

Abstract
Current methods of plasmid preparation do not allow for large capacity automated processing. We have developed an automated high-throughput system that prepares plasmid DNA for large-scale sequencing. This system is based on our previously reported filtration method. In this method, cell harvesting, alkaline lysis, and plasmid purification occur in a single 96-well microtiter plate from which sequence-ready DNA samples are collected. The plates are designed to allow all reagents to be injected from above the wells and the spent reagents to be aspirated from below. This design has enabled us to build a linear process plasmid preparation system consisting of an automated filter plate stacker and a 21-stage automated plasmid preparator. The 96-well plates used are outfitted with glass-filters that trap Escherichia coli before the plates are stacked in the automated stacker. The plates move from the stacker to each of the 21 stages of the preparator. At specific stages, various reagents or chemicals are injected into the wells from above. Finally, the plates are collected in the second stacker. The optimal throughput of the preparator is 40,000 samples in 17.5 hr. Here, we describe a pilot experiment preparing 15,360 templates in 160 specially designed 96-well glass-filter plates. The prepared plasmids were subjected to restriction digestion, DNA sequencing, and transcriptional sequencing.
Recent progress in genome technology has created abundant opportunities for development of new areas of research based on genome information. Improvements in sequencing reaction chemistries and detection systems has allowed for a new generation of large-scale sequencing, one requiring larger-scale template DNA preparation. Expanded automated systems for PCR address the requirement for large-scale preparation of template DNA. Direct cell PCR is an especially useful technology for preparing template DNAs. However, the accuracy and length bias of PCR present serious problems. The use of plasmid DNAs can overcome these problems because the amplification of plasmid DNA is based on Escherichia coli proliferation processes which achieve accurate DNA replication without any size bias. Various length inserts can be incorporated into plasmids which can then be prepared for large-scale cDNA sequencing.
Conventional plasmid preparation of sequencing template DNA requires many steps such as centrifugation, organic solvent extraction, and ethanol precipitation (Birnboim and Doly 1979; Sambrook et al. 1989). These steps present significant hurdles in the development of a large-scale plasmid preparation system. Recently, many commercial kits for plasmid preparation have become available. Most kits use a silica matrix or ion-exchange resin (Boom et al. 1990). These methods can eliminate purification steps such as organic solvent extraction. However, these conventional methods still require centrifugation to remove cell debris produced by alkaline lysis. The need for centrifugation makes automation of the procedure difficult. Several commercially available instruments based on the traditional preparation protocol possess a centrifuge and robot arm, allowing for some automation. However, the centrifugation steps limit throughput.
Recently, we developed a transcriptional sequencing technique based on the chain termination method and using T7 RNA polymerase (Sasaki et al. 1998a,b; Izawa et al. 1998). For this method, T7 RNA polymerase transcribes the DNA template from the T7 promoter and produces RNA chain termination products. Most plasmid preparation protocols cannot be used for transciptional sequencing, however, because they require RNase treatment to remove contaminating E. coli RNA. Even a trace of RNase in the final plasmid DNA sample would digest transcriptional sequencing products, thus destroying the sequencing pattern.
Peviously, we reported a new method for simple high-throughput plasmid preparation (Itoh et al. 1997). In this method, the template plasmids for sequencing were prepared in a single 96-well filter plate by use of a one-way flow system that eliminated centrifugation, extraction with organic solvents such as phenol, ethanol precipitation, and RNase treatment. Here, we describe an automated plasmid preparation system based on this one-way filtration method to reduce cost, time, and labor. High-throughput equipment can prepare 40,000 plasmid samples in 17.5 hr.
