

Abstract
Single-nucleotide polymorphisms (SNPs) are the most frequently found DNA sequence variations in the human genome. It has been argued that a dense set of SNP markers can be used to identify genetic factors associated with complex disease traits. Because all high-throughput genotyping methods require precise sequence knowledge of the SNPs, any SNP discovery approach must involve both the determination of DNA sequence and allele frequencies. Furthermore, high-throughput genotyping also requires a genomic DNA amplification step, making it necessary to develop sequence-tagged sites (STSs) that amplify only the DNA fragment containing the SNP and nothing else from the rest of the genome. In this report, we demonstrate the utility of a SNP-screening approach that yields the DNA sequence and allele frequency information while screening out duplications with minimal cost and effort. Our approach is based on the use of a homozygous complete hydatidiform mole (CHM) as the reference. With this homozygous reference, one can identify and estimate the allele frequencies of common SNPs with a pooled DNA-sequencing approach (rather than having to sequence numerous individuals as is commonly done). More importantly, the CHM reference is preferable to a single individual reference because it reveals readily any duplicated regions of the genome amplified by the PCR assay before the duplicated sequences are found in GenBank. This approach reduces the cost of SNP discovery by 60% and eliminates the costly development of SNP markers that cannot be amplified uniquely from the genome.
[Sequence data for this article were deposited with the NCBI dbSTS and dbSNP data libraries under accession nos. G42862–G42905]
Single-nucleotide polymorphisms (SNPs) are the most frequently found DNA sequence variations in the human genome (Taillon-Miller et al. 1998). It has been argued that a dense set of SNP markers can be used to identify genetic factors associated with complex disease traits (Risch and Merikangas 1996; Collins et al. 1997). Advocates of these approaches suggest that some 100,000 or more SNP markers (at 30-kb intervals or up to five markers per gene) will be needed in population studies to detect genetic factors with moderate effects in the complex traits being investigated (Collins et al. 1997). Several efforts, sponsored by both the National Human Genome Research Institute and private industry, have been launched to develop SNP markers with the goal of achieving the numbers needed for association studies within the next 3 years (Marshall 1997, 1998; Wang et al. 1998).
Because all high-throughput genotyping methods capable of handling large numbers of markers and samples require precise knowledge of the DNA sequence surrounding the SNP markers, and the usefulness of the markers is determined by their heterozygosity in the population, any SNP discovery approach must involve the determination of DNA sequence and allele frequencies. Furthermore, most high-throughput genotyping methods also require a genomic DNA amplification step, making it necessary to develop sequence-tagged sites (STSs) that amplify only the DNA fragments containing the SNPs and nothing else from the rest of the genome.
This is not a trivial concern because there are many repetitive elements and duplicated regions in the genome in which near identical sequences are found on different chromosomal regions. If the DNA fragments amplified by the PCR (Saiki et al. 1988) came from different parts of the genome but were near identical, the DNA sequence differences might be erroneously considered alleles of an SNP, leading to highly confusing results when the genotyping experiments were performed. The use of computer programs such as REPEAT MASKER (A.F.A. Smit and P. Green, unpubl.) has made it a simple task to avoid developing SNP markers from common repetitive regions such as those containing Alu or L1 elements. What is more difficult to detect is the presence of a putative SNP in a duplicated region of the genome. In this report, we demonstrate the utility of a SNP screening approach that yields the DNA sequence and allele frequency information while screening out duplications with minimal cost and effort.
The approach combines the use of a complete hydatidiform mole (CHM) as a homozygous DNA reference sample and a pooled DNA sequencing strategy for SNP identification and allele frequency estimation (Kwok et al. 1994). A CHM is usually a 46, XX homozygote formed by the fertilization of an empty ovum by a single haploid sperm, which later duplicates its chromosomes to give a diploid tumor (Lawler et al. 1991). The worldwide incidence of hydatidiform moles is 1/1000 pregnancies (Grimes 1984). We have reported previously that the CHM can be used as a homozygous DNA reference in SNP marker development (Taillon-Miller et al. 1997). In the course of screening anonymous STSs for SNPs, we have noticed that the DNA from this CHM1 can be used to identify false-positive SNPs that are the result of amplification of duplicated regions of the genome as in the case of multigene families or low-frequency repeats. In this study we show that in every case in which the CHM sequence contains a heterozygous base, it is the result of duplication, and the sequence differences are not in fact allelic.
