

Abstract
The Epstein-Barr virus (EBV) glycoprotein 42 (gp42) is a type II membrane protein essential for entry into B cells but inhibits entry into epithelial cells. X-ray crystallography suggests that gp42 may form dimers when bound to human leukocyte antigen (HLA) class II receptor (Mullen et al., 2002) or multimerize when not bound to HLA class II (Kirschner et al., 2009). We investigated this self-association of gp42 using several different approaches. We generated soluble mutants of gp42 containing mutations within the self-association site and found these mutants have a defect in fusion. The gp42 mutants bound to gH/gL and HLA class II, but were unable to bind wild-type gp42 or a cleavage mutant of gp42. Using purified gp42, gH/gL, and HLA, we found these proteins associate 1:1:1 by gel filtration suggesting gp42 dimerization or multimerization does not occur or is a transient event undetectable by our methods.
Introduction
Epstein-Barr virus (EBV), a member of the human γ-herpesvirus, is a large DNA virus infecting the majority of the adult population worldwide. EBV predominantly infects B cells and epithelial cells. EBV is transmitted via the saliva, enters the host via the oral epithelium and subsequently infects human leukocyte antigen (HLA) class II expressing B cells where it establishes a lifelong latent infection (Rickinson, 2007). Primary infection during early childhood is relatively benign but during adulthood infectious mononucleosis can result. Significantly, EBV has been associated with several human B cell malignancies, including endemic Burkitt’s Lymphoma (BL), Hodgkin’s Lymphoma (HL) and non-Hodgkin’s Lymphoma (NHL), as well as epithelial malignancies such as nasopharyngeal carcinoma, oral hairy leukoplakia and gastric carcinoma (Bieging et al., 2010; Longnecker, 1998; Rickinson, 2007; Takada, 2001; Thompson and Kurzrock, 2004; Wei and Sham, 2005). Immunodeficient patients, including those with AIDS and those undergoing organ transplant and cancer treatment, are particularly at risk for developing EBV associated lymphoproliferative diseases (Rickinson, 2007).
EBV attaches to B cells via viral glycoprotein gp350 and complement receptor type 2 (CR2/CD21), (Fingeroth et al., 1984; Nemerow et al., 1987; Tanner et al., 1987). Following attachment, glycoprotein 42 (gp42) interacts with its receptor, human leukocyte antigen (HLA) class II (DR, DP and most DQ forms) (Haan et al., 2000; Li et al., 1997; McShane et al., 2003; Spriggs et al., 1996). It has been hypothesized that binding of gp42 to HLA class II causes a conformational change in gp42 triggering the core fusion machinery of gH/gL and gB to mediate fusion between the viral envelope and a cellular membrane (Kirschner et al., 2009). EBV entry into epithelial cells occurs by a different mechanism since HLA class II is not typically expressed on epithelial cells and soluble gp42 promotes B cell fusion but inhibits epithelial cell fusion. Interestingly, recent studies have shown that CR2/CD21 is expressed on tonsilar epithelial cells indicating that, in some cases, EBV attachment to epithelial cells may be similar to B cells (Jiang et al., 2008). Finally, integrins have been found to serve as gH/gL receptors, allowing efficient EBV entry into epithelial cells independent of CR2/CD21 and HLA class II (Chesnokova et al., 2009). The use of different glycoprotein-receptor combinations to enter epithelial and B cells allows EBV to regulate cell tropism. EBV virions exiting B cells contain less gp42 and more efficiently infect epithelial cells, whereas virions exiting epithelial cells contain more gp42 and more efficiently infect B cells (Borza and Hutt-Fletcher, 2002).
gp42, encoded in the BZLF2 open reading frame, is a type II membrane glycoprotein, containing a short cytoplasmic tail, a transmembrane domain and an extracellular domain. In infected B cells, two forms of gp42 are produced: a full length 223 amino acid transmembrane protein and a truncated soluble form (Ressing et al., 2005). Soluble gp42 is generated in the endoplasmic reticulum posttranslationally by host proteases, which cleave at amino acids 40–42, downstream of the transmembrane spanning domain (amino acids 9–22) (Ressing et al., 2005). Due to inefficient cleavage, however, the full length transmembrane form is also produced (Ressing et al., 2005). Both forms of gp42 bind gH/gL and HLA class II and both forms are present in gH-gL-gp42 complexes (Kirschner et al., 2007; Kirschner et al., 2006; Liu et al., 2010; McShane et al., 2003; Ressing et al., 2005). Deletion of the cleavage site (residues 37–41) renders gp42 unable to mediate fusion, suggesting soluble gp42 is the functional form required for B cell fusion (Sorem et al., 2009).