RESULTS
Plasmid Preparation Method
The plasmid preparation method described below is a modification of the filtration method we reported previously. This method is based on the principle of silica surface adsorption of DNA in the presence of guanidine-HCl. In addition, at the high concentration of guanidine-HCl used, insoluble proteins are dissolved. E. coli cells stored in a 384-well microtiter plate are inoculated into a 96-deep-well plate containing 700 μl of HT medium. After shaking at 37°C, 275 rpm, and a 30°C angle for 17 hr, each culture is transferred to the corresponding well of a filter plate. The cells are harvested by filtration at a 10-inch Hg vacuum pressure. In the preparator, 25 μl of solution 1 containing egg white lysozyme is added to each well of the filter plate and the samples left for 2.5 min. Then, 50 μl of solution 2 is added. After 2.5 min, 200 μl of neutralization solution containing guanidine-HCl is added and the reaction left for 2.5 min. At this neutralization step, Tris-HCl (pH 8.0) is added to adjust the pH to pH 8.8 after mixing. Under these conditions, DNA but into RNA, is adsorbed onto the silica surface. Then, the mixture is filtered at a 16-inch Hg vacuum pressure, and 600 μl of washing solution containing 50% ethanol and 0.2 m NaCl is added to remove the guanidine salts or other soluble contaminants. The solution is filtered and washing solution added again. After filtration, 300 μl of 80% ethanol is added and the mixture is filtered at a 20-inch Hg vacuum pressure to remove residual salts from the washing solution. The residual ethanol is vacuum dried at 60°C for 5 min. After the above treatment, 100 μl of 1 mm Tris-HCl (pH 8.5) is added by a Quadra 96 multi-channel dispenser (Tomtec, Canada), and the filter plate is put on top of a microtiter plate. Then, the two plates are centrifuged at 1000g, for 10 min to elute the DNA and transfer it into microtiter plate. To concentrate the eluted DNA, the solution is dried overnight at 60°C. The dried DNA is dissolved in an appropriate volume of distilled water and used for sequencing. These processes are summarized in Table 1.
Table 1.
The Process of Plasmid Preparation
| A. Cultivation | ||||
| Process | Person | Tools | Vacuum pressure (inch Hg) | Time |
|---|---|---|---|---|
| Medium dispensing | 1 | HT medium | 1.75 | |
| dispenser | min/plate | |||
| Inoculation | 1 | 384 microtiter plate | 3 min/plate | |
| 96 pins | ||||
| 70% ethanol | ||||
| Cultivation | 1 | shaker (275 rpm) | 17 hr | |
| (for setting) | platform (30° angle) | |||
| 37°C room | ||||
| Culture transfer | 2 | 96-channel dispenser | 0.5 min/plate | |
| Vacuum filtration | 4 | vacuum manifold | 10 | 3 min/plate |
| vacuum pump (160 Torr) |
| B. Plasmid preparation | |||||
| Process | Stage | Solution | Volume (μl) | Vacuum pressure (inch Hg) | Time (min) |
|---|---|---|---|---|---|
| Lysozyme treatment | 2 | 1 | 25 | 2.5 | |
| Alkaline lysis | 3 | 2 | 50 | 2.5 | |
| Neutralization | 4 | neutralization | 200 | 2.5 | |
| Filtration | 5, 6, 7, 8 | 16 | 10 | ||
| Washing | 9,10 | washing | 600 | 16 | 5 |
| 11, 12 | 600 | 16 | 5 | ||
| Salt washing | 13, 14 | 80% ethanol | 300 | 20 | 5 |
| Drying | 15, 16 | 20 | 15 | ||
| 17, 18 | |||||
| 19, 20 |
| C. Elution | |||
| Process | Person | Tools | Time |
|---|---|---|---|
| Eluant dispensing | 1 | 96-channel dispenser | 0.5 min/plate |
| Centrifugation | 2 | centrifuge (1000g, for microtiter plate) | 10 min/16 |
| microtiter plate | plates | ||
| Drying | 1 | 60°C drying oven | overnight |
Filter Plate
This system requires a newly designed 96-well plate. We developed this 96-well filter plate for plasmid preparation (Fig. 1) to facilitate injection of reagents and other chemicals from above the wells and extraction of spent reagents and chemicals from below. The volume of each well is 600 μl and there is a nozzle at the bottom for efficient suction extraction. The bottom of each well has a polyvinylidene difluoride membrane (PVDF) 0.45-μm pore size filter referred to as the bottom membrane, which retains reagents before the vacuum is applied. Two glass filters are placed on top of the bottom membrane to harvest the E. coli cells and capture the plasmid DNA.
