

Abstract
Homeobox genes encode transcription factors that dictate developmental identity, including that of the Mullerian tract. These genes also direct differential Mullerian transformation of the ovarian cancer cells. The homeobox gene HOXA10 controls uterine organogenesis during embryonic development and similarly is expressed in endometroid epithelial ovarian cancer. Here we confirmed aberrant regulation of HOXA10 expression in epithelial uterine and ovarian carcinomas. We identified a HOXA10 epithelial regulatory element containing an enhancer that drove HOXA10 expression specifically in gynaecologic epithelium. We further identified an adjoining dominant repressor element that restricted regulation by the epithelial enhancer to a subset of epithelial cell types. The repressor contained two functional WT1 binding sites. We identified a strong inverse correlation between HOXA10 expression and that of the Wilms’ Tumour 1 (WT1) gene in multiple benign and malignant gynaecologic tissues, suggesting functionality of the WT1 sites in the repressor. Mutation of the two WT1 binding sites abolished WT1 binding to the element as well as the ability to affect epithelial enhancer activity in reporter assays. Similarly, decreased expression of WT1 using siRNA prevented repressor activity. The Mullerian phenotype seen in ovarian cancer is dependent on gain of HOX gene expression secondary to the loss of WT1-mediated HOX repression. This suggests that Gynaecologic epithelial histologic type is regulated by WT1 expression through its selective repression of HOX genes.
Keywords: WT1, HOXA10, uterine cancer, ovarian cancer
Introduction
Human gynaecologic tumours are common neoplasias that typically arise from the epithelium of the uterus, cervix and ovary. Typically uterine tumours are adenocarcinomas that histologically resemble uterine endometrium. Although the ovary is not derived from the Mullerian system during embryogenesis, tumours of the ovary typically demonstrate Mullerian features. Serous ovarian tumours histologically resemble the fallopian tube, mucinous tumours resemble the cervix and endometriod ovarian tumours resemble the endometrium. Interestingly, the same genes that assign developmental identity to the embryonic Mullerian duct are expressed in ovarian tumours in a fashion that correlates with their histologic identity [1, 2]. Homeobox genes are highly evolutionary conserved regulators of embryonic morphogenesis and differentiation [3–5]. HOX genes assign developmental identity to many undifferentiated axis during embryogenesis, including the Mullerian tract [6–11]. HOXA9 is normally expressed in the developing fallopian tube and is also expressed in serous tumours of the ovary; similarly, HOXA10 is expressed in the embryonic uterus and in endometriod ovarian tumours, and HOXA11 is expressed in the developing cervix and in mucinous ovarian tumours. Homeobox genes are closely associated with histologic type in both normal gynaecologic tissues and malignancies [1, 2, 12].
Here we investigated the mechanism of altered HOXA10 expression in epithelial endometrial and ovarian tumours. We demonstrate a negative correlation between WT1 expression and HOXA10 expression in several gynaecologic tumours. We demonstrate direct repression of HOXA10 by WT1 acting through two WT1 binding sites in the HOXA10 promoter. We suggest that WT1 directly regulates HOX gene expression in normal and neoplastic gynaecologic tissues and that this expression determines tissue identity.
Materials and methods
Cell culture
The human endometrial adenocarcinoma cell line (Ishikawa), human embryonal kidney cell (293T cell lines) and human endometrial stromal cells (HESC) were used. These cells were used as a model of a Mullerian epithelium, kidney and Mullerian stroma, respectively. Ishikawa cells were maintained in Minimum Essential Medium (MEM) (Invitrogen, Carlsbad, CA, USA), supplemented with 10% foetal bovine serum, 1% penicillin-streptomycin-amphotericin B. HESC and 293 T cell lines were cultured in a Dulbecco's Modified Eagle Medium with high glucose content (Invitrogen), supplemented with 10% foetal bovine serum and 1% penicillin-streptomycin-amphotericin B.
Western blot
Nuclear protein was extracted from 293 T, Ishikawa and HESC using Nuclear Extract Kit (Active Motif, Carlsbad CA, USA) according to manufacturer’s protocol. Twenty-five μg of protein was electrophoresed through 4–15% polyacryl-amide gels (Bio-Rad, Hercules, CA, USA) at 150 V for 60 min. and transferred onto Immun-Blot polyvindylidene difluoride membranes (Bio-Rad) in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) at 100 V for 1 hr. After incubation in blocking buffer (1 × PBS, 0.2% Tween 20, 5% milk), the blot was incubated individually with WT1 (C19) rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) dilution 1:1000 and goat polyclonal actin antibody (Santa Cruz Biotechnology) dilution 1:1000 overnight at 4°C. After washing, the membranes were incubated for 1 hr with anti-goat IgG or anti-rabbit IgG secondary antibody, respectively (Vector Laboratories, Burlingame, CA, USA) diluted in the blocking buffer.