The HLA binding site residues of gp42 lie within the C-terminus (residues 94–221) which contains a canonical C-type lectin domain (CTLD). The CTLD of gp42 is structurally similar to that of natural killer (NK) cell receptors, however gp42 does not utilize the “canonical” binding site to bind HLA class II. Instead, this domain of gp42 constitutes a hydrophobic pocket and the HLA binding site lies outside of this pocket (Mullen et al., 2002; Shaw et al., 2010). Significantly, soluble gp42 can inhibit HLA class II-restricted antigen presentation to T cells and block TCR-class II peptide interactions by steric hindrance (Ressing et al., 2003; Ressing et al., 2005) based on TCR:MHC class II crystal structures (Hennecke et al., 2000; Hennecke and Wiley, 2002; Reinherz et al., 1999). Thus, binding of gp42 to HLA is not only necessary for viral entry but also may play a role in EBV immune invasion.
gp42 forms a multimer in the crystal structure of unbound gp42 (Kirschner et al., 2009) and a dimer in the crystal structure of gp42 when bound to HLA class II (Mullen et al., 2002). Although the precise interface between these two forms differ, they share a key region of gp42 that is important in both forms for gp42 self-association. In the unbound form, the N- terminal region extends away from the gp42 core and contacts an adjacent gp42 molecule, making interlayer connections in the crystal lattice. Residues 87–93 in one gp42 interacts with residues 104–111 and 154–155 of an adjacent gp42 (Fig. 1A). In the bound crystal structure, two gp42 molecules bound to two HLA class II molecules contact each other. The N- terminal residues 86–95 of both gp42 molecules form a symmetric antiparallel two-stranded B sheet and residue F88 of one gp42 molecule docks into a shallow hydrophobic pocket on the CTLD surface of the adjacent gp42, formed by W125, F129 and Y167 (Fig. 1B). Thus F88, and the residues surrounding F88, are key in the self-association of gp42 observed in the two crystal forms of gp42 in the absence of HLA class II and in the presence of HLA class II.
Fig. 1. Putative multimer/dimer formation of gp42.

Mutated gp42 residues F88 (yellow) and 87–93 (green), also residues W125, F129, and Y167 (cyan) are displayed as sticks. (A) gp42 multimer packing in the crystal lattice (PDB ID 3FD4). Residues 87–93 (green) in one gp42 interacts with residues 104–111 and 154–155 (red) of an adjacent gp42. The gp42 crystal structure contains two molecules in the asymmetric unit, chain A (residues 83–222, colored blue-gray), and chain B (residues 76–222, colored wheat). (B) gp42:HLA class II complex dimer formation in the crystal structure (PDB ID 1KG0). Residues 86–95 of two gp42 molecules form a symmetric antiparallel two-stranded B sheet and residue F88 (yellow and labeled) of one gp42 molecule docks into a shallow hydrophobic pocket on the CTLD surface of the adjacent gp42, formed by W125, F129 and Y167 (cyan and labeled). gp42 molecules are colored wheat, HLA molecules are colored gray. The N-terminus of each molecule is labeled N.