Figure 1.

Microtiter glass-filter plate used for plasmid preparation. The volume of each well is 600 μl. At the bottom of each plate is a glass filter and hydrophilic PVDF membrane with a pore size of 0.45 μm. The glass filters are designed to trap and harvest E. coli cells and to adsorb the plasmid DNA in the presence of guanidine-HCl after the lysis. The PVDF membrane is installed to keep the solutions on the stages. When the samples are aspirated the membrane filters the samples to maintain adherence of the plasmid DNAs to the glass filter.
Plasmid Preparation System Structure
This system consists of two main parts: (1) an automated plate stacker that can hold 216 96-well filter plates; (2) an automated plasmid preparator consisting of 21 stages with six 96 multichannel dispensers for adding reagents.
The plate stacker is used to supply plates to the preparator, for storage, and for transport of the plates. As shown in Figure 2,A and B, the automated plate stackers for plate supply and collection are interchangeable. The stacker has 18 stages onto which a set of 12 plates can be stacked. The stacker can be connected to the plasmid preparator and supplies filter plates for the automated process. Sets of 12 plates are transferred from stage to stage by a pair of bars under the stages. During the transfer, these bars raise the plates up and move then to the next stage. After plasmid preparation, the plates are stacked and transferred to the collection stacker. The collected plates are removed from the stacker and transferred to a Quadra 96 multiple dispenser where elution buffer is added. Then, the plates are placed in a centrifuge where the DNA is removed from the glass-filters and transferred to microtiter plates.
Figure 2.

Structure of the automated plasmid preparation system. (A) Automated plate stacker for plate supply and collection. (B) Schematic representation of plate stacker. (C) Automated plasmid preparator. (D) Schematic representation of the stages of plasmid preparator. The supply stacker and collection stacker are connected to the plasmid preparator. These two stackers have fundamentally the same structure. In B, the numbered squares indicate stages. In the supply stacker, a set of 12 glass-filter plates is piled up on each stage. The 18 stages are designed on a single stacker. The stacked plates are sent from larger to smaller numbered stages and are supplied a set by set from stage 1 of the stacker to the plasmid preparator. For collection, the plates treated in the plasmid preparator are transferred to stage 18. After treatment of the 216 plates, the empty supply stacker can be detached from the plasmid preparator and a new stacker carrying the next 216 plates can be connected. After collection of the first 216 plates by the collection stacker, the empty collection stacker has to be reset. (D) Numbered squares indicate stages. As shown in C and D, the plasmid preparator receives a set of plates onto the plate sorter [referred to as the dividing apparatus (DA)]. Here, the lowest plate is sorted and sent to stage 1 by conveyor belts. At stages 2 and 3, respectively, solutions 1 (I) and 2 (II) are dispensed by cylinder-type dispensers (blue stages). The neutralization solution (N) is dispensed at stage 4 by a replaceable-tip cylinder-type dispenser (green stage). Between stages 5 and 20, the vacuum pads are lifted up and vacuum filtration is achieved. At stages 9 and 11, washing solution (W) is dispensed by solenoid dispensers as described in Results (yellow stages). At stage 13, 80% ethanol (E) is dispensed by a cylinder-type dispenser. Residual liquid in each well after vacuum filtration is aspirated from above each plate by use of an automatic 96-nozzle aspirator [stages 8, 10, 12, and 14 (red stages)]. Warm air (60°C) is supplied at stages 19 and 20 to dry each well. The plate carrying the plasmid DNA is stacked in the stacking apparatus (SA). Then, a set of plates is sent and collected by the collection stacker.