In regions of the genome in which high-quality, large-scale sequencing is being performed, we have shown that the most efficient and cost-effective approach to SNP identification is comparison of the consensus sequences of the overlapping regions of the large-insert clones being sequenced (Taillon-Miller et al. 1998). In regions in which no such overlapping sequences are available, one has to develop STSs and screen the DNA fragments amplified from multiple individuals for DNA sequence variations (Kwok et al. 1996; Wang et al. 1998). We have advocated a sequence comparison approach consisting of obtaining the DNA sequences from four individuals (eight chromosomes) plus a pooled DNA sample for allele frequency estimation (Kwok et al. 1994, 1996). This strategy allows one to identify, with >85% probability all the SNPs with >20% allele frequency for the minor allele (Kwok et al. 1994). SNPs developed by use of the population pool method of estimating frequencies have been confirmed by subsequent genotyping of the markers in every individual present in the pool and the frequencies have been shown to be accurate (±5%) (Kwok et al. 1994).
With the advent of two new classes of dye-labeled dideoxy chain terminators (the dRhodamine and the energy transfer, BigDye terminator) that have improved spectral properties and give more even peaks in cycle sequencing (Zakeri et al. 1998), we show in this study that one can reduce the number of samples used in each screening experiment from five to just two (CHM and pooled sample) and still identify all the SNPs found with the previous approach. Reducing the number of sequencing reactions required to identify SNPs from anonymous STSs and screening out duplications undetected by computer filters greatly lowers the reagent and labor cost of SNP development.
RESULTS
In the first set of experiments, we compared the outcome of the two polymorphism scanning methods to see whether SNPs could be identified by analyzing the sequences of just two samples. Thirty-six STSs developed from genomic sequences (that were not annotated as containing common repetitive elements) with unique STS primers (i.e., no match when a BLAST search was performed against the GenBank nonredundant database) were scanned for SNPs. In one arm of the study, genomic DNA samples from 4 unrelated individuals and a pooled sample consisting of equal amounts of DNA from 80 individuals were sequenced with the dRhodamine or BigDye terminators. The sequences from the four individuals were compared to identify changes in the peak pattern followed by the estimation of allele frequencies with the pooled DNA sequence (method 1, Kwok et al. 1994). In the second arm of the study, the homozygous CHM1 DNA and the same 80 individual pooled DNA samples were sequenced with the BigDye terminators and the sequences were compared to look for significant changes between the two sequences. The SNP identification and allele frequency estimation were done simultaneously (method 2). The DNA sequences in the two arms of the study were analyzed independently and the pooled sample was sequenced independently for each part of the study so that the results would not be biased. In addition, care was taken to make sure that excellent sequence data were available for both methods in cases in which dRhodamine sequences were compared with BigDye terminator sequences. In all other cases, BigDye terminator sequence data were used in both methods. The DNA samples used were derived from the 80 parents of the Centre d’Etude Polymorphisme Humain (CEPH) pedigrees (Dausset et al. 1990).
In all, 25,777 bp of DNA sequence in 36 STSs were scanned for polymorphisms by both method 1 (four individuals and the pool) and method 2 (CHM1 and the pool). Figure 1 shows the sequencing results by use of methods 1 and 2 with the STS sWXD3868. The polymorphism is easily identified by use of both methods. In A, representing results from method 1 sequenced with dRhodamine dye terminators, individuals 1 and 3 are T homozygotes in the TGAG[C/T]CTCC string, individual 2 is a C/T heterozygote, and individual 4 is a C homozygote. The pooled sample (sequence 5) showed that it is an informative SNP with allele frequencies of 60% T, 40% C. In B, representing results from method 2 sequenced with BigDye terminators, the CHM1 is a T homozygote (sequence 6), whereas the pooled sample (sequence 5) contains a compound C/T peak in the TGAG[C/T]CTCC. Allele frequency estimates with the CEPH pool sequence and the CHM1 sequence gave similar results of 70% T and 30% C.