Construction of plasmid for promoter analysis and in vitro mutagenesis
Two kb of the HOXA10 promoter was amplified by PCR and cloned into pGL3-basic vector (Promega, Madison, WI, USA). The fragment was generated by PCR from human genomic DNA (Promega) using HOXA10 5′ regulatory region-specific primers. The primers were designed using the GenBank database (NT_079592). PCR was performed as follows: 95°C for 60 sec.; 60°C for 60 sec.; 72°C for 90 sec., 35 cycles.
Transcription factor binding sites were identified using Genomatix database (Genomatix, München, Germany).
The QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) was used for in vitro mutagenesis. Mutation design was performed in this manner neither to create nor delete other known transcription factor bindings. The HOXA10 promoter was mutated using a two 17-oligomer oligonucleotides: the first −98/−82 bp of HOXA10 promoter, 5′-CGGTGCGGGGGGATTGC-3′ and the second −316/−300 bp of HOXA10 promoter, 5′-CCAGGCCCCCCACCAGC-3′. The mutagenesis reaction was carried out according to the manufacturer’s protocol and the mutation was confirmed by sequencing.
Transfection and luciferase assays
293T, Ishikawa and HESC were grown to 50–60% confluence in 24-well plates. 293T and Ishikawa cell lines were transfected with the 0.8 μg per well of appropriate plasmid using Lipofectamine 2000 Reagent (Invitrogen). After 6 hours, the media were changed and the cultures allowed growing for an additional 20–24 hrs. We used TransIT-LT1 (Mirus, Madison, WI, USA) for HESC transfection and 0.5 μg per well of the appropriate plasmid according to transfection protocol. HESC were harvested 24–30 hrs after transfection. All cells were cotransfected with 20 ng per well of pRL-TK to control transfection efficiency. The cells were rinsed with cold PBS and lysed with Reporter Lysis Buffer (Promega). The lysate was collected after two freeze/thaw cycles. Luciferase activity was measured using the Luciferase Reagent Kit (Promega) and luminometer. A Renilla luciferase activity was used for normalization. All transfections were performed in triplicate and experiments were repeated three to four times.
Transfection of siRNA
293T cells were seeded onto 24 well plates and grew to 40–50% confluence. Transfections were as detailed in the Lifopectamine 2000 for siRNA transfection protocol (Invitrogen), using 120 pmol per well of WT1 siRNA (Santa Cruz Biotechnology) or 120 pmol per well of control siRNA (Santa Cruz Biotechnology) and 0.8 μg per well of appropriate plasmid. Cells were harvested 50 hrs after transfection and rinsed with cold PBS and lysed with Reporter Lysis Buffer. The lysate was used for both Western blotting and Luciferase assay as described above.
EMSA
The double stranded oligonucleotides were used for electrophoritic mobility shift assay (EMSA). We created two probes corresponding to two putative WT1 binding sites and the flanking sequences (probe 1 and probe 2), and two probes with WT1 mutated binding sites and the flanking sequences as well (probe M1 and M2). Mutated binding sites are designated in boldface. The sequences were as follows:
Probe 1: 5′- cccgccgcggtgcggggggattgctaatcg-3′ (−105/−76 bp)
Probe M1: 5′- cccgccgcggtgcagggggaatcctaatcg -3′ (−105/−76 bp)
Probe 2: 5′-aatggccaggccccccaccagccacgttgg -3′ (−321/−292 bp)
Probe M2: 5′- aatggccaggcacaccaccagccacgttgg -3′ (−321/−292 bp)
Complementary oligonucleotides were annealed and end labelled with 32P-dATP (PerkinElmer Life Sciences, Waltham, MA, USA) using T4 polynucleotide kinase (New England BioLabs, Ipswich, MA, USA) and purified with MicroSpin G-25 columns (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Nuclear extract was obtained from 293T cell lines using Nuclear Extract Kit (Active Motif) according to the manufacturer’s protocol. Binding reactions consisted of 30 μl mixture of 10 μg nuclear extract and 80,000 cpm 32P-labelled oligonucleotides, which were incubated for 1 hr at 4°C. The resultant protein-DNA complexes were separated on a 5% polyacrylamide gel (acrylamide/bisacrylamide, 29:1 for 3 hrs at 180 V in 0.5 × TBE buffer (1 × TBE is 50 mM Tris, 50 mM boric acid, and 1 mM EDTA) at 4°C. To confirm the identity of WT1 in the shifted complex, 10 μg nuclear extract protein were incubated with 2 μg WT1 (180) rabbit polyclonal antibody (Santa Cruz Biotechnology) or 2 μg rabbit IgG at room temperature for 1 hr, followed by a 1-hr incubation with labelled oligonucleotides at 4°C. The gel was dried under vacuum at 80°C, exposed on X-OMAT film and subsequently developed.