Deletion of portions of the putative dimerization domain or point mutations at residue F88 reduce the fusion activity of gp42 (Kirschner et al., 2007; Kirschner et al., 2009). These studies demonstrate that the region encompassing F88 region is critical for gp42 function, however whether this phenotype is attributable to a loss of multimerization or dimerization is unknown since this was not analyzed in earlier studies. Interestingly, conformational differences between the bound and unbound forms of gp42, specifically movement of the “158 loop” towards HLA class II in the bound form (Kirschner et al., 2009), results in widening of the canonical CTLD binding pocket. Changes in gp42 dimerization upon binding HLA class II may contribute to this conformational change and potentially promote the interaction of the gp42 binding pocket with gH/gL or an unknown ligand to trigger the core fusion machinery of gH/gL and gB. Thus far, gp42 dimers have never been detected and purified soluble gp42 is monomeric in solution when analyzed in the absence of HLA class II by gel filtration chromotography (Kirschner et al., 2006). Our study sought to investigate the interesting possibility of gp42 multimerization and/or dimerization and determine if this gp42 self-association occurs in the presence of HLA class II, with or without the gH/gL complex, and if it is important for EBV fusion and entry.
Results
Construction of gp42 Flag Tagged Mutants
Several EBV gp42 functional domains have been characterized previously, including a transmembrane domain, a gp42 cleavage site, two gH/gL binding regions separated by a linker, and the C-terminal C-type lectin domain (CTLD) which binds HLA class II (Fig. 2A). Previous studies have shown that deletion of the first 36 residues of gp42, including the transmembrane domain, creates a soluble form of gp42 which is efficiently expressed and secreted and mediates B cell fusion in cell-cell fusion assays, whereas a cleavage mutant in which residues 37–41 are deleted does not (Sorem et al., 2009). Therefore, to study whether the putative gp42 region, spanning amino acids 87–93, is involved in self-association of gp42 molecules, as suggested by X-ray crystallography (Fig. 1A, B), we first constructed soluble N-terminal variants of gp42 incorporating the Flag epitope. Mutant d36Flag has the first 36 residues of gp42 replaced with the Flag tag but is otherwise wild type (Fig. 2B). We chose to use the d36 mutant since our earlier studies had shown that this mutant retained full fusion function and the presence of the N-terminal Flag tag provided a convenient means to monitor expression and binding to gH/gL. Since residues 87–93, and in particular residue F88, were shown in the gp42 unbound and gp42:HLA class II structures to mediate gp42 self association (Fig. 1A, B), we generated the mutant F88AFlag and d87-93Flag to study this in more detail. F88AFlag is identical to d36Flag with the exception of one point mutation at residue 88 from a phenylalanine to an alanine. d87-93Flag is identical to d36Flag with the exception of a deletion spanning the entire putative dimerization domain.
Fig. 2. Schematic representation of wild type gp42 and gp42 Flag-tagged mutants.

(A) Approximate location of gp42 functional domains are shown including the transmembrane domain (residues 9–22), gp42 cleavage site (residues 40–42), the two gH/gL binding regions (residues 47–61 and 67–87) separated by a linker region, and the C-terminal C-type lectin domain (CTLD) which is responsible for binding HLA class II. The hydrophobic pocket is indicated by HP. (B) gp42 Flag tagged mutants, d36Flag, F88AFlag and d87-93Flag, all have the N-term 36 a.a of gp42 deleted, thus deleting the transmembrane domain and rendering them secreted forms of gp42. F88AFlag is a point mutant, d87-93 deletes the entire putative self-association site.
Gp42 mutants are expressed and secreted into the supernatant
To ensure that the flag tagged gp42 mutants were expressed similar to wild type gp42 and produced soluble forms of gp42, each of the mutants was transfected into CHOKI cells and their expression was analyzed 48 hrs post-transfection by Western Blot. We found that all of the flag tagged gp42 mutants were expressed and secreted into the supernatant, as determined by probing with both an anti-flag antibody and an anti-gp42 antibody (Fig. 3A, 3B). Soluble gp42 and the gp42 mutants were readily detected in media supernatants as well as cell lysates, presumably as intracellular protein undergoing transport to the surface (compare Fig. 3A, lanes 5–7 with lanes 12–14 and Fig. 3B, lanes 5–7 with lanes 12–14). All forms of gp42 displayed some heterogeneity in size compatible with gp42 being glycoslyated. The cell lysates of wild type gp42 and d37-41 showed a broader size range compared to the mutants, reflective of retention of the various glycosylated forms of gp42 by the transmembrane domain (compare Fig. 3B, lanes 2–3 with 5–7). However, the difference in glycosylation was not as significant in the supernatant, likely due to the fact that the soluble products represent fully glycosylated gp42 (compare Fig. 3B, lane 9 with lanes 12–14). As predicted, d87-93 migrated faster than the other Flag mutants due to deletion of amino acids 87–93 (Fig. 3A, B lane 7 and 14). Previous work has shown wild type gp42 is inefficiently cleaved resulting in some gp42 being bound at the cell surface and some being soluble (Fig. 3B, lane 2 and 9). This was also readily observed in our experiments. The mutant d37-41, which lacks the cleavage site, remains entirely in cell lysates (compare Fig. 3B, lane 3 and 10), similar to our previous finding (Sorem et al., 2009). This mutant is a useful tool for studying whether dimerization of gp42 can occur at the cell surface when gp42 is not cleaved.
