine 1001, and serine 1022, labeled by an P underneath the alignment and their vicinity are conserved.
Fugu Clathrin Coat Assembly Protein AP19 and U2AF1-RS2
The AP19 protein is evolutionarily highly conserved. The high conservation of the Fugu protein (Table 1) is reflected even at the nucleotide level showing 80% and 76% identity to the mouse and human homologs, respectively (data not shown). Intron/exon structure comparison revealed that all splice sites are identical. Comparison of the Fugu and human U2AF1-RS2 genes showed that position and length of Fugu exons 6 and 8–10 correspond to their human counterparts. Because we were not able to assign the intron/exon boundaries at the 3′ end of the gene by sequence comparison, we used exon prediction instead. In this way, an unusually long terminal exon 11 was detected, encompassing 875 bp compared with 509 bp in humans. The human U2AF1-RS2 gene is characterized by an increased CG content >50% and by CpG islands surrounding the coding region. The CG content of the corresponding Fugu regions is slightly increased (44%) with >60% at the 3′ end, but these values do not indicate the existence of CpG islands. A statistical analysis of the protein sequence by the program SAPS (Brendel et al. 1992) revealed a high usage of arginine (13.2%) and glutamic acid (13.3%), classifying the protein significantly charged.
Intron/Exon Organization, Base Composition, and Repetitive Elements
Comparison of the coding regions of Fugu and human homologs revealed 60% to 76% nucleotide identity. Although exon sizes were mostly similar, sizes of Fugu introns showed a range from 61 to 2115 bp, giving an average of 308 ± 343 bp for 82 introns examined. This result concurs with previous observations that Fugu introns are at least 50 bp in size and small in average (Brenner et al. 1993). The overall size of the Fugu genome is reduced by the factor 6–8 when compared with the human genome. Analysis of individual genes revealed size reductions ranging between factors of 7 and 22 for Fugu XLRS1 and PPEF-1, respectively.
By analyzing the base composition of 164 intron/exon boundaries, we determined the 5′ splice site consensus sequence as C39A61G84g100t99a48a51g59t41 and 3′ splice site consensus sequence as c/t77c/t80c/t80c/t83g/t67c74a100g100G44T44. Numbers indicate the percentage of the respective nucleotide at this position. The Fugu consensus sequences equal those of mammals (Shapiro et al. 1987).
Base composition analysis revealed a GC content of 42.7%, which is slightly less than the 44.2% reported (Brenner et al. 1993). The search for repeated sequences using the program CENSOR (Jurka et al. 1996) identified a few short tandem repeats but neither highly nor moderately repetitive elements.
DISCUSSION
We have sequenced 68 kb of Fugu genomic DNA corresponding to >600 kb of human Xp22.2–p22.1. The comparative analysis of Fugu genes demonstrated the feasibility of gene identification and characterization in silico. Gene structures were deduced by comparing stretches of amino acids with >60% identity to homologous proteins and partly by sequence comparison with Fugu RT–PCR products. High conservation of splice sites were found in evolutionarily conserved domains, however, additional splice sites were detected in regions accompanied by low amino acid identity and in loop regions or protein ends. For the subsequent functional characterization of the putative proteins, we used various methods, including statistical analysis of the amino acid composition, motif and signal search, multialignment, as well as phylogenetic studies.
The gene order in human Xp22.2–p22.1 had been established for the five-gene cluster SCML2–STK9, XLRS1, PPEF–PHKA2 (Sanger Centre; Davidson et al. 1992; Montini et al. 1997, 1998; Sauer et al. 1997) and for the two gene-cluster AP19, U2AF1-RS2 (accession no. AC004106) (Kitagawa et al. 1995) showing conserved linkage and transcriptional orientation. However, in contrast to the order in Fugu, human AP19 and U2AF1-RS2 are located telomeric to SCML2. This result indicates the existence of an evolutionary breakpoint, separating AP19 and U2AF1-RS2 from PHKA2. Other examples of fragmented gene order in Fugu include the Surfeit locus, which is composed of at least six housekeeping genes, whose organization and juxtaposition is conserved between mouse, humans, and chicken, but in Fugu are found at three loci (Gilley et al. 1997). Fugu vasotocin/isotocin locus has undergone a localized reorganization during vertebrate evolution. The simplest model to explain the rearrangement includes a duplication of the ancestral vasotocin gene of cyclostomes, followed by an inversion in the lineage that led to the Fugu (Venkatesh et al. 1997). Compared with the situation in humans, the gene order in mouse is less well defined. STK9, Ppef, U2af1-rs2, and Phka2 map to the telomeric third of the X chromosome (Montini et al. 1998, http://www.informatics.jax.org/).