Immunohistochemistry
Tumour samples from 15 patients with endometrial epithelial cancer and 15 patients with ovarian epithelial cancer were collected for immunohistochemical evaluation. Normal endometrial and ovarian tissues from 23 pre- and post-menopausal women who underwent operative treatment due to pelvic organ prolapse were sampled as a control. We also collected as control endometrial samples from 10 normally cycling women with no history of gynaecologic diseases. Normal kidney tissue was used as positive control for WT1 expression. Haematoxylin and eosin slides were reviewed in each case and histologic diagnosis was confirmed using accepted criteria. Immunohistochemical staining was performed on formalin-fixed paraffin sections using streptavidin-biotin-peroxidase complex method. The primary antibodies used in this study were WT1 (C19) rabbit polyclonal antibody (Santa Cruz Biotechnology) and HOXA10 (sc-17159) goat polyclonal antibody (Santa Cruz Biotechnology). WT1 and HOXA10 antibody were used at a dilution 1:500 and 1:2500, respectively. Samples were deparaffinized, rehydrated and boiled in 0.01 M citrate buffer (ph 6.0). After endogenous peroxidase activity had been blocked, the sections were incubated with normal goat or normal horse serum to reduce non-specific binding. They were then incubated with a primary antibody or control non-immunized animal serum at 4°C overnight. After washing, the sections were incubated with biotinylated goat anti-rabbit or horse anti-goat IgG, then with peroxidase-conjugated streptavidin. They were stained with diaminobenzidine and counterstained with haematoxylin.
Statistical analysis
The Student’s t-test was used for statistical evaluation.
Results
HOXA10 expression in normal endometrium, kidney and in epithelial uterine and ovarian tumours
To evaluate HOXA10 expression in above-mentioned tissues, we performed immunohistochemistry. As previous reported, HOXA10 expression in normal endometrium undergoes characteristic changes through the menstrual cycle [13, 14]. Glandular expression was low in the proliferative phase (Fig. 1A) and increased in the secretory phase (Fig. 1B). However, there was no HOXA10 expression in endometrium obtained from post-menopausal subjects nor in normal kidney (Fig. 1C and D, respectively). Further immunostaining of endometrioid uterine tumours revealed HOXA10 expression in low grade (G1) tumours (Fig. 1E). In contrast, G3 (high grade) endometrioid tumours did not express HOXA10 (Fig. 1F). In mucinous ovarian carcinomas, we observed HOXA10 expression, but not in serous adenocarcinoma (Fig. 1G and H, respectively). These results confirm previous reports of HOXA10 expression in benign endometrium, in well differentiated endometrial carcinomas and in those ovarian tumours which histologically resemble tissues that normally expresses HOXA10 [11].
Figure 1.
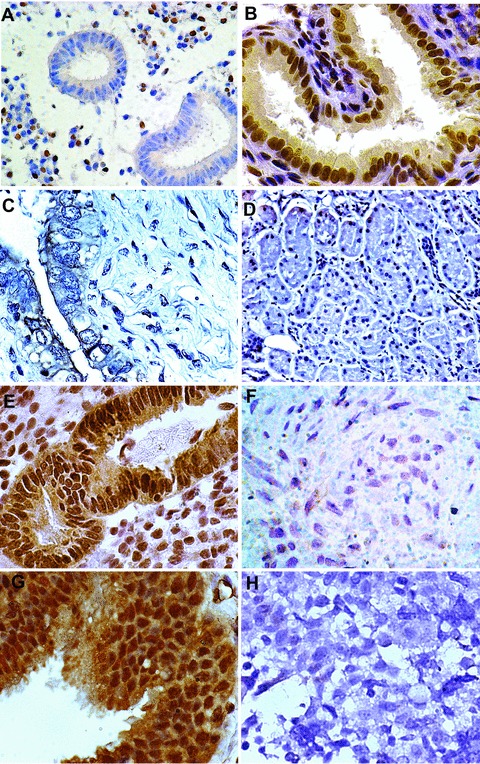
Nuclear HOXA10 expression in normal genitourinary tract tissues and in endometriod and ovarian carcinomas. (A) Proliferative endometrium; (B) secretory endometrium; (C) post-menopausal atrophic endometrium; (D) kidney; (E) G1 endometrioid carcinoma; (F) G3 endometrioid carcinoma; (G) mucinous ovarian carcinoma; (H) serous ovarian carcinoma. Photographs taken at 400–600 × magnification.
Defining an epithelial Müllerian HOXA10 enhancer
Despite the known function of HOXA10 as transcription factors, few cis- or trans-regulators of HOX gene expression in the reproductive tract are known. Based on our correlative immunohistochemical data, we analysed the promoter region of HOXA10 for a potential enhancer that drove the expression of HOXA10 in epithelial cells, including endometrioid and ovarian carcinomas. We utilized transient transfection and luciferase assays using pGL3 basic to search for potential epithelial enhancer elements. Ishikawa, 293 T and HESC lines were used for the transfection and luciferase assay [15]. The cells were also transfected with a Renilla luciferase expression construct as a control for transfection efficiency; Luciferase activity was normalized to Renilla activity.