The automated plasmid preparator (Fig. 2C,D) consists of a plate sorter, 21 treatment stages, and a plate stacker. Filter plates are supplied and sorted by the plate sorter. In this apparatus, a lifter under the plates lifts up and a plate stopper opens. One plate passes through the stopper, the lifter descends, and, then, the stopper closes to support the rest of the plates. The sorted filter plate is transferred by conveyer belts positioned at both sides. When the plate is transferred to the next stage, the stopper lifts up and secures the plate on the stage. Ninety-six-channel liquid dispensers are located at the second, third, fourth, ninth, eleventh, and thirteenth stages. At the second, third, and thirteenth stages, there are 96-cylinder-type dispensers, in which the liquids are provided from a reservoir. The accuracy of the dispensing volume is ∼5%. At the fourth stage there is a 96-cylinder-type dispenser with replaceable tips. For this dispencer, the liquid is supplied by a reservoir tray. The accuracy of this dispenser is also ∼5%. At the ninth and eleventh stages there are solenoid dispensers composed of a solenoid and valves, which lead liquid from a reservoir to nozzles at fixed times. The accuracy of these dispensers is ∼10%. Vacuum extraction takes place from the fifth to the twentieth stage. When a plate reaches the vacuum stage, a sealed rubber tray lifter beneath the plate, which is connected to the vacuum reservoir, lifts up to aspirate liquid from the plate. Occasionally, some reagents remain after the vacuuming process. In such cases, the residual liquid is removed from above the plate by a 96-nozzle vacuum aspirator. These nozzles descend 30 sec before the completion of the treatment and lift up automatically at the end of the treatment. These aspirators are located on the eighth, tenth, twelfth, and fourteenth stages. On the first and twenty-first stages, bar code readers identify the plate number and record it on a personal computer. The filter plates move from stage to stage at 2.5-min intervals, which include reaction or treatment times and transport of plates.
This preparator has a treatment time-allocation management system, in which the 2.5-min interval is the minimal time among the processes of plasmid preparation and is thus designated as the unit time. Longer processes are assigned unit values. For example, the vacuum filtration that follows neutralization takes 10 min and, is therefore worth four units. Thus, the fifth through the eighth stages were used for vacuum filtration. In this time management system, the filter plates carrying the final 96 DNA samples could be recovered at the collection stacker every 2.5 min.
The system is controlled by a PLC, with the control software in C language. This PLC controls the intervals, dispensing timing, transfer of plates, lifting and descending of vacuumn pads, vacuum timing, sorting, and stacking. It also controls the automated plate stackers.
Plasmid Preparation
To test the plasmid preparation system, we used mouse cDNA libraries prepared by the cap-trapper method (Carninci et al. 1996, 1997). These libraries contain full-length cDNAs (Carninci et al. 1996). Mouse small intestine and stomach cDNA libraries were used as samples. As described in Methods and Materials, we purified ∼15,000 plasmids in 8 hr with this system. To evaluate the yield and purity of template DNA for large-scale sequencing we subjected prepared plasmid DNAs to agarose gel electrophoresis and sequencing.
Purity of Plasmid DNA
To measure the purity of DNA by optical density, test plasmid preparations were dried in 96-well microtiter plates and then dissolved in TE buffer. The ratio of the OD readings at 260 nm and 280 nm was 1.9–2.2, with 2.0 as an average. These data indicate that the prepared plasmids were highly purified and suggested that the plasmids could be used for restriction digestion or sequencing.
Agarose Gel Electrophoresis
The prepared plasmids were subjected to restriction enzyme digestion and agarose gel electrophoresis without any adjustment of the DNA concentration to evaluate the availability for enzyme reactions and the yields (Fig. 3). On average, the yield of plasmid DNA was ∼0.5 μg and, at maximum, 1.0 μg as calculated from the intensity on an agarose gel. The success rate as determined by the final product on an agarose gel was 76.8% (295/384) of the total wells.
Figure 3.

Agarose gel electrophoretic patterns of plasmid DNAs prepared by this system. cDNAs were cloned into λ-ZAPII and excised in E. coli SOLR cells. The plasmids were digested with PvuII, and subjected to electrophoresis. (Arrows) Locations of DNA size marker (λ -HindIII digest).