Fig. 3. gp42 mutants are expressed and secreted into the supernatant.
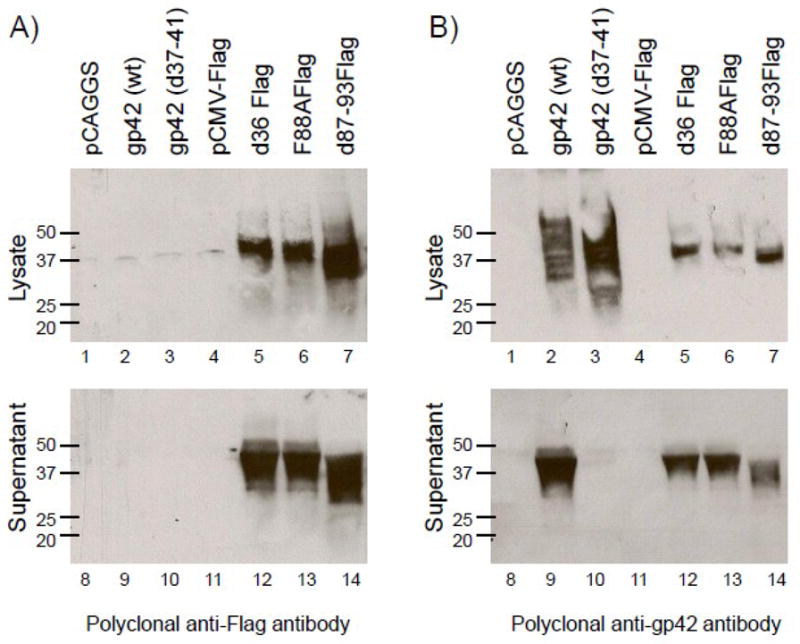
CHO-KI cells were transiently transfected with empty vector (pCAGGS and pCMV-Flag), wild type gp42, d37-41 gp42, d36Flag, F88AFlag and d87-93Flag. Forty eight hours post transfection supernatant was collected and cells were lysed in triton X-100 lysis buffer containing protease inhibitors. Cell lysates and culture supernatants were analyzed by 12% SDS-PAGE and Western blotting with (A) polyclonal anti-Flag antibody (F7425-Sigma) and (B) polyclonal anti-gp42 antibody (PB114). Size markers in kDa are noted to the left of the blots.
Deletion of residues 87–93 alters cell-cell fusion
To study the effect of gp42 N-terminal truncation and replacement with a Flag tag on membrane fusion with B cells, CHO-KI cells transfected with gB, gH/gL, and wild type or Flag tagged gp42 were overlaid with Daudi cells. CELISA (Cell-based Enzyme-Linked Immunosorbent Assay) analysis showed all of the soluble Flag tagged mutants are present at the cell surface at wild-type levels when co-expressed with gH/gL, thus confirming their ability to bind gH/gL which tether these soluble proteins to the cell (Fig. 4A). In the absence of gH/gL, the Flag-tagged mutants were not detected bound to cell the surface (data not shown), as expected from our previous studies (Sorem et al., 2009). Whereas d36Flag had a modest 20% decrease in cell-cell fusion compared to wild type gp42, F88AFlag had a 40–50% decrease in fusion and d87-93Flag was severely impaired, exhibiting an approximately 90% reduction in its ability to mediate fusion (Fig. 4B). Our data is consistent with fusion data obtained for the untagged version of F88A (Kirschner et al., 2009) and the untagged version of a similar deletion mutant, d87-91, (Kirschner et al., 2007). As in our case, the reduced fusion of these untagged versions was not due to a defect in gH/gL binding.