Figure 1.
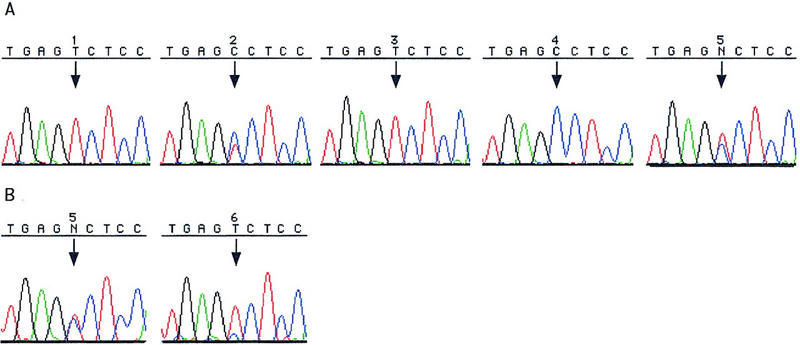
The results of scanning sWXD3868 for SNPs by method 1 is shown in A. CEPH parents are 1, 2, 3, 4, and the CEPH population pool is 5 (additional detail about DNAs used are included in Methods). Sequencing was done with the dRhodamine terminators in A. The results for method 2 are shown in B. The CHM1 is sample 6 and the CEPH population pool is sample 5. Sequencing was done with the BigDye terminators in B. (↓) SNP locations. The small blue underhand for the T peak (↓) in B, sample 6 is a common sequencing artifact and was seen in a number of T peaks in this sequencing trace.
The results of scanning all 36 STSs by both methods are shown in Table 1. No SNPs were found by either method in 18 of the 36 STSs tested (50%). Three STSs (8.3%) gave inconclusive results with method 1. With method 2, two of the inconclusive STS’s were shown to be from duplicated regions and the third contained no SNPs. In 9 STSs (25%), 10 SNPs were found by both methods. In the remaining six STSs (16.7%), two SNPs were found by both methods and an additional seven SNPs were found by method 2 alone. All of the additional SNPs found by method 2 alone were uninformative polymorphisms (frequency <0.20) in the CEPH population and were differences between the CHM1 sequence and the consensus sequences found in the CEPH pool that were identical to the GenBank sequence from which these STSs were developed. All informative SNPs have been confirmed by genotyping >100 individuals as part of a different study.
Table 1.
Results of Scanning for SNPs using Methods 1 and 2
| Xq | GenBank STS accession no. | No. of bases sequenced | Method 1 | Method 2 | ||
|---|---|---|---|---|---|---|
| rare SNPsa | informative SNPsb | rare SNPsa | informative SNPsb | |||
| 3840 | G42862 | 870 | 0 | 1 | 1 | 1 |
| 3841 | G42863 | 755 | 0 | 1 | 0 | 1 |
| 3843 | N.S. | 200 | N.U. | |||
| 3844 | G42864 | 790 | 0 | 0 | 0 | 0 |
| 3845 | G42865 | 750 | 0 | 0 | 0 | 0 |
| 3846 | G42866 | 865 | 0 | 1 | 1 | 1 |
| 3847 | G42867 | 850 | 0 | 1 | 0 | 1 |
| 3849 | G42868 | 829 | 0 | 1 | 0 | 1 |
| 3851 | G42869 | 650 | 0 | 0 | 0 | 0 |
| 3852 | G42870 | 773 | 0 | 0 | 0 | 0 |
| 3853 | G42871 | 500 | 0 | 0 | 0 | 0 |
| 3855 | G42872 | 790 | 0 | 1 | 0 | 1 |
| 3856 | G42873 | 830 | 0 | 0 | 0 | 0 |
| 3857 | N.S. | 500 | N.U. | |||
| 3858 | G42874 | 820 | 0 | 0 | 1 | 0 |
| 3859 | G42875 | 680 | 0 | 0 | 0 | 0 |
| 3860 | G42876 | 470 | 0 | 0 | 0 | 0 |
| 3861 | G42877 | 520 | 0 | 0 | 0 | 0 |
| 3862 | G42878 | 485 | 0 | 1 | 0 | 1 |
| 3863 | G42879 | 880 | 0 | 0 | 0 | 0 |
| 3864 | G42880 | 725 | 0 | 0 | 0 | 0 |
| 3865 | G42881 | 445 | 0 | 0 | 0 | 0 |
| 3866 | G42882 | 605 | 0 | 0 | 0 | 0 |
| 3867 | G42902 | 585 | 0 | 0 | 0 | 0 |
| 3868 | G42883 | 730 | 0 | 1 | 0 | 1 |
| 3870 | G42884 | 850 | 0 | 0 | 0 | 0 |
| 3871 | G42885 | 930 | 0 | 0 | 1 | 0 |
| 3872 | G42886 | 900 | 0 | 0 | 0 | 0 |
| 3875 | G42887 | 900 | 0 | 2 | 0 | 2 |
| 3879 | G42888 | 860 | 0 | 1 | 0 | 1 |
| 3880 | G42903 | 700 | 0 | 0 | 0 | 0 |