DNA Sequencing
To test whether or not the quality of the plasmid DNAs prepared by this system was appropriate for sequencing, some of the DNAs were subjected to dye-terminator sequencing with an ABI 377 DNA sequencer (Perkin-Elmer). To obtain a good yield, whole prepared plasmid DNA was used. A typical electropherogram is shown in Figure 4. The pattern showed low background and enough resolution for proper sequence analysis. The sequencing success rate was 71.9% (276/384) of total sequenced samples and 93.6% (276/295) of samples that should successful plasmid preparation.
Figure 4.
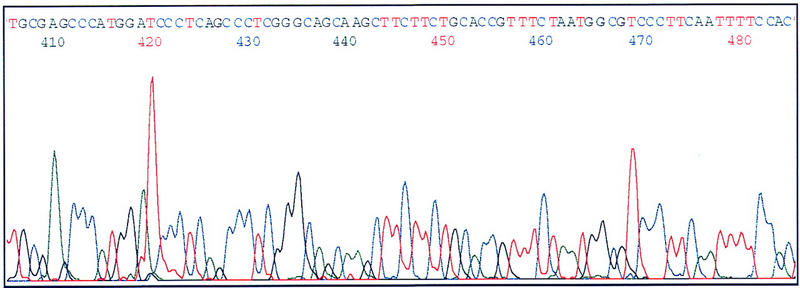
Electropherogram of cDNA-containing plasmids prepared by this system. The prepared plasmids were dissolved in TE buffer, precipitated with ethanol, and, then, sequenced. The dye-terminator sequencing kit (Perkin-Elmer) and an ABI 377 DNA sequencer were used.
Transcriptional Sequencing
As an additional step, we examined whether or not the prepared plasmids were useful for our previously described transcriptional sequencing method (Sasaki et al. 1998a,b; Izawa et al. 1998). Transcriptional sequencing is based on RNA transcription from a promoter with T7 RNA polymerase without the need for sequencing primers. Because our automated plasmid preparation system does not require RNase treatment, we expected we could use the prepared DNA for transcriptional sequencing.
As shown in Figure 5 , the transcriptional sequencing with the plasmid prepared by the system described in this paper was successful. Therefore, this system is an effective means for preparing templates.
Figure 5.
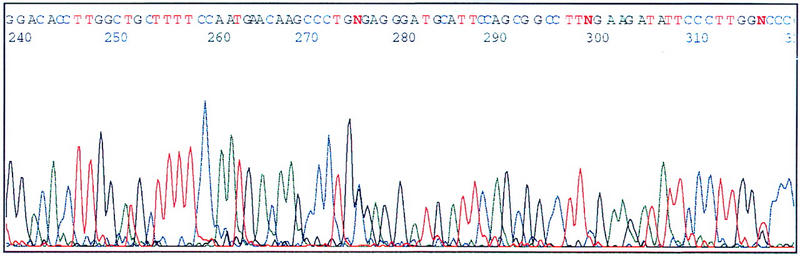
Electropherogram of insert cDNA-containing plasmids prepared by this system and sequenced by transcriptional sequencing. DNAs were dissolved in TE buffer, precipitated with ethanol, and, then, sequenced. Sequencing was as described previously (Sasaki et al. 1998a,b; Izawa et al. 1998). An ABI 377 DNA sequencer was used to anlayze the sequence.