Fig. 4. Secreted mutants bind gH/gL. Mutation or deletion of the gp42 putative dimerization site reduces fusion in B cells.
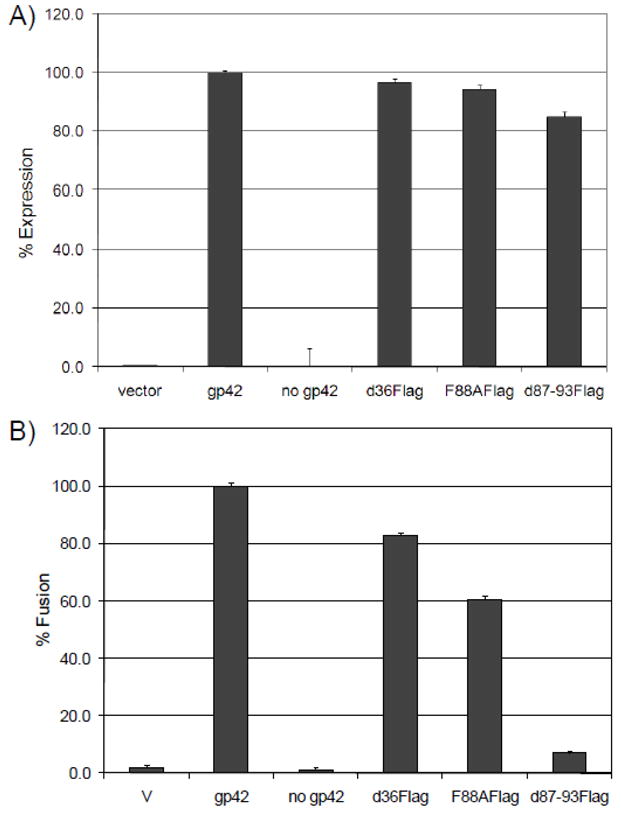
(A) Surface expression of gp42 constructs co-transfected with gH, gL, and gB measured by CELISA using monoclonal anti-gp42 antibody (3H3), secondary biotinylated anti-mouse IgG antibody, tertiary streptavidin-HRP and TMB substrate. Color development was measured by absorbance at 370nm. Cell surface expression was normalized to wild type levels, which were set to 100%. Data shown are representative results of three independent experiments. (B) Fusion of Daudi cells (target) overlayed with CHO-KI cells (effector cells) co-transfected with gp42 mutant constructs, gH, gL and gB. Luciferase activity was normalized to wild type levels. Data shown are representative results of three independent experiments. Error bars represent standard deviations for the normalized values.
Secreted gp42 mutants bind gH/gL but are not able to bind wild type gp42 nor cleavage mutant gp42 (d37-41), when added exogenously
To assess the abilities of the secreted gp42 flag mutants to bind (dimerize or multimerize) wild type gp42 or cleavage mutant gp42 (d37-41), we performed a monolayer binding assay in which exogenously expressed and normalized protein produced from each of the flag tagged mutants (d36Flag, F88AFlag and d87-93Flag) was overlaid for 1hr at 4°C on CHO-K1 monolayers transfected with wild type gp42, d37-41 or gH/gL. Western blot analysis of the cell lysates, including any bound proteins, with anti-flag antibody showed all of the flag tagged gp42 mutants, regardless of the point mutation at residue F88 or deletion 87–93, were able to bind gH/gL (Fig. 5A), as expected since the gH/gL binding sites are retained in these mutants. However, d36Flag was not able to stably bind wild type gp42 nor was it able to stably bind cleavage mutant gp42 (d37-41) (Fig. 5A, d36Flag), suggesting soluble gp42 does not form dimers or multimers with membrane bound gp42. Similar results were found when F88AFlag and d87-93Flag were overlaid on transfected cells; binding to gHgL was detected but no binding to wild type gp42 nor membrane bound gp42 could be detected (Figure 5A, F88AFlag and d87-93Flag). If dimerization or multimerization occurred, we would have expected d36Flag to bind to gp42, F88AFlag to have reduced dimerization and d87-93Flag to be greatly altered in dimerization ability. In a control duplicate blot probed with anti-gp42 antibody (Fig. 5B), we detected abundant amounts of wild type gp42 and cleavage mutant gp42 (d37-41) protein, suggesting the inability of the soluble forms of gp42 to bind membrane bound gp42 was not due to insufficient gp42 at the cell surface.