| 3881 | G42889 | 860 | 0 | 0 | 0 | 0 |
| 3884 | G42890 | 525 | 0 | 0 | 0 | 0 |
| 3885 | G42891 | 750 | 1 | 0 | 1 | 0 |
| 3886 | G42892 | 800 | 0 | 0 | 2 | 0 |
| 3888 | G42893 | 750 | 0 | 0 | 1 | 0 |
(N.S.) Not submitted; (N.U.) not unique.
Rare SNPs are those with allelic frequencies <0.20.
Informative SNPs are those with allelic frequencies >0.20.
In the course of our SNP development efforts using method 1 over the past several years, we have encountered situations (∼5% of 373 STSs examined) in which the results were suspicious because multiple SNPs were found within 100 bp or heterozygous peaks were found in all four individuals and in the pooled sample (see Fig. 2, sequences 1–5). In such cases, it was difficult to determine whether the sequence differences were true SNPs, sequencing artifacts, or differences in duplicated sequences as a result of amplification of near-identical DNA sequences found in various parts of the genome. In the second set of experiments, we investigated the utility of the homozygous CHM as a standard to resolve these difficult cases.
Figure 2.
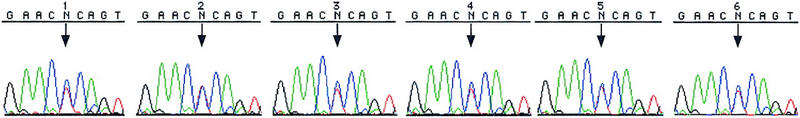
The results of scanning sWXD3654 for SNPs by methods 1 and 2 are shown. (Method 1) CEPH parents are 1, 2, 3, and 4 and the CEPH population pool is 5 (additional detail about DNAs used is included in Methods); (method 2) CHM1 is sample 6, and the CEPH population pool is sample 5. The CEPH population pool is shown only once. Sequencing was done with the BigDye terminators. (↓) SNP locations.
We selected 14 STSs (which include the three problematic STSs in the first set of experiments) that gave inconclusive results with method 1 in the past (mostly with the original rhodamine dye terminators) for analysis with method 2. The majority of these STS had been sequenced multiple times by the method 1 approach without yielding conclusive results. The DNA sequences of all 14 STSs were searched against GenBank, and no homologous sequences were found at the time when they were first developed. These 14 STSs were amplified against the CHM1 DNA and the CEPH population pool and the sequencing results are shown in Table 2. Five of the 14 STSs were found to contain no SNPs when analyzed with method 2, thereby proving that the heterozygous peaks seen in method 1 were sequencing artifacts. These results reflect the superior data quality associated with the new dye terminator chemistry used in method 2. Six STSs were found to contain SNPs unambiguously with method 2. In these cases, the CHM sequence was homozygous throughout the entire sequence, but the pooled DNA samples gave composite peaks at the SNP locations. In the remaining three STSs, heterozygous peaks were seen not only in the individuals screened, but also in the CHM sequence (e.g., see Fig. 2, sequence 6). Because the CHM DNA is completely homozygous, heterozygous peaks represent sequences from two simultaneously amplified DNA fragments from duplicated regions of the genome.