A series of nested deletions identified a potential epithelial enhancer element, which drove expression of HOXA10 in gynaecologic epithelium. This enhancer element occupied the −965/−563 bp region of the HOXA10 promoter. In Ishikawa cells, normalized luciferase expression was increased five-fold when driven by the enhancer element relative to pGL3 (P < 0.0001). (Fig. 2A). The enhancer element also drove reporter gene expression in 293 T cells. Normalized luciferase activity driven by the enhancer element was increased four-fold (P < 0.0001). (Fig. 2A). There was no difference between reporter activity driven by pGL3 basic and the epithelial enhancer in HESC, a non-epithelial uterine cell line (Fig. 2A). We identified a HOXA10 enhancer that drove constitutive expression in epithelial cells.
Figure 2.
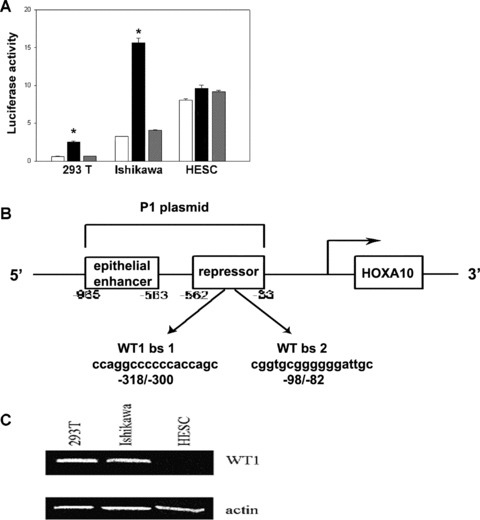
(A) Normalized luciferase/renilla ratio after pGL3 basic, enhancer element and P1 plasmid transfection of 293T, Ishikawa and HESC lines. The unshaded bars represent luciferase activity after transfection with the pGL3 basic vector as a control. The black bars demonstrate significantly increased expression driven by the enhancer element. The grey bars represent luciferase activity driven by the P1 element. (B) The human HOXA10 5′ regulatory elements were amplified by PCR and cloned into pGL3 basic vector. The HOXA10 epithelial enhancer is closely linked to a WT1 inducible repressor. Binding site 1 (bs 1, −98/−82): 5′-CGGTGCGGGGGGATTGC-3′ Binding site 2 (bs 2, −316/−300): 5′-CCAGGCCCCCCACCAGC-3′. (C) WT1 protein expression in 293T, Ishikawa and HESC lines. Western blot was performed using rabbit polyclonal WT1 antibody (C19). Actin was used as control for loading.
Inclusion of larger regions of the HOXA10 promotor resulted in loss of epithelial enhancer activity due to the presence of a putative repressor site or sites. A plasmid consisting of the larger −965/−33 bp region of the HOXA10 promoter, designated P1, containing both the epithelial enhancer as well as the putative repressor region, prevented and repressed enhancer element-driven gene expression in 293T cell lines (Fig. 2A). In 293T cells, inclusion of this repressor region diminished expression to one-third of that obtained using the epithelial enhancer construct (P < 0.0001). Similarly P1 repressed enhancer element-driven expression in Ishikawa cells by approximately 75% (P < 0.0001). There was no difference between lucifease expression driven by P1 and that driven by the enhancer element in HESC (where the epithelial enhancer was inactive alone) (P > 0.05) (Fig. 2A). These data identify a repressor region, located between −562 and −33 bp of HOXA10 promoter. The repressor was activated in Ishikawa and 293 T cell line, but not in the HESC line. Although the enhancer element resulted in promiscuous expression in epithelial cells, the repressor element restricted this expression to the cell types that normally expresses HOXA10 in vivo. Taken together the results suggested that restriction of HOXA10 expression to a subset of gynaecologic epithelial malignancies is regulated by a repressor that binds this element.
Identification of a HOXA10 repressor
To identify the putative repressor of HOXA10 that binds the −562/−33 region of HOXA10 promoter, we performed sequence analysis. Using MatInspector browser (Genomatix), we identified two putative binding sites for Wilms’ tumour suppressor (WT1) in the repressor region located in −98/−82 (bs 1) and −316/−300 (bs 2) of HOXA 10 promoter (Fig. 2B).
One putative binding site (BS1) had a core similarity 95.3% and matrix similarity 95.4%. The second binding site (BS2) possessed similar characteristics; the core similarity of BS2 was 100% and matrix similarity 93.7%.
To evaluate WT1 expression, we performed Western blotting. WT1 protein was expressed in both the Ishikawa and 293 T cell lines, but not in HESC (Fig. 2C). WT1 expression correlated with the cell lines that showed both HOXA10 expression and repression of P1-driven reporter gene activity and suggested the involvement of WT1 in HOXA10 repression.
WT1 expression profile in normal endometrium, kidney and in epithelial uterine, ovarian tumours
We evaluated WT1 expression in normal endometrium, kidney and in gynaecologic malignancies. In proliferative endometrium, WT1 was expressed in epithelial cells (Fig. 3A). In normal secretory endometrium, there was no glandular WT1 staining (Fig. 3B). In normal endometrium from post-menopausal subjects and in normal kidney WT1 immunoreactivity was prominent (Fig. 3C and D). In G1 endometrioid carcinoma, there was no WT1 staining, in contrast to G3 endometrioid carcinomas, where there was abundant WT1 expression (Fig. 3E and F). In serous adenocarcinoma, WT1 was also expressed, but not in mucinous adenocarcinomas (Fig 3G and H). These immunohistochemical data inversely correlated with our previous HOXA10 immunostaining results. There was a clear inverse relationship between HOXA10 and WT1 expression in epithelial tissues, suggesting a role for the putative WT1 binding sites identified above in HOXA10 repression.