DISCUSSION
In this report we describe an automated plasmid preparation system based on a plasmid preparation method reported previously (Itoh et al. 1997). For that method we eliminated the organic solvent extraction, centrifugation to separate the precipitate and supernatant, labor intensive transfer of the solutions from tube to tube, or RNase treatment. The elimination of these steps enabled us to automate all steps for plasmid preparation with a linear process of 21 steps using 96-well plates to transfer samples from stage to stage. To design the high-throughput system by the linear process, we used a treatment time-allocation management system. This management system is based on a set interval time to transfer the sample plates from stage to stage, which includes reaction or treatment time. The preparator was engineered such that each stage was established to allow one process to be completed per stage. Samples are transferred from stage to stage to achieve the linear process. However, it is crucial that the interval at each stage be exactly the same period of time for the linear process to function efficiently. Otherwise, complications would arise from samples backing up in the process. For this reason, the interval time is usually limited by the longest time required for any step in the whole procedure. In the case of this plasmid preparation, the needed time for dispensing and treatment of solutions 1, 2, and neutralization solution is 2.5 min, although the vacuum filtration spends 10 min and drying of the DNA-adsorbed glass-filters 15 min. In the linear process, each plate is followed by the next plate, so that the shorter process must be adjusted to the time of the longer process because plates cannot be transferred downstream before the longer process is finished. If the longest time was used for the interval time, in this case, there would be 11 processes and the interval time including the reaction time would be 15 min as dictated by the time needed for drying of the DNA-adsorbed glass-filters. Hence, the whole process for one plate would be 165 min. On the other hand, the treatment time-allocation management system employs the minimal reaction time as an interval period. The reaction steps requiring the longest time are divided into two or more stages. On the basis of this system, although the number of stages is larger than the number of steps, the entire time necessary to complete all steps can be decreased. In this case, 21 stages are essential and the interval time is 2.5 min. Therefore, the time for processing each plate is 52.5 min. In such a way, various treatments requiring different times can be achieved in the same linear process.
Our data demonstrate that the quality of prepared plasmid DNAs was high enough to be used for enzyme reactions such as restriction digestion or sequencing. In addition, the plasmid DNA is less contaminated with chromosomal DNA as compared with DNA prepared by the manual process described previously (Itoh et al. 1997). One reason is that insoluble precipitates after alkaline lysis and neutralization were stable even at the elution step.
For the evaluation of the yield and quality of the plasmid DNA, 384 samples were subjected to agarose gel electrophoresis without any adjustment of the DNA concentration or additional purification steps to eliminate the salt contaminants (Fig. 3). These data indicate that the final yield of plasmid (0.5–1.0 μg of 3–5 kb of plasmid DNA) was enough for one trial sequencing reaction. Some wells failed to obtain sufficient levels of plasmid DNA because of the absence of or poor growth of E. coli during the cultivation step, but not because of a failure in the plasmid preparation. About 76.8% of plasmids (295/384) were successfully prepared among the samples analyzed. The yield could be improved by the use of a mild washing condition before the elution step.
Of 384 samples, 276 (71.9%) were used successfully for sequencing. Of 384 samples, 295 were successful for preparation of DNA, and 93.6% of the prepared DNAs produced successful sequencing data. Subsequent experiments revealed that most of the unsuccessful samples could improved by further purification of the DNA by spin column. This step eliminated low-molecular-weight contaminants, such as guanidine-HCl or salt in the washing solution, which inhibits the sequencing reaction.
The average readable length of sequenced DNA after desalting by spin column was ∼450 bp even when a 36-cm gel apparatus and the dye-terminator reaction were used. If an energy-transfer primer or terminator were to be used, the signal strength and readable length would be improved. However, for cDNA analysis and classification of the terminal, sequences should be sufficient.
This system could be improved by changes in the cultivation and preparation conditions. The low success rate of plasmid preparation could be attributable to the cultivation step. For efficient cultivation, the volume of each well in the 96 format deep-well plate should be increased as the present volume does not allow good aeration. To refine the preparation, the adsorbed plasmid DNAs should be washed efficiently, and the culture volume should be increased. Currently, however, the area of the filters limits the size of the culture volume. To increase the culture volume, the 96-well microtiter plate format must be changed to expand the filtration area. Alternatively, we could use a 24-well format, which would provide a large enough filtration area. This approach, however, would increase the number of filter plates needed.