Table 2.
Results of Using Method 2 to Resolve Troublesome Sequence Data
| Xq | GenBank STS accession no. | Resolution |
|---|---|---|
| 3474 | G42894 | STS was unique but no SNPs found |
| 3504 | G42895 | STS was unique but no SNPs found |
| 3661 | G42896 | STS was unique but no SNPs found |
| 3806 | G42904 | STS was unique but no SNPs found |
| 3864 | G42880 | STS was unique but no SNPs found |
| 3555 | G42897 | STS was unique and 5 SNPs found |
| 3695 | G42898 | STS was unique and 4 SNPs found |
| 3696 | G42899 | STS was unique and 5 SNPs found |
| 3705 | G42900 | STS was unique and 2 SNPs found |
| 3812 | G42901 | STS was unique and 4 SNPs found |
| 3813 | G42905 | STS was unique and 2 SNPs found |
| 3654 | N.S. | STS not unique |
| 3843 | N.S. | STS not unique |
| 3857 | N.S. | STS not unique |
(N.S.) Not submitted.
To prove that this was the case, the nine STSs containing sequence differences were tested against the somatic cell hybrid chromosome panel II, v.2 (Coriell Institute, Camden, NJ). Each of the 24 cell lines in this panel contains one of the 24 human chromosomes in a mouse or hamster background. As expected, all six STSs containing real SNPs were found to map to only one chromosome, whereas the three STSs in which heterozygous peaks were found in the CHM sequence mapped to multiple chromosomes, with PCR products of the same size found in more than two chromosome pools. Figure 3 shows two examples of these experiments. In Figure 3A, the STS sWXD3555 amplified only the two pools containing the X chromosome plus the human control, lending strong evidence that a unique DNA fragment was amplified. In Figure 3B, the duplicated STS sWXD3857 amplified all eight chromosomal pools, proving that multiple DNA loci from various parts of the genome had been amplified. Although these STSs were not matching to any known sequences several months before these experiments were performed, two of these STSs (3843 and 3857) turned out to be retroviral elements that were unannotated in the original GenBank entry, and the sequencing of the STS was not unique even though a Blast search with the primer sequences gave unique results. The third DNA sequence is found within a few thousand bases from the adrenoleukodystrophy (ALD) gene in the Xq28 region in which a recent BLAST search yielded two homologous sequences on chromosome 2p11 and chromosome 16.
Figure 3.

Results of testing STSs sWXD3555 (A) and sWXD3857 (B) against the chromosome panel pools (1–8), human genomic DNA (9), hamster genomic DNA (10), and mouse genomic DNA (11). sWXD3555 amplified only pools 2 and 8 and the human control, indicating that it was found on the X chromosome. sWXD3857 amplified all eight pools and the human control, indicating that it was found on multiple chromosomes. (Shaded arrows) The 700- and 1000-bp molecular weight standards (50–2000 bp, Bio-Rad, Hercules, CA); (black arrows) the specific PCR product. Detail descriptions of the chromosome panel pools are included in Methods.
DISCUSSION
The results from the studies reported here show the two main advantages of utilizing the homozygous complete hydatidiform mole as the standard in a comparative sequencing approach with a pooled DNA sample and the new dye terminators in SNP marker development. The first advantage is the 60% reduction in the cost and effort needed in this approach. The second is the ease of weeding out those SNPs that are really duplications of near identical sequences in the genome when heterozygous peaks are seen in the complete hydatidiform mole sequence.
The pooled DNA sequencing approach is a useful approach to SNP discovery because the goal is to identify informative markers and not rare polymorphisms. With a homozygous standard, SNPs with the minor allele of >15% allele frequency can be found very easily by use of the new dye terminators (Zakeri et al. 1998). By analyzing only two DNA samples with the new approach instead of five in the old approach, scanning for SNP is much faster and has a 60% savings in labor and reagent cost. On the basis of our current costs, this represents $30 in savings per marker. With the goal of developing 100,000 SNPs, even a saving of $30 per marker developed translates to $3 million being made available for other projects.