Figure 3.
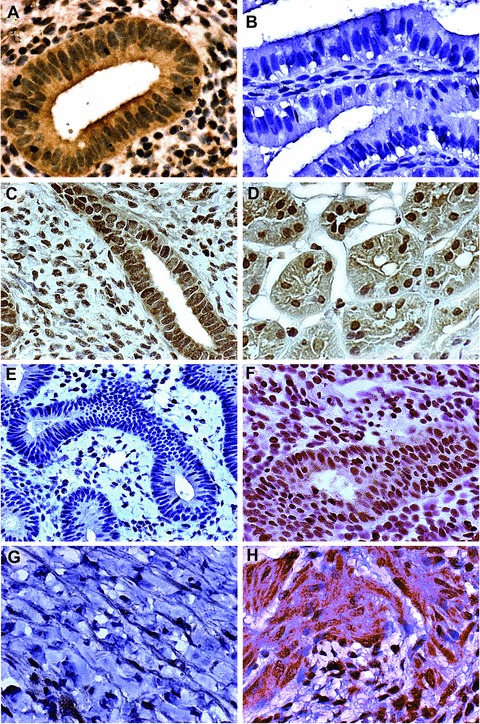
Nuclear WT1 expression in normal genitourinary tract tissues and in endometriod and ovarian carcinomas. (A) Proliferative endometrium; (B) secretory endometrium; (C) post-menopausal atrophic endometrium; (D) kidney; (E) G1 endometrioid carcinoma; (F) G3 endometrioid carcinoma; (G) mucinous ovarian carcinoma; (H) serous ovarian carcinoma. Photographs taken at 400–600 × magnification.
WT1 binds and represses the HOXA10 promoter
To determine whether WT1 binds the putative sites identified above, we performed EMSA. We created four 30 bp probes. Two probes contained each of putative WT1 binding sites (probe 1 and probe 2); in addition to two probes, contained a mutated version of each of the WT1 binding sites (probe 1M and probe 2M). Mutation design was performed using GEMS Launcher (Genomatix) in a manner that neither created nor deleted other known transcription factor binding sites. In binding site 1, we created three-point mutations, G-92A, T-85A, G-83C. In binding site 2, two point mutations were created, C-308A, C-310A. Mutations were confirmed by sequencing.
Probes 1 and 2, each containing one of the native WT1 putative binding sites, bound to nuclear extract (N) and cause shifted complex as shown in Fig. 4. To confirm the specificity of binding, we added 50-fold molar excess of unlabelled probe, which resulted in decreased binding (Fig. 4 lanes labelled ‘C’). To determine the presence of WT1 in protein-DNA complexes, we added 2 μg WT1 (180) rabbit polyclonal antibody and used 2 μg rabbit IgG as a control. A supershifted complex was absent when using the control rabbit IgG, however, was observed after adding WT1 antibody (Fig. 4: Ig indicates rabbit IgG control; W indicates anti-WT1). The mutated probes 1M and 2M lost the ability to bind to nuclear extract and failed to result in a shifted complex in the presence of nuclear extract (Fig. 4). These findings confirm the binding of WT1 to sites identified −98/−82 bp and −316/−300 bp upstream of HOXA10 promoter.
Figure 4.
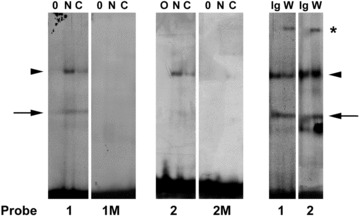
Radiolabelled Probe 1 (WT1 binding site 1) and probe 2 (WT1 binding sites 2) were used to demonstrate WT1 binding to the HOXA10 regulatory element using EMSA. 0 represents probe alone, N indicates addition of nuclear extract, whereas C represents addition of 50-fold molar excess unlabelled (cold) competitor to the probe and nuclear extract. Both probes 1 and 2 bound a protein from the WT1 containing nuclear extract. Probe 1M (mutated WT1 binding site 1) and probe 2M (mutated WT1 binding site 2) each failed to bind to any component of the nuclear extract. Probe 1 (WT1 binding site 1) and probe 2 (WT1 binding site 2) each bind WT1. Rabbit IgG was used as a control (Ig). We demonstrated a supershifted complex (*) with the addition of anti-WT1 IgG. Arrow indicates non-specific shift. Arrowhead indicated WT1 binding. * indicates supershifted complex in the presence of anti-WT1 antibody.