Fig. 5. Secreted gp42 mutants bind gH/gL but not wild type gp42 nor gp42 cleavage mutant d37-41.
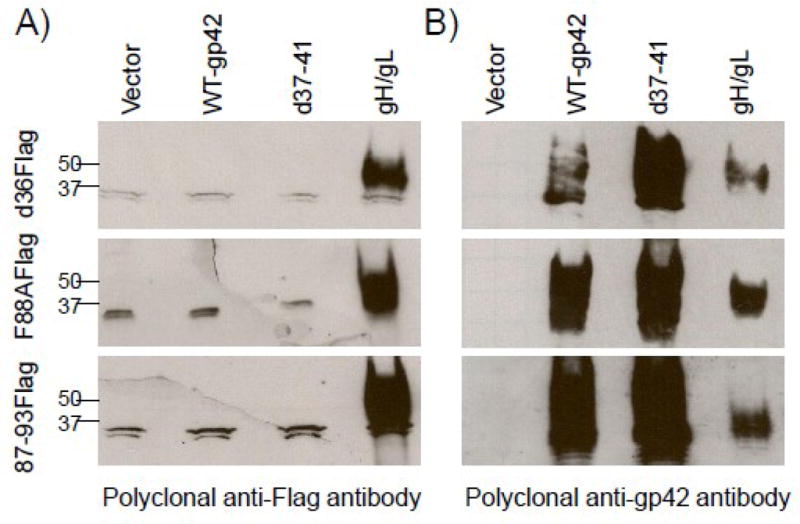
CHO-K1 cells were transiently transfected with d36Flag, F88AFlag and d87-93Flag. Secreted protein was isolated 48 hrs post transfection, normalized by Western Blot and then overlayed onto CHO-K1 cells transiently transfected with WT-gp42, d37-41 gp42, or gH/gL for 1 hour at 4°C. Cells were washed, lysed with TritonX-100 and analyzed by 12% SDS-PAGE and Western blotting with A) polyclonal anti-flag antibody (F7425-Sigma) and B) polyclonal anti-gp42 antibody (PB114). Size markers in kDa are noted to the left of the blots. Data shown are representative results from two independent experiments.
Gp42 mutants bind HLA class II at the cell surface but are not able to bind wild type gp42 nor cleavage mutant gp42 (d37-41) when co-expressed in cells
We next investigated whether soluble gp42 binds membrane bound gp42 or HLA class II at the cell surface when expressed endogenously and analyzed by CELISA using anti-Flag antibody. CHO-KI cells were co-transfected with each of the soluble mutants and either wild-type gp42, d37-41 gp42 or HLA class II. We found d36Flag, F88AFlag and d87-93Flag had similar abilites to bind HLA, however, in agreement with the monolayer binding assay, none of the soluble proteins could bind wild-type gp42 or d37-41 gp42 (Fig. 6), indicating that soluble gp42, when co-expressed with a membrane bound form of gp42, is not able to bind membrane bound gp42 at the cell surface. A duplicate CELISA using anti-gp42 antibody showed approximately equal levels of gp42 in all but the vector alone transfections (data not shown).
Fig. 6. gp42 mutants bind HLA but not wild type gp42 nor the cleavage mutant d37-41.