Although one can argue that a homozygous individual or a clone that contains the STS can be used as the reference in a pooled DNA sequencing approach to SNP development and realize the same savings, the real advantage of utilizing the CHM as a reference is the ease with which one can identify duplications in the genome before the duplications are found in GenBank. This is an important advantage because the ultimate goal of identifying SNPs is to develop genetic markers for genetic analysis. By definition, genetic markers must be unique signposts in the genome. For this reason, STSs are developed from regions devoid of repetitive sequences. Specifically, homology searches are performed to ensure that primers for STSs are not selected from repeat sequences. Although computer filtering leads to the production of high-quality STSs in most cases, this filtering process fails occasionally. If care is not taken, much time, effort, and expense are wasted when an SNP is developed and used in genetic mapping before it has been determined that the alleles are actually sequence differences in near-identical sequences found in duplicated regions of the genome.
For example, in the three cases in which the candidate SNPs were found to be heterozygous peaks in the CHM, two contained retroviral element sequences, which accounted for the lack of specificity in these cases. These retroviral elements are only now starting to be included in the computer filters used to annotate genomic sequence and in these two cases retroviral elements were not annotated in the GenBank entries. SNPs will be developed in duplicated regions and other low-frequency repeats creating useless SNP markers until computer filters contain all repeat elements.
A more interesting case is the third nonunique STS, sWXD3654. This STS contained >10 candidate SNPs as well as many bases that were different from the original published sequence. Although we suspected that the STS was not amplifying a unique region because all four individuals were heterozygous at all locations, with the pool sample giving exactly the same pattern, it was not until we repeated the sequencing with the CHM1 DNA that we were able to fully explain the results. The STS sWXD3654 was developed from DNA sequence within a few thousand bases from the ALD gene in the Xq28 region (GenBank accession no. U52111). Although the particular sequence of sWXD3654 found no homologies when a BLAST search was performed when the STS was developed originally, a recent BLAST search yielded two additional sequences found on chromosome 2p11 and chromosome 16. This is consistent with the recent observations that a 9.7-kb segment of the ALD locus has been duplicated to chromosomes 2p11, 10p11, 16p11, and 22q11 (Eichler et al. 1996, 1997). It is clear that the sWXD3654 primers are amplifying multiple regions of the genome in this multigene family.
Although our approach is superior to the current methods, it is not without limitations. Any compromise in sequence data quality will create severe problems in the pooled sequencing approach. Furthermore, it is unlikely that an SNP with the minor allele frequency of <10% can be detected with confidence. However, although the use of a CHM reference will not identify all gene duplications, and one may argue that a 5% drop-out rate is tolerable, the ability to remove from further development and testing SNPs that are not real or are impossible to amplify uniquely will save a substantial amount of resources when developing thousands of SNP markers. One may also contend that with the ever-expanding DNA sequence database available, computer filtering will identify all duplicated regions in the genome. Whereas this is certainly the case when the reference human genome is completely sequenced in 2003, the use of the homozygous CHM standard (after initial filtering on the basis of database searches) provides the best chance of exposing duplications of near-identical sequences over the next few years if costly development of genotyping assays of these false SNPs are to be avoided.
METHODS
A group of 36 STSs from the Xq28 (sWXD3840 and sWXD3841), Xq27 (sWXD3885–sWXD3888), Xq26 (sWXD3870–sWXD3884), and Xq25 (sWXD3844–sWXD3868) regions of the X chromosome were scanned for SNPs by the following two methods.
Method 1
Genomic DNA samples from 4 female CEPH parents (individual 1, CEPH K102-02, Coriell NA04479; individual 2, CEPH K1340-02, Coriell NA07019; individual 3, CEPH K1345-02, Coriell NA07348A; individual 4, CEPH K13294-02, Coriell NA07434 Coriell Institute; 8 chromosomes) and a pooled genomic DNA sample consisting of 40 female and 40 male CEPH parents (120 X chromosomes) were amplified by PCR. The PCR products were gel purified and sequenced with either dRhodamine or BigDye terminators. The DNA sequence data were compared to identify changes in the peak pattern as described below.