In order to test the effect of loss of WT1 binding on expression driven by this element, we evaluated the mutated constructs in luciferase reporter assays. We termed the mutated construct P2 which was identical to P1 plasmid except for mutation of both WT1 binding sites. Transfection results demonstrated that the mutated plasmid P2 drove luciferase activity to a level more than double that driven by P1 in the 293 T cell line (P < 0.0001) (Fig. 5A). In Ishikawa cells, the loss of repression after mutation of the WT1 binding sites was less pronounced than in 293 T cells; however, the same pattern was observed (P < 0.0001). Loss of WT1 binding resulted increased reporter activity.
Figure 5.
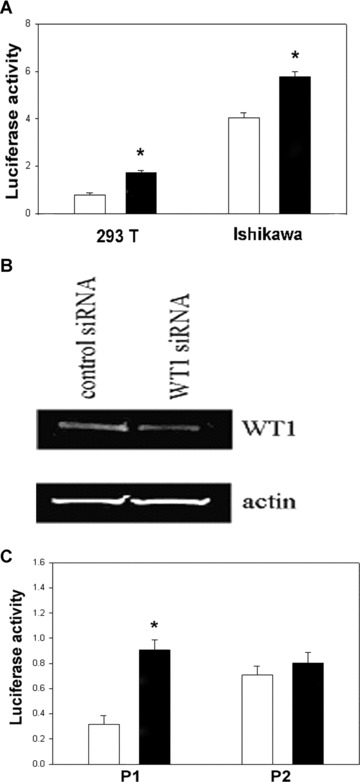
(A) Normalized luciferase/renilla ratio after P1 and mutated (M1) plasmid transfection of 293T and Ishikawa cell lines. The unshaded bars represent luciferase activity after transfection with the P1 element. The black bars demonstrate significantly increased expression driven by the mutated (M1) enhancer element. (B) WT1 protein expression in 293T cell lines after WT1 and control siRNA transfection. (C) Normalized luciferase/renilla ratio after co-transfection of P1 and P2 plasmid with WT1 and control siRNA in 293T cell lines. The unshaded bars represent luciferase activity after transfection with the control siRNA. The black bars demonstrate significantly increased activity after cotransfection with WT1 siRNA.
To further determine the functional activity of WT1 on the HOXA10 repression, we co-transfected cells with either the P1 or P2 plasmid and with either control or WT1 siRNA. WT1 SiRNA treatment resulted in a profound decrease of WT1 expression in the 293 T cell line (Fig. 5B). Transient transfection, using the P1 reporter construct containing both the epithelial enhancer and the native WT1 repressor, resulted in increased normalized luciferase activity when using WT1 siRNA compared to control siRNA (P < 0.0001) (Fig. 5C). As P1 comprises −965/−33 bp of HOXA10 promoter containing both WT1 binding sites and the epithelial enhancer, the data demonstrating the functional regulation of this element by WT1. The P2 plasmid, with both WT1 binding sites mutated, did not demonstrate differential luciferase activity after WT1 siRNA transfection (P > 0.05). Taken together, these data demonstrate that both WT1 and the WT1 binding sites are required for repression of the HOXA10 promoter.
Discussion
HOX genes encode regulatory proteins containing a highly conserved 61-amino acid motif, the homeodomain, which enables HOX proteins to bind to DNA specifically and transcriptionally activate or repress their target genes [16, 17]. HOX genes control cell growth and differentiation during embryonic development. Aberrant regulation of many homeobox genes has been demonstrated in a wide variety of tumours including haematologic, gynaecologic and breast malignancies [12, 18–20]. This abnormal regulation leads to altered cell differentiation.
HOXA10 is one of the Abdominal-B-like HOX genes. In addition to regulating the developing uterus, HOXA10 expression is important for adult endometrial development and suggests a dynamic role of HOXA10 in differentiation of adult tissues as well. In endometrial carcinoma, loss of HOXA10 expression in poorly differentiated endometrial carcinomas strongly and inversely correlates with increasing tumour grade [12]. Tumour dissemination in nude mice and invasive behaviour of endometrial carcinoma cells in vitro can be inhibited by enforced expression of HOXA10. These data suggest a role for HOXA10 in regulating tumour differentiation. Here we identified the molecular pathway by which HOXA10 is regulated in these tumours by WT1.
The Wilms’ tumour suppressor gene was the second tumour suppressor gene cloned after the retinoblastoma gene [21]. Approximately 10–20% of all Wilms’ tumours carry mutations in WT1 [21–24]. The WT1 gene is approximately 50 kb and encodes multiple 52–56 kD protein isoforms generated via alternative splicing, RNA editing and non-AUG translational initiation. The proteins have a proline and glutamine-rich N-terminal domain and a DNA/RNA-binding C-terminal domain containing four zinc finger motifs that are similar to those found in the early growth response family of transcription factors [21]. Depending on the tumour type, WT1 can either function as tumour suppressor protein or as survival factor, preventing differentiation and promoting neoplastic progression [22]. Alterations in the WT1 gene expression have been reported in several types of tumours, including endometrial and ovarian cancer [25, 26]. There is a significant direct correlation between WT1 expression and high histological grade as well as with a trend towards a worse clinical outcome [26]. Similarly, several investigators have demonstrated WT1 expression in ovarian cancer. Shimizu et al. showed high expression of WT1 in serous ovarian tumours and lower expression in endometrioid ovarian adenocarcinoma [25]. Serous ovarian tumours do not express HOXA10, whereas endometrioid do. These studies are consistent with ours, showing a strong inverse relationship between WT1 and HOXA10 expression. Here we demonstrated a repressor region in HOXA10 promoter that contains two functional WT1 binding sites, linking the expression of these two genes. Here we show that WT1 functions not simply as a tumour suppressor or survival factor, but rather as a determinant of tissue identity. It performs this function by regulating HOX genes, well known as the prototypical regulators of developmental identity.