The transcriptional sequencing method developed in our laboratory is a powerful tool for large-scale sequencing. Previous and recent plasmid extraction methods and kits include RNase treatment. However, trace RNase in the prepared plasmid inhibits transcriptional sequencing by digesting the sequencing product. For this report, we tested the filtration method and the plasmid preparation system for generation of template for transcriptional sequencing (Fig. 5). The results indicated that the prepared plasmid could be used for transcriptional sequencing because this method does not require RNase treatment. Thus, for large-scale sequencing, this plasmid preparation system and transcriptional sequencing are effective tools.
We plan to develop an automated system for medium dispensing and E. coli inoculation, which will be installed upstream of the E. coli cultivation step. This new system will dispense medium in each well of 96 deep-well plates and transfer a clone into the corresponding well from a 384-storage plate automatically. We are also developing a system for automated transfer of cultivated broth and for filtration to harvest cultivated E. coli cells with the glass-filter plates.
METHODS
Reagents and Medium
Most reagents were purchased from Wako Pure Chemicals (Japan). Bacteria were cultured in HT medium [2.5% Bacto-tryptone (DIFCO), 0.25% Bacto-yeast extract (DIFCO), 0.75% NaCl]. For plasmid preparation, solution 1 contained 50 mm glucose, 25 mm Tris-HCl (pH 8.0), and 10 mm EDTA (pH 8.0). For lysozyme treatment, freshly prepared 25 mg/ml egg white lysozyme, (Seikagaku Kogyo, Japan), was used. Solution 2 contained 0.4 n NaCl and 1% SDS. The neutralization solution [1 m Tris-HCl (pH 8.0), 7 m guanidine-HCl] was used for alkaline lysis, neutralization, and DNA adsorption to the glass filters. Solution 1 and the neutralization solution were filtered through a hydrophilic PVDF membrane, with a 0.22-μm pore size (Millipore). The washing solution (100 mm Tris-HCl (pH 8.8), 0.2 m NaCl, 50% ethanol), and 80% ethanol were used to remove contaminants after alkaline lysis and adsorption.
cDNA Library Construction
The cDNA libraries used in this paper were prepared by the cap-trapper method (Carninci et al. 1996, 1997). The cDNAs were inserted into the λ-ZAPII vector, transferred to E. coli SOLR, and excised in vivo. The resultant colonies were picked by Q-bot (Genetix, Christchurch, UK), and inoculated into wells of a 384-well microtiter plates containing 40 μl of LB medium with 7% glycerol. The plates were incubated at 30°C overnight, then stored at −80°C.
Cultivation of E. coli Cells
For cultivation of E. coli SOLR cells carrying cloned cDNA plasmids, stored culture liquids in 384-well microtiter plates were thawed at room temperature and inoculated with 96 pins into deep-well plates with 0.7 ml of HT medium in each well. The inoculated plates were shaken at 37°C, 275 rpm and a 30° angle for 17 hr.
Pretreatment of Glass-filter Plates
After cultivation, the culture in each well was transferred into the corresponding well of a filter plate with a Quadra 96 multichannel pipetter (Tomtec, Canada). Then, the liquid was filtered by use of a vacuum manifold (Millipore) and adaptor for the filter plate.
Treatment
In this preparation system, lysozyme treatment, alkaline lysis, neutralization, wash, and drying were completed. From the second through the fourth stages, 25 μl of solution 1, 50 μl of solution 2, and 200 μl of neutralization solution were added, respectively. At the ninth and eleventh stages, 600 μl of washing solution was added. At the thirteenth stage, 300 μl of 80% ethanol was added. Between the fifth and twentieth stages, the filter plates were subjected to vacuum suction via bottom nozzles fitted with vacuum pads. The filter plates were transferred from stage to stage at 2.5-min intervals.
Elution
To elute the plasmid DNA, 100 μl of 1 mm Tris-HCl (pH 8.5) was added to each well of the treated filter plates. Then, the plates were centrifuged at 1000g for 10 min. The eluted plasmid DNA solutions were dried overnight at 60 °C.