Method 2
The genomic DNA sample from CHM1 and the same pooled DNA sample from 80 CEPH parents detailed above were amplified by PCR. The PCR products were gel purified and sequenced as in method 1. The DNA sequence data were analyzed as described below.
PCR
Reaction conditions were optimized for products of ∼1 kb (modified from published long-range PCR conditions; Barnes et al. 1994). Specifically, 20 ng of genomic DNA was amplified in a 50-μl reaction containing 50 mm Tris (pH 9.2), 16 mm ammonium sulfate, 2.5 mm MgCl2, 8% glycerol, 0.25 mm dNTP, 1 μm each primer, 0.05 units of Amplitaq (Perkin-Elmer Corp.), 0.000125 units of cloned Pfu DNA polymerase (Stratagene, La Jolla, CA). Thermal cycling conditions were initial denaturation at 96°C for 5 min followed by 35 cycles of 92°C for 15 sec, 55°C for 22 sec, 68°C for 1 min; with a final extension period of 10 min at 68°C. The reaction mixture was then held at 4°C until used.
PCR Product Purification
The PCR reaction mixture was run on a 1% low melting point agarose gel (FMC, Rockland, ME) and the excised gel band was purified with the Wizard PCR Purification Kit (Promega Corp., Madison, WI) and eluted in 50 μl of water.
DNA Sequencing
The gel-purified PCR product was sequenced as described in detail previously (Zakeri et al. 1998) by use of the ABI Prism dRhodamine terminators or the ABI Prism BigDye terminators (Perkin-Elmer Biosystems, Foster City, CA). For method 2, the BigDye terminators were used exclusively. In all cases, the reaction volume was cut in half (5.2 μl of purified PCR product in a 10-μl reaction). The excess dye terminators were removed by ethanol precipitation before loading an aliquot (1.6 μl/6 μl of loading buffer, deionized formamide, 50 mm EDTA) on a 5% polyacrylamide gel containing 8 m of urea in TBE run on an ABI 377 automatic DNA sequencer (or an ABI 373A automatic DNA sequencer, 3 μl loaded). For dRhodamine terminators, sequencing reactions are resuspended in one-half the volume before loading on the gel.
Sequence Analysis
The sequencing traces were aligned with Sequencer 3.0 (Gene Codes Corp., Ann Arbor, MI) and SNPs were identified first by looking at mismatch bases marked by the sequencer program followed by visually inspecting each base in all the aligned sequencing traces. In method 1, a drop in peak height of >40% in a base in a sequencing trace as compared with the same base in another sequencing trace with the appearance of a second peak underneath signifies the presence of a heterozygous locus (Kwok et al. 1994; Zakeri et al. 1998). The allele frequencies were estimated by comparing the ratios of heterozygous peaks in the sequence of the pooled DNA sample with the ratios of the same peaks in the sequence of an individual as described in detail previously (Kwok et al. 1994). In method 2, any significant drop in the peak height of a base in the pooled DNA sample as compared with the homozygous CHM sequence (>10%) with a new peak found under the same base peak is scored as an SNP. The allele frequencies are estimated as in method 1 above.
Chromosomal Assignment of STSs
Genomic DNA from 24 somatic hybrid cell lines each containing 1 of the 22 autosome and the X and Y chromosomes (Coriell Institute, Mapping Panel II, v. 2) were grouped into 8 pools (pool 1, chromosomes 1, 8, 14, 19, 22, and Y; pool 2, chromosomes 1, 2, 9, 15, 20, and X; pool 3, chromosomes 2, 3, 8, 10, 16, and 21; pool 4, chromosomes 3, 4, 9, 11, 14, and 17; pool 5, chromosomes 5, 10, 12, 15, 18, and 19; pool 6, chromosomes 4, 5, 6, 13, 20, and 22; pool 7, chromosomes 6, 7, 11, 12, 16, and Y; pool 8, chromosomes 7, 13, 17, 18, 21, and X. The primers of the STSs being mapped were used to amplify DNA from the eight chromosome pools, together with the human, hamster, and mouse controls by use of the conditions detailed above. The PCR products were analyzed on 1% agarose gels.
Acknowledgments
We thank Qun Li for technical assistance and David G. Mutch for the CHM specimen. This work is supported in part by grants from the National Human Genome Research Institute (RO1 HG1439).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kwok@im.wustl.edu; FAX (314) 362-8159.
REFERENCES
- Barnes WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci. 1994;91:2216–2220. doi: 10.1073/pnas.91.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Chakravarti A. Variations on a theme: Cataloging human DNA sequence variation. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Dausset J, Cann H, Cohen D, Lathrop M, Lalouel JM, White R. Centre d’etude du polymorphisme humain (CEPH): Collaborative genetic mapping of the human genome. Genomics. 1990;6:575–577. doi: 10.1016/0888-7543(90)90491-c. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Lu F, Shen Y, Antonacci R, Jurecic V, Doggett NA, Moyzis RK, Baldini A, Gibbs RA, Nelson DL. Duplication of a gene-rich cluster between 16p11.1 and Xq28: A novel pericentromeric-directed mechanism for paralogous genome evolution. Hum Mol Genet. 1996;5:899–912. doi: 10.1093/hmg/5.7.899. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Budarf ML, Rocchi M, Deaven LL, Doggett NA, Baldini A, Nelson DL, Mohrenweiser HW. Interchromosomal duplications of the adrenoleukodystrophy locus: A phenomenon of pericentromeric plasticity. Hum Mol Genet. 1997;6:991–1002. doi: 10.1093/hmg/6.7.991. [DOI] [PubMed] [Google Scholar]
- Grimes DA. Epidemiology of gestational trophoblastic disease. Am J Obset Gynecol. 1984;150:309–318. doi: 10.1016/s0002-9378(84)90370-3. [DOI] [PubMed] [Google Scholar]
- Kwok P-Y, Carlson C, Yager T, Ankener W, Nickerson DA. Comparative analysis of human DNA variations by fluorescence-based sequencing of PCR products. Genomics. 1994;23:138–144. doi: 10.1006/geno.1994.1469. [DOI] [PubMed] [Google Scholar]
- Kwok P-Y, Deng Q, Zakeri H, Nickerson DA. Increasing the information content of STS-based genome maps: Identifying polymorphisms in mapped STSs. Genomics. 1996;31:123–126. doi: 10.1006/geno.1996.0019. [DOI] [PubMed] [Google Scholar]
- Lawler SD, Fisher RA, Dent J. A prospective genetic study of complete and partial hydatidiform moles. Am J Obstet Gynecol. 1991;164:1270–1277. doi: 10.1016/0002-9378(91)90698-q. [DOI] [PubMed] [Google Scholar]
- Marshall E. “Playing Chicken” over gene markers. Science. 1997;278:2046–2048. doi: 10.1126/science.278.5346.2046. [DOI] [PubMed] [Google Scholar]
- ————— A second private genome project. Science. 1998;281:1121. doi: 10.1126/science.281.5380.1121a. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Bauer-Sardiña I, Zakeri H, Hillier L, Mutch DG, Kwok P-Y. The homozygous complete hydatidiform mole: A unique resource for genome studies. Genomics. 1997;46:307–310. doi: 10.1006/geno.1997.5042. [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Gu Z, Li Q, Hillier L, Kwok P-Y. Overlapping genomic sequences: A treasure trove of single nucleotide polymorphisms. Genome Res. 1998;8:748–754. doi: 10.1101/gr.8.7.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- Zakeri H, Amparo G, Chen S-M, Spurgeon S, Kwok P-Y. Peak height pattern in dichloro-rhodamine and energy transfer dye terminator sequencing. BioTechniques. 1998;35:406–414. doi: 10.2144/98253st01. [DOI] [PubMed] [Google Scholar]