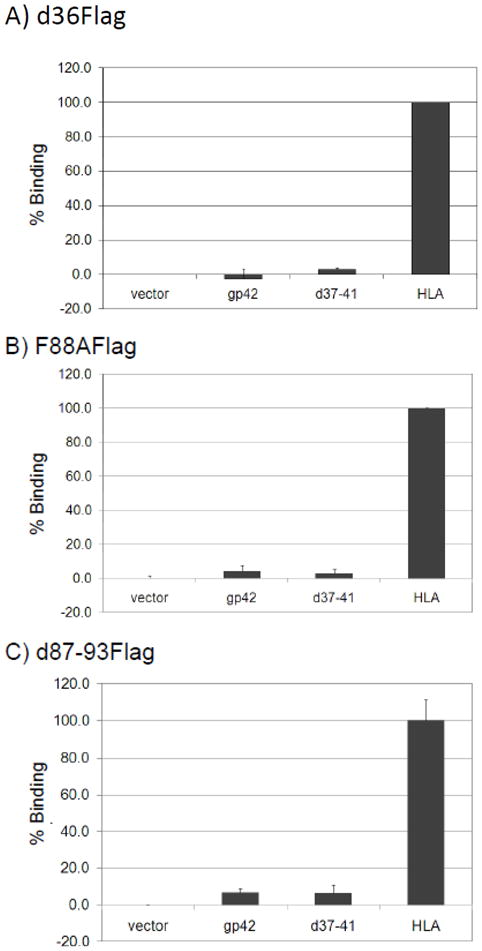
Binding of soluble gp42 mutants was determined by CELISA using polyclonal anti-Flag antibody (F7425-Sigma), secondary biotinylated anti-rabbit IgG antibody, tertiary streptavidin-HRP and TMB substrate. Color development was measured by absorbance at 370nm. Cell surface expression was normalized to HLA binding, which was set to 100%. (A) d36Flag co-transfected into CHO-K1 cells with vector alone, wild type gp42, d37-41 gp42 or HLA. (B) F88AFlag co-transfected into CHO-K1 cells with vector alone, wild type gp42, d37-41 gp42 or HLA. (C) d87-93Flag co-transfected into CHO-K1 cells with vector alone, wild type-gp42, d37-41 gp42 or HLA. Data shown are representative results from three independent experiments. Error bars represent standard deviations for the normalized values.
Gel filtration analysis of gp42-gHgL-HLA-DR1 complex formation
Previous data has shown the soluble forms of gp42 and gHgL form a stable complex with a ratio of 1:1 by gel filtration analysis (Kirschner et al., 2006). We sought to determine whether the addition of soluble HLA class II may be important for gp42 self-association. Soluble forms of gp42 (residue 33–223), gH (residue 18–679) and gL (residue 24–137) were expressed and purified as previously described (Kirschner et al., 2006) (Mullen et al., 2002). HLA-DR1 (residue 1–192 α chain and residue 1–198 of β chain), was expressed in S2 insect cells and purified as previously described (Sloan et al., 1995). A2 peptide derived from HLA-A2 was loaded in the peptide binding grove of HLA-DR1, as this peptide is known to be a predominant peptide bound to HLA-DR1 in B cells (Murthy and Stern, 1997).
Purified and concentrated gp42, gHgL and HLA-DR1 proteins were initially injected on an S200 gel filtration column individually to confirm the molecular weight of each protein compared to known molecular weight standards run simultaneously. The gH/gL complex, which is stable under physiological conditions, was expressed and purified together and as result was injected as a complex. gp42 was observed at a retention volume corresponding to 43 kDa (yellow), HLA-DR1 at 62 kDa (blue), and gHgL at 116 kDa (magenta), consistent with their predicted molecular weights (Fig. 7A-C, and Table 1).
Fig. 7. Gel filtration elution traces (overlaid) showing the soluble gp42-gHgL-HLAD-R1, complex in a 1:1:1 ratio as determined by size.
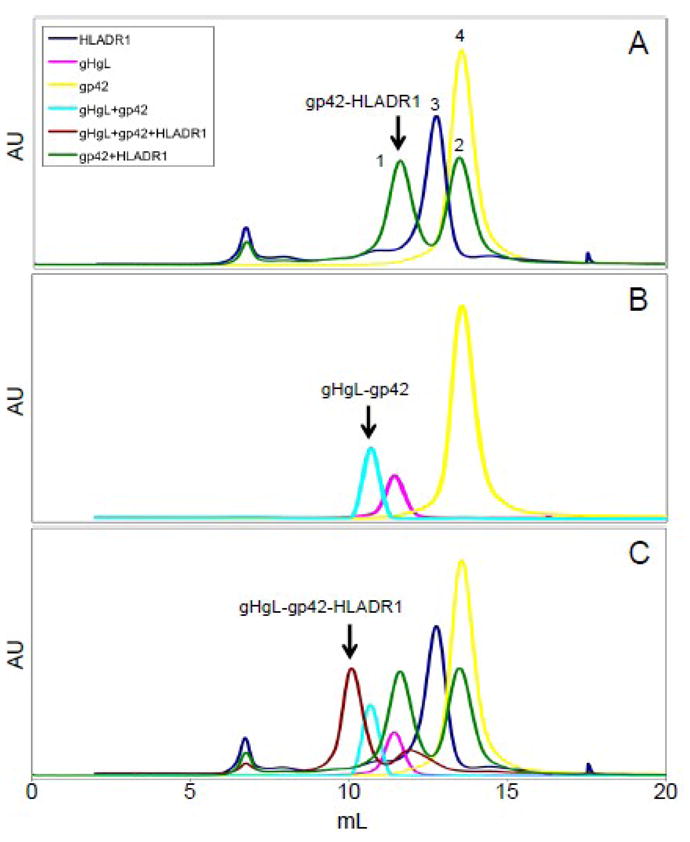
(A) Elution traces of gp42 (yellow, Peak 4), HLADR1 (blue, Peak 3), and gp42-HLA-DR1 complex (2:1 mixture, green, Peak 1 and 2). (B) Elution traces of gp42 (yellow), gHgL (magenta), and gHgL-gp42 complex (1:1:1 mixture, cyan). (C) Elution traces of gp42 (yellow), HLA-DR1 (blue), gp42-HLA-DR1 (2:1 mixture, green), gHgL (magenta), gHgL-gp42 (1:1 mixture, cyan) and gHgL-gp42-HLA-DR1 complex (brown) are overlaid. The soluble gHgL-gp42-HLA-DR1 complex (indicated by arrow) corresponds to a 1:1:1 molar ratio. Elution of each protein from an S200 column was monitored with 280 nm absorbance unit.
Table 1. Soluble protein(s) injected on S200 gel filtration column, their retention values and calculated molecular weights.
Each molecular weight value was calculated using the equation y = −0.2026x + 4.3768 where y is log10 of the molecular weight (in kDa) and x is the retention (in mL). The equation was calculated using 8 different protein samples with known molecular weight distributed between 670 and 13.7 kDa.
| Protein | Retention (mL) | Calculated Molecular Weight (kDa) |
|---|---|---|
| gHgL-gp42-HLADR1 | 10.07 | 217.08 |
| gHgL-gp42 | 10.65 | 165.62 |
| gp42-HLADR1 | 11.61 | 105.83 |
| gHgL | 11.41 | 116.18 |
| HLADR1 | 12.75 | 62.18 |
| gp42 | 13.54 | 43.01 |
Next, gp42 and HLA-DR1 were combined at a 2:1 molar ratio (gp42:HLA-DR1). After a 2 hr incubation at room temperature the combined sample was loaded onto the S200 column (Fig. 7A and 7C, green line). We observed two elution peaks, one at 106 kDa (Fig. 7A, peak 1 and Table 1), consistent with one gp42 bound to one HLA-DR1 in a 1:1 molar ratio and a second peak at 42 kDa (Figure 7A, peak 2 and Table 1) consistent with gp42 monomers. Because gp42 was added in excess, we did not observe a peak for HLA-DR1 alone. We confirmed by western blot that elution peak 1 contains both gp42 and HLA-DR1 (data not shown). The stability of the gp42:HLA-DR1 complexes appears