The Fugu SCML2 gene is homologous to Drosophila Sex comb on midleg and its human homolog has not been published to date other than by database sequences of two overlapping PAC clones, each containing only part of the gene, which may have led its incomplete annotation. Sequence comparison revealed that intron/exon junctions of all coding exons are conserved. These findings strongly sustain the existence of a complete human SCML2 in human Xp22, which is further supported by two human ESTs, one containing exons 1–5 and the other exon 7 and the 3′ UTR. Thus, comparative genomics is a useful tool to establish the genomic structure of new human genes, of which sequences are generated in large scale sequencing projects.
The Fugu STK9 protein is a new member of the CMGC group of serine/threonine kinases that are all related by the presence of highly conserved kinase domains consisting of ∼250–300 amino acid residues. Highest homology has been found to human STK9 protein. Less homologous proteins include serine/threonine kinases of Dictyostelium and yeast, but as in Fugu, these contain a polyglutamine stretch that is absent in the human counterpart, suggesting that it might have been lost during evolution.
Fugu PPEF-1 protein belongs to a subfamily of protein serine/threonine phosphatases that are characterized by the presence of at least two EF-hand motifs near the carboxyl terminus, suggesting a regulation of the enzymatic activity by intracellular calcium. Reversible phosphorylation of proteins on serine and threonine residues plays a crucial role in the regulation of a variety of cellular processes. In the mouse, Ppef-1 transcripts are restricted to primary somatosensory neurons and to the inner ear (Montini et al. 1997) and Ppef-2 transcripts have been found in retinal rods and in the pineal (Sherman et al. 1997) suggesting that the transcripts might play analogous roles in different sensory systems, but their roles seem to be distinct from that described for the Drosophila member retinal degeneration C (rdgC) (Steele and O’Tousa 1990: Steele et al. 1992). The Fugu homolog is closely related to human PPEF-1 as indicated by gene position and phylogenetic tree.
Fugu KELCH1 and KELCH2 exhibit high nucleotide identity and an identical gene structure. Up to now we could not identify human or mouse orthologs by comparison with databases or by Southern hybridization. Both Fugu proteins are members of a family containing at least one Kelch domain. Characteristic for this domain is the presence of up to six repeated segments of ∼50 amino acids each. Noteworthy, in Fugu, corresponding repeats of both proteins are identical in length. Although both Fugu proteins share 48% amino acid identity only a few selected residues within the Kelch domain are conserved. These are presumably implicated in the correct folding. Through sequence analysis, Bork and Doolittle (1994) have shown that the six repeats in Kelch proteins form a β-propeller or super-barrel structure. This structural fold is also found in several nonKelch proteins with repeat sequences, including bacterial and fungal, as well as influenza virus enzymes such as neuraminidase, galactose oxidase, or the sialidases. The question remains open whether all propeller folds share a remote ancestor or whether the possibility of structural convergence must be taken into consideration (Bork and Doolittle 1994; Robinson and Cooley 1997). Generating phylogenetic trees from the Kelch region showed various topologies in which only subbranches were reproducible (not shown). Although Kelch proteins form a large family, there are only a few hints toward the biochemical functions of this family. In the case of the Drosophila Kelch protein, a simple model suggests that the protein might bind to ring canal actin filaments through the repeat motif and might cross-link the actin filaments by dimerization through the BTB domain. Caenorhabditis elegans genome sequencing revealed at least six hypothetical Kelch proteins. One of these, Spe26, interacts with actin through the repeat motif and is required for a normal actin cytoskeleton during spermatogenesis (Varkey et al. 1995). At present, we cannot make any reliable prediction about the function(s) of the two Fugu Kelch proteins. However, our data suggest that the Fugu genes represent paralogs that may have originated from a common ancestor and evolved separately after duplication.
Until recently, only one AP19 locus had been reported in humans. Our EST database search identified three distinct groups of homologous transcripts and subsequent fluorescence in situ hybridization with one cDNA highlighted at the known locus on 17q25 and an additional locus on Xp22 (not shown). The presence of an AP19 locus on Xp22 has now been confirmed by the recently published genomic sequence AC004106, in which it is annotated. The AP19 protein is the smallest component of AP-1, the clathrin-associated protein complex found in clathrin-coated vesicles of the Golgi apparatus (Kirchhausen et al. 1991). Disruption of this gene in yeast elicits no detectable mutant phenotype. Fugu AP19 polypeptide shares 96% identity with the human protein. This high conservation suggests that the structure and function of these proteins is under stringent selective pressure. Hence, we consider Fugu AP19 as the true ortholog. Sequence comparison of mouse and Fugu AP19 revealed 86% identity. However, as the mouse gene has not been mapped yet, it is presently unknown whether they are true homologs or only members of the same family.
Taking into account that intergenic regions are compressed in Fugu, identification of regulatory elements should be much easier in this species than in mammals. Because sequence comparisons did not identify significantly matching regions, we applied pattern searching and promoter prediction programs (like TESS, PROMOTORSCAN). Depending on the programs used, several possible regulatory elements could be identified, but results were contradictory. In addition, we obtained a long list of putative transcription factor binding sites, the status of which is uncertain because most of the programs available use pattern information derived from mammalian transcription factor binding sites and their degree of conservation in Fugu is not yet known. Functional studies with Fugu clones that harbor intergenic regions may be a more direct approach toward identifying regulatory elements in the Fugu genome.
In summary, we have identified one of the largest regions of conserved gene order between Fugu and humans known to date. Given the much smaller size of Fugu genes and their generally conserved structure, for which this study provides further evidence, the pufferfish is an excellent model organism to identify and characterize new genes and to predict their order in the human genome. Moreover, the small intergenic and intronic distances should greatly facilitate the detection of regulatory elements once improved sequence recognition software is available or, alternatively, through functional studies involving gene transfer experiments.
METHODS
Probe Generation
Total human liver RNA was reverse transcribed exactly as described previously (Kalscheuer et al. 1993). Amplification of three partially overlapping PHKA2 cDNAs (position 381–1547, 1440–2580, and 2487–3746 bp of accession no. D38616) was carried out with primers Le α12 + Le α13 and Le α15 + Le α27 with a final MgCl2 concentration of 1.5 mm and Le α6 + Le α4* with a final MgCl2 concentration of 3 mm (Burwinkel et al. 1996). Initial denaturation was for 3 min at 96°C, followed by 45 cycles each consisting of 94°C for 1 min, 58°C (Le α6 + Le α4*) or 56°C (Le α12 + Le α13 and Le α15 + Le α27) for 2 min and 72°C for 3 min, and a final extension step of 72°C for 7 min. Total human brain RNA was reverse transcribed to generate a 355-bp RT–PCR product of the PPEF-1 cDNA. Primers 41 (5′-GCAGCAATCGAGGAGCTTAC-3′) and 42 (5′-AATGCGGATAATTCTG-GAAGC-3′) were used for amplification by PCR in the presence of 3 mm MgCl2. The amplification conditions were as described above except for the annealing temperature, which was at 60°C.
A 675-bp probe encompassing the coding region of the human XLRS1 gene was generated by amplifying ∼106 PFU of a retina cDNA library cloned in λgt10 (J. Nathans, Johns Hopkins University, Baltimore, MD) with primers 206 (5′-ATGTCACGCAAGATAGAAGGC-3′) and 207 (5′-TCAGGCACAGTTGCTGACG-3′) with a final MgCl2 concentration of 0.5 mm. Initial denaturation was at 94°C for 10 min, followed by 40 cycles of 94°C for 1 min, 58°C for 1 min, and 72°C for 1 min, and a final elongation step for 10 min at 72°C.
To generate hybridization probes, PCR fragments were separated by agarose gel electrophoresis. The respective bands were cut out off the gel and the DNA was isolated by centrifugal filtration with either an Ultrafree-MC 0.45 μm column (Millipore) or by the Gene Clean Kit (BIO 101), exactly following the suppliers instruction. Labeling was performed with random hexamer priming in the presence of [α-32P]dCTP.
Total mouse eye RNA was reverse transcribed with random hexamers and amplified with a primer set that spans exons 4–6 of the human XLRS1 gene (209 5′-CAGAATGCCCATATCACAAGCCTC-3′ and 210 5′-GCTCCATCCGGATGGCAATGCG-3′) under the following conditions: initial denaturation for 10 min at 95°C, 40 cycles including 1 min at 94°C, 1 min at 65°C, and 3 min at 72°C and a final extension step of 7 min at 72°C. The resulting product of 465 bp was ligated into the pUAG-Vector (Ingenius) and subsequently sequenced. To complete the mouse cDNA, 5′and 3′ RACE were performed on cDNA synthesized by SMART PCR (Clontech) starting with 0.75 μg of total eye RNA. Both amplifications were carried out with primers 210 and 209, respectively, in combination with one adapter primer complementary to part of the SMART linkers. Subsequently, 20 ng of the SMART PCR cDNA were used as template for the RACE experiments in a sample volume of 50 μl containing 50 pmoles of primer 210 or 209 and 10 pmoles of the nested SMART primer. Cycling conditions were as above, except for the decreased annealing temperature of 60°C.
Isolation of Fugu Cosmids
A gridded high-density Fugu genomic Lawrist 4 cosmid library (36,864 clones equivalent to 3.7 haploid Fugu genomes) was screened with the three human PHKA2 cDNAs. Filters were hybridized in 0.5 m Na2PO4, 7% SDS, 1 mm EDTA at 55°C. Washing was in 2× SSC/0.1% SDS for 2 × 10 min and in 1 × SSC/0.1% SDS for 1 × 30 min at 55°C. Exposure was for 6 hr at −70°C. Candidate positive clones exhibiting duplicate hybridization signals were isolated, miniprepared, and digested with EcoRI. DNA fragments were separated in an 0.8% agarose gel. Following Southern transfer of cosmid DNA onto Genescreen Plus (NEN), blots were probed as above.
Cosmid Walking
Both end fragments of Fugu cosmid ICRFc66A2095Q1.2 were generated by PCR on 80 ng of EcoRI-digested cosmid DNA with sequence specific primers E1f (5′-ATGAAGAGCTGGACTCTTGTG-3′) and E1r (5′-TCTCATCGGCGTCGGAGTG-3′) amplifying 719 bp and with primers E2f (5′-CTAGTAGACAGGTTATTGGAC-3′) and E2r (5′-ATGAGTAGATACAAGAGCAGG-3′) amplifying 602 bp. PCRs were performed in the presence of 1.5 mm MgCl2 with an initial denaturation at 96°C for 3 min, followed by 45 cycles at 94°C for 1 min, 58°C for 2 min, and 72°C for 3 min in case of E1 and 35 cycles with annealing at 56°C for 1 min in case of E2.
Direct sequencing of Fugu cosmid ICRFc66L2390Q1.2 with primer Lawrist 4 forward (5′-CGCCTCGAGGTGGCTTATC-3′) enabled us to determine sequence-specific primers 147 (5′-TCGAAACCGAGAGGCCTGTG-3′) and 147′ (5′-ACCCTGTGATGATGACTGAGG-3′) that were used to amplify an end fragment of 758 bp in the presence of 1.5 mm MgCl2. The starting template was 20 ng of EcoRI-digested cosmid DNA, and the PCR cycles consisted of an initial denaturation step of 96°C for 3 min, 35 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min, and a final extension step of 72°C for 7 min. The 758-bp long PCR product was used for screening a second Fugu genomic Lawrist 4 cosmid library (G. Elgar). Hybridization was performed in PEG buffer (125 mm Na2PO4, 250 mm NaCl, 1 mm EDTA, 10% PEG 6000, 7% SDS) at 65°C. Filters were washed in 2× SSC/0.1% SDS for 10 min and in 2× SSC/0.1% SDS for 25 min at hybridization temperature. Exposure was for 4 hr at −70°C.
FISH
Human AP19 cDNA (accession no. AA262073) was labeled by nick translation with biotin-16–dUTP. A total of 40 ng of this probe was then coprecipitated with human Cot-1 competitor DNA and herring sperm DNA, followed by resuspension in 50% formamide, 10% dextran sulfate, 2× SSC. Denaturation for 10 min at 80°C preceeded 1 hr of preannealing. Slides were treated with 100 μg/ml RNase A and 0.01% pepsin, then dehydrated through an ethanol series. Denaturation with 70% formamide/2× SSC at 80°C was followed by dehydration in cold ethanol. The probe was then applied to the slides and allowed to hybridize for 72 hr at 37°C. Slides were washed three times in 50% formamide/2× SSC, once in 2× SSC, and finally in 0.2× SSC at 42°C with little agitation. Detection was performed with fluorescein isothiocyanate (FITC)-conjugated avidin and counterstaining with DAPI.
DNA Sequencing Strategy
Fugu cosmids ICRFc66A2095Q1.2, ICRFc66L2390Q1.2, and F089J4 were digested with EcoRI, HindIII, and PstI, respectively. The resulting fragments were randomly subcloned into appropriately cut and dephosphorylated pBluescript II KS(+) (Stratagene) or pT7T3 18U (Pharmacia) vector. DNA sequences were determined by dideoxy chain termination with the thermo Sequenase fluorescent-labeled primer cycle sequencing kit containing 7-deaza-dGTP (Amersham), primers complementary to the vector arms (5′ labeled with IRD700 and IRD800, respectively) on an automated gel reader (LICOR 4000L and LONG READIR 4200, MWG). Gaps were closed by a combination of primer walking and deletion cloning of larger subclones. All sequences were processed and assembled with the Staden package.
Sequence Analysis
Potential coding regions were identified with the exon prediction programs FGENES, GENSCAN, and GRAIL 2. The putative genes were searched for homologies by protein (SWISSPROT, PIR) and nucleotide databases (GenBank, EMBL), with the BLAST program. Database entries with a score >10−10 were selected and grouped into distinct sets by the criteria homology and position. The putative intron/exon organization of each of the Fugu gene homologs was deduced either from the structure of the corresponding human genes or from sequence comparison to homologs. This procedure was assisted by the PGSEARCH program (Birney et al. 1996). Predicted splice sites were compared with consensus 5′ donor and 3′ acceptor sequences of mammals (Shapiro and Senapathy 1987) to confirm the results. The sets of homologous proteins were aligned by the program PILEUP, with sequences gapped for optimization. Polyadenylation site predictions were performed by the programs FGENES, GENSCAN, and GRAIL 2 and extended by the program POLYAH.
Phylogenetic trees were generated from the aligned nucleotide sequences with the maximum likelihood method as implemented in the DNAML program, available in the PHYLIP (Felsenstein 1981) computer software package. Regions in which some sequences had alignment gaps or that had ambiguous alignments were excluded. Phylogenetic trees were generated with a different starting parameter. Bootstrap resampling was conducted by the method of Felsenstein (1985). One-hundred bootstrap replicates for the DNAML method were conducted.
Acknowledgments
We thank A. Tominaga and M.L. Yaspo for the gift of Fugu genomic DNA and RNA, respectively, and the RZPD for libraries and clones.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kalscheuer@mpimg-berlin-dahlem.mpg.de; FAX +49-30-8413-1383.
REFERENCES
- Aparicio S, Brenner S. How good a model is the Fugu genome. Nature. 1997;387:140. doi: 10.1038/387140a0. [DOI] [PubMed] [Google Scholar]
- Armes N, Gilley J, Fried M. The comparative genomic structure and sequence of the surfeit gene homologs in the puffer fish Fugu rubripes and their association with CpG-rich islands. Genome Res. 1997;7:138–152. doi: 10.1101/gr.7.12.1138. [DOI] [PubMed] [Google Scholar]
- Baxendale S, Abdulla S, Elgar G, Buck D, Berks M, Micklem G, Durbin R, Bates G, Brenner S, Beck S. Comparative sequence analysis of the human and pufferfish Huntington’s disease genes. Nat Genet. 1995;10:67–76. doi: 10.1038/ng0595-67. [DOI] [PubMed] [Google Scholar]
- Birney E, Thompson JD, Gibson TJ. Pairwise and searchwise—Finding the optimal alignment in a simultaneous comparison of a protein profile against all DNA translation frames. Nucleic Acids Res. 1996;24:2730–2739. doi: 10.1093/nar/24.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Doolittle RF. Drosophila kelch motif is derived from a common enzyme fold. J Mol Biol. 1994;236:1277–1282. doi: 10.1016/0022-2836(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Brendel V, Bucher P, Nourbakhsh IR, Blaisdell BE, Karlin S. Methods and algorithms for statistical analysis of protein sequences. Proc Natl Acad Sci. 1992;89:2002–2006. doi: 10.1073/pnas.89.6.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, Elgar G, Sandford R, Macrae A, Venkatesh B, Aparicio S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature. 1993;18:265–268. doi: 10.1038/366265a0. [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Shin YS, Bakker HD, Deutsch J, Lozano MJ, Maire I, Kilimann MW. Mutation hotspots in the PHKA2 gene in X-linked liver glycogenosis due to phophorylase kinase deficiency with atypical activity in blood cells (XLG2) Hum Mol Genet. 1996;5:653–658. doi: 10.1093/hmg/5.5.653. [DOI] [PubMed] [Google Scholar]
- Davidson JJ, Ozcelik T, Hamacher C, Willems PJ, Francke U, Kilimann MW. cDNA cloning of a liver isoform of the phosphorylase kinase alpha subunit and mapping of the gene to Xp22.2-p22.1, the region of human X-linked liver glycogenosis. Proc Natl Acad Sci. 1992;89:2096–2100. doi: 10.1073/pnas.89.6.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgar G, Rattray F, Greystrong J, Brenner S. Genomic structure and nucleotide sequence of the p55 gene of the puffer fish Fugu rubripes. Genomics. 1995;27:442–446. doi: 10.1006/geno.1995.1075. [DOI] [PubMed] [Google Scholar]
- Elgar G, Clark M, Green A, Sandford R. How good a model is the Fugu genome. Nature. 1997;387:140. doi: 10.1038/387140b0. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. The statistical approach to inferring evolutionary trees and what it tells us about parsimony and compatibility. In: Duncan T, Stues TF, editors. Cladistics: Perspectives on the reconstruction of evolutionary history; workshop on the theory and application of cladistic methodology. New York, NY: Columbia University Press; 1981. pp. 22–28. [Google Scholar]
- ———. Confidence limits on phylogenies an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed]
- Gilley J, Armes N, Fried M. Fugu genome is not a good mammalian model. Nature. 1997;385:305–306. doi: 10.1038/385305a0. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hendrickx J, Coucke P, Dams E, Lee P, Odievre M, Corbeel L, Fernandes JF, Willems PJ. Mutations in the phosphorylase kinase gene PHKA2 are responsible for X-linked liver glycogen storage disease. Hum Mol Genet. 1995;4:77–83. doi: 10.1093/hmg/4.1.77. [DOI] [PubMed] [Google Scholar]
- How GF, Venkatesh B, Brenner S. Conserved linkage between the puffer fish (Fugu rubripes) and human genes for platelet-derived growth factor receptor and macrophage colony-stimulating factor receptor. Genome Res. 1996;6:1185–1191. doi: 10.1101/gr.6.12.1185. [DOI] [PubMed] [Google Scholar]
- Jurka J, Klonowski P, Dagman V, Pelton P. Censor—a program for identification and elimination of repetitive elements from DNA sequences. Comput & Chem. 1996;20:119–121. doi: 10.1016/s0097-8485(96)80013-1. [DOI] [PubMed] [Google Scholar]
- Kalscheuer VM, Mariman EC, Schepens MT, Rehder H, Roperes H-H. The insulin-like growth factor type-2 receptor gene is imprinted in the mouse but not in humans. Nat Genet. 1993;5:74–78. doi: 10.1038/ng0993-74. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T, Davis AC, Frucht S, Greco BO, Payne GS, Tubb B. AP17 and AP19 the mammalian small chains of the clathrin-associated protein complexes show homology to YAP17P their putative homolog in yeast. J Biol Chem. 1991;266:11153–11157. [PubMed] [Google Scholar]
- Kitagawa K, Wang X, Hatada I, Yamaoka T, Nojima H, Inazawa J, Abe T, Mitsuya K, Oshimura M, Murata A, Monden M, Mukai T. Isolation and mapping of human homologues of an imprinted mouse gene U2AF1-RS-1. Genomics. 1995;30:257–263. doi: 10.1006/geno.1995.9879. [DOI] [PubMed] [Google Scholar]
- Macrae AD, Brenner S. Analysis of the dopamine receptor family in the compact genome of the puffer fish Fugu rubripes. Genomics. 1995;25:436–446. doi: 10.1016/0888-7543(95)80044-m. [DOI] [PubMed] [Google Scholar]
- Mason PJ, Stevens DJ, Luzzatto L, Brenner S, Aparicio S. Genomic structure and sequence of the Fugu rubripes glucose-6-phosphate dehydrogenase gene (G6PD) Genomics. 1995;26:587–591. doi: 10.1016/0888-7543(95)80179-p. [DOI] [PubMed] [Google Scholar]
- Miles C, Elgar G, Coles E, Kleinjan D-J, van Heyningen V, Hastie N. Complete sequencing of the Fugu WAGR region from the WT1 to PAX 6: Dramatic compaction and conservation of synteny with human chromosome 11p13. Proc Natl Acad Sci. 1998;95:13068–13072. doi: 10.1073/pnas.95.22.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Rugarli EI, van de Vosse E, Andolfi G, Mariani M, Puca AA, Consalez GG, den Dunnen JT, Ballabio A, Franco B. A novel human serine-threonine phosphatase related to the Drosophila retinal degeneration C (rdgC) gene is selectively expressed in sensory neurons of neural crest origin. Hum Mol Genet. 1997;6:1137–1145. doi: 10.1093/hmg/6.7.1137. [DOI] [PubMed] [Google Scholar]
- Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M, Consalez G, Trump D, Ballabio A, Franco B. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics. 1998;51:427–433. doi: 10.1006/geno.1998.5391. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Robinson DN, Cooley L. Drosophila kelch is an oligomeric ring canal actin organizer. J Cell Biol. 1997;138:799–810. doi: 10.1083/jcb.138.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer CG, Gehrig A, Warnekewittstock R, Marquardt A, Ewing CC, Gibson A, Lorenz B, Jurklies B, Weber BHF. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997;17:164–170. doi: 10.1038/ng1097-164. [DOI] [PubMed] [Google Scholar]
- Schofield JP, Elgar G, Greystrong J, Lye G, Deadman R, Micklem G, King A, Brenner S, Vaudin M. Regions of human chromosome 2 (2q32-q35) and mouse chromosome 1 show synteny with the pufferfish genome (Fugu rubripes) Genomics. 1997;45:158–167. doi: 10.1006/geno.1997.4913. [DOI] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman PM, Sun H, Macke JP, Williams J, Smallwood PM, Nathans J. Identification and characterization of a conserved family of protein serine/threonine phosphatases homologous to Drosophila retinal degeneration C. Proc Natl Acad Sci. 1997;94:11639–11644. doi: 10.1073/pnas.94.21.11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele F, O’Tousa JE. Rhodopsin activation causes retinal degeneration in drosophila rdgC mutant. Neuron. 1990;4:883–890. doi: 10.1016/0896-6273(90)90141-2. [DOI] [PubMed] [Google Scholar]
- Steele FR, Washburn T, Rieger R, O’Tousa JE. Drosophila retinal degeneration C (rdgC) encodes a novel serine/threonine protein phosphatase. Cell. 1992;69:669–676. doi: 10.1016/0092-8674(92)90230-a. [DOI] [PubMed] [Google Scholar]
- Trower MK, Orton SM, Purvis IJ, Sanseau P, Riley J, Christodoulou C, Burt D, See CG, Elgar G, Sherrington R, Rogaev EL, St. George-Hyslop P, Brenner S, Dykes CW. Conservation of synteny between the genome of the pufferfish (Fugu rubripes) and the region on the human chromosome 14 (14q24.3) associated with familial Alzheimer disease (AD3 locus) Proc Natl Acad Sci. 1996;93:1366–1369. doi: 10.1073/pnas.93.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vosse E, Walpole SM, Nicolaou A, van der Bent P, Cahn A, Vaudin M, Ross MT, Durham J, Pavitt R, Wilkinson J, Grafham D, Bergen AAB, van Ommen G-J B, Yates JRW, den Dunnen JT, Trump D. Characterization of SCML1, a new gene in Xp22, with homology to developmental polycomb genes. Genomics. 1998;49:96–102. doi: 10.1006/geno.1998.5224. [DOI] [PubMed] [Google Scholar]
- Varkey JP, Muhlrad PJ, Minniti AN, Do B, Ward S. The Caenorhabditis elegans spe-26 gene is necessary to form spermatids and encodes a protein similar to the actin-associated proteins Kelch and scruin. Genes & Dev. 1995;9:1074–1086. doi: 10.1101/gad.9.9.1074. [DOI] [PubMed] [Google Scholar]
- Venkatesh B, Si-Hoe SL, Murphy D, Brenner S. Transgenic rats reveal functional conservation of regulatory controls between the Fugu isotocin and rat oxytocin genes. Proc Natl Acad Sci. 1997;94:12462–12466. doi: 10.1073/pnas.94.23.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GS, Elgar G, Sandford R, Brenner S. Cloning and sequencing of complement component C9 and its linkage to DOC-2 in the pufferfish Fugu rubripes. Gene. 1997;200:203–211. doi: 10.1016/s0378-1119(97)00423-x. [DOI] [PubMed] [Google Scholar]