In addition to a role in neoplasia, WT1 may also be required for normal reproductive tract development. Patients with WAGR syndrome have mutations in WT1 and also have genital tract malformations [27]. Similarly, alternative splicing of WT1 mRNA can lead to Frasier syndrome, manifestations of which include male pseudohermaphroditism [28, 29]. Failure to repress the expression of HOX genes required for female reproductive tract development would be expected to lead to anomalies in males and females. WT1 is highly expressed in the genitourinary tract during embryogenesis, however is down-regulated in many of these tissues in the adult [30, 31]. This suggests that WT1 may initially repress HOX gene expression until the onset of HOX-driven reproductive tract patterning. WT1 expression in some reproductive tract tumours may suppress epithelial HOX gene expression, allowing developmental plasticity and altering mesenchymal/epithelial identity.
Taken together, these findings suggest a novel regulatory relationship in which HOXA10 is directly transcriptionally repressed by WT1. Regulation of HOXA10 gene expression by WT1 contributes to the differentiation of several tumour types including endometrial and ovarian cancer. Although epithelial ovarian cancers have traditionally been thought to derive from the ovary, these tumours typically demonstrate histologic and morphologic features of the Mullerian tract. This may reflect Mullerian transformation of the ovarian surface epithelium or alternatively a Mullerian origin of ovarian cancer [32]. Regardless of the origin of these cells, HOX genes have been shown to regulate the morphologic differentiation of ovarian tumours [33]. Inappropriate developmental programming by altered regulation of HOX genes leads to ovarian and uterine tissue gaining or loosing a Mullerian phenotype, respectively. Here we demonstrate that WT1 regulates HOX gene expression in these tissues, both physiologically and in cancer. WT1 regulates tissue differentiation through direct regulation of developmental control genes including HOX genes.
Acknowledgments
This study was supported by NIH HD036887 and HD052668.
References
- 1.Samuel S, Naora H. Homeobox gene expression in cancer: insights from developmental regulation and deregulation. Eur J Cancer. 2005;41:2428–37. doi: 10.1016/j.ejca.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Cheng W, Liu J, Yoshida H, et al. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract. Nat Med. 2005;11:531–7. doi: 10.1038/nm1230. [DOI] [PubMed] [Google Scholar]
- 3.Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–70. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 4.McGinnis W, Krumlauf R. Homeobox genes and axial patterning. Cell. 1992;68:283–302. doi: 10.1016/0092-8674(92)90471-n. [DOI] [PubMed] [Google Scholar]
- 5.Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 6.Taylor H. The role of HOX genes in the development and function of the female reproductive tract. Semin. Reprod. Med. 2000;18:81–9. doi: 10.1055/s-2000-13478. [DOI] [PubMed] [Google Scholar]
- 7.Taylor H, VandenHuvel G, Igarashi P. A conserved HOX axis in the mouse and human reproductive system; late establishment and persistent expression of the HOXA cluster genes. Biol Reprod. 1997;57:1338–45. doi: 10.1095/biolreprod57.6.1338. [DOI] [PubMed] [Google Scholar]
- 8.Satokata I, Benson G, Maas R. Sexually dimorphic sterility phenotypes in HOXA10 deficient mice. Nature. 1995;374:460–3. doi: 10.1038/374460a0. [DOI] [PubMed] [Google Scholar]
- 9.Hsiehli HM, Witte DP, Weinstein M, et al. HOXA11 structure, extensive antisense transcription, and function in male and female fertility. Development. 1995;121:1373–85. doi: 10.1242/dev.121.5.1373. [DOI] [PubMed] [Google Scholar]
- 10.Mortlock DP, Post LC, Innis JW. The molecular basis of hypodactyly (Hd): a deletion in HOXA13 leads to arrest of digital arch formation. Nat Genet. 1996;13:284–9. doi: 10.1038/ng0796-284. [DOI] [PubMed] [Google Scholar]
- 11.Podsalek CA, Duboule D, Bushman W. Male accessory sex organ morphogenesis is altered by loss of function of HOXD13. Dev Dynam. 1997;208:454–65. doi: 10.1002/(SICI)1097-0177(199704)208:4<454::AID-AJA2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida H, Broaddus R, Cheng W, et al. Deregulation of the HOXA10 homeobox gene in endometrial carcinoma: role in epithelial-mesenchymal transition. Cancer Res. 2006;66:889–97. doi: 10.1158/0008-5472.CAN-05-2828. [DOI] [PubMed] [Google Scholar]
- 13.Taylor HS, Arici A, Olive D, et al. HOXA10 is expressed in response to sex steroids at the time of implantation in the human endometrium. J Clin Invest. 1998;101:1379–84. doi: 10.1172/JCI1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarno JL, Kliman HJ, Taylor HS. HOXA10, Pbx2, and Meis1 protein expression in the human endometrium: formation of multimeric complexes on HOXA10 target genes. J Clin Endocrinol Metab. 2005;90:522–8. doi: 10.1210/jc.2004-0817. [DOI] [PubMed] [Google Scholar]
- 15.Krikun G, Mor G, Alvero A, et al. A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology. 2004;145:2291–6. doi: 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
- 16.Mann RS, Affolter M. Hox proteins meet more partners. Curr Opin Genet Dev. 1998;8:423–9. doi: 10.1016/s0959-437x(98)80113-5. [DOI] [PubMed] [Google Scholar]
- 17.Chariot A, Gielen J, Merville MP, et al. The homeodomain-containing proteins: an update on their interacting partners. Biochem Pharmacol. 1999;58:1851–7. doi: 10.1016/s0006-2952(99)00234-8. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence HJ, Largman C. Homeobox genes in normal hemotopoiesis and leukemia. Blood. 1992;80:2445–53. [PubMed] [Google Scholar]
- 19.Chu MC, Selam F, Taylor H. HOXA10 regulates p53 expression and matrigel invasion in human breast cancer cells. Cancer Biol Ther. 2004;3:568–72. doi: 10.4161/cbt.3.6.848. [DOI] [PubMed] [Google Scholar]
- 20.Taylor H, Bagot C, Kardana A, et al. HOX gene expression is altered in the endometrium of women with endometriosis. Hum Reprod. 1999;14:1328–31. doi: 10.1093/humrep/14.5.1328. [DOI] [PubMed] [Google Scholar]
- 21.Brown KW, Malik KT. The molecular biology of Wilms tumour. Expert Rev Mol Med. 2001;2001:1–16. doi: 10.1017/S1462399401003027. [DOI] [PubMed] [Google Scholar]
- 22.Scharnhorst V, Van Der Eb AJ, Jochemsen AG. WT1 proteins: functions in growth and differentiation. Gene. 2001;273:141–61. doi: 10.1016/s0378-1119(01)00593-5. [DOI] [PubMed] [Google Scholar]
- 23.Fischbach BV, Trout KL, Lewis J, et al. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005;116:984–8. doi: 10.1542/peds.2004-0467. [DOI] [PubMed] [Google Scholar]
- 24.McTaggart SJ, Algar E, Chow CW, et al. Clinical spectrum of Denys-Drash and Frasier syndrome. Pediatr Nephrol. 2001;16:335–9. doi: 10.1007/s004670000541. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu M, Toki T, Takagi Y, et al. Immunohistochemical detection of the Wilms’ tumor gene (WT1) in epithelial ovarian tumors. Int J Gynecol Pathol. 2000;19:158–63. doi: 10.1097/00004347-200004000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Dupont J, Wang X, Marshall DS, et al. Wilms Tumor Gene (WT1) and p53 expression in endometrial carcinomas: a study of 130 cases using a tissue microarray. Gynecol Oncol. 2004;94:449–55. doi: 10.1016/j.ygyno.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Baird PN, Groves N, Haber DA, et al. Identification of mutations in the WT1 gene in tumours from patients with the WAGR syndrome. Oncogene. 1992;7:2141–9. [PubMed] [Google Scholar]
- 28.Klamt B, Koziell A, Poulat F, et al. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/−KTS splice isoforms. Hum Mol Genet. 1998;7:709–14. doi: 10.1093/hmg/7.4.709. [DOI] [PubMed] [Google Scholar]
- 29.Saylam K, Simon P. WT1 gene mutation responsible for male sex reversal and renal failure: the Frasier syndrome. Eur J Obstet Gynecol Reprod Biol. 2003;110:111–3. doi: 10.1016/s0301-2115(03)00088-5. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong JF, Pritchard-Jones K, Bickmore WA, et al. The expression of the Wilms’ tumour gene, WT1, in the developing mammalian embryo. Mech Dev. 1993;40:85–97. doi: 10.1016/0925-4773(93)90090-k. [DOI] [PubMed] [Google Scholar]
- 31.Pritchard-Jones K, Fleming S, Davidson D, et al. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature. 1990;346:194–7. doi: 10.1038/346194a0. [DOI] [PubMed] [Google Scholar]
- 32.Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes. Gynecol Oncol. 1999;72:437–42. doi: 10.1006/gyno.1998.5275. [DOI] [PubMed] [Google Scholar]
- 33.Ota T, Gilks CB, Longacre T, et al. HOXA7 in epithelial ovarian cancer: interrelationships between differentiation and clinical features. Reprod Sci. 2007;14:605–14. doi: 10.1177/1933719107307781. [DOI] [PubMed] [Google Scholar]