DNA Sequencing
For sequencing, dye-terminator sequencing (Perkin-Elmer) was used. Dried plasmids in 96-well microtiter plates were dissolved in 10 μl of distilled water and applied to a spin column for desalting. Ten microliters of the sequencing reaction mixture was added to the desalted plasmid solutions, which were then subjected to thermal cycle sequencing. After the reaction, each mixture was applied to Sephadex G-50 (Amersham Pharmacia Biotech) gel-filtration matrix in a 96-well filter plate (Whatman) to remove the residual free dye terminator. Sequences were analyzed by use of an ABI 377 DNA sequencer.
Transcriptional Sequencing
The dried DNAs were dissolved in 10 μl of distilled water and then applied to a spin column for desalting. Transcriptional sequencing was performed by a previously reported method (Izawa et al. 1998; Sasaki et al. 1998a,b). The residual free dye terminator was removed as described above. Sequences were also analyzed by use of an ABI 377 DNA sequencer.
Acknowledgments
We thank Lifetech Co. (Japan), for production of the automated plasmid preparation system, and Fuji Technical Industry Co. (Japan), for production of the glass-filter plates. We also thank Hiromi Takano, Saiko Akahira, Ayako Hara, Noriko Kikuchi, and Mari Itoh for their assistance preparing the data for this paper. We thank all members of the Genome Science Laboratory, Tsukuba Life Science Center, and the Genome Exploration Research Group of the GSC. This study was supported by a research grant for the Genome Exploration Research Project, special coordination funds to Y.H. (RIKEN) and CREST Program funding to Y.H. (JST) from the Science and Technology Agency of the Japanese Government. Y.H is also supplemented by a grant-in-aid for Scientific Research on Priority Areas and the Human Genome Program from The Ministry of Education and Culture, a grant for Research on Aging and Health, and a grant-in-aid for a Second Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health and Welfare of the Japanese Government.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL yosihide@rtc.riken.go.jp; FAX 81 298 36 9098.
REFERENCES
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics. 1996;37:327–336. doi: 10.1006/geno.1996.0567. [DOI] [PubMed] [Google Scholar]
- Carninci P, Westover A, Nishiyama Y, Ohsumi T, Itoh M, Nagaoka S, Sasaki N, Okazaki Y, Muramatsu M, Schneider C, Hayashizaki Y. High efficiency selection of fulllength cDNA by improved biotinylated cap trapper. DNA Res. 1997;28:61–66. doi: 10.1093/dnares/4.1.61. [DOI] [PubMed] [Google Scholar]
- Itoh M, Carninci P, Nagaoka S, Sasaki N, Okazaki Y, Ohsumi T, Muramatsu M, Hayashizaki Y. Simple and rapid preparation of plasmid template by a filtration method using microtiter filter plates. Nucleic Acids Res. 1997;25:1315–1316. doi: 10.1093/nar/25.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa M, Sasaki N, Watahiki M, Ohara E, Yoneda Y, Muramatsu M, Okazaki Y, Hayashizaki Y. Recognition sites of 3′-OH group by T7 RNA polymerase and its application to transcriptional sequencing. J Biol Chem. 1998;273:14242–14246. doi: 10.1074/jbc.273.23.14242. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Izawa M, Watahiki M, Ozawa K, Tanaka T, Yoneda Y, Matsuura S, Carninci P, Muramatsu M, Okazaki Y, Hayashizaki Y. Transcriptional sequencing: A method for DNA sequencing using RNA polymerase. Proc Natl Acad Sci. 1998a;95:3455–3460. doi: 10.1073/pnas.95.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Izawa M, Sugahara Y, Tanaka T, Watahiki M, Ohara E, Funaki H, Yoneda Y, Ozawa K, Matsuura S, Muramatsu M, Okazaki Y, Hayashizaki Y. Identification of stable RNA hairpins causing band compression in transcriptional sequencing and their elimination by use of inosine triphosphate. Gene. 1998b;222:17–23. doi: 10.1016/s0378-1119(98)00447-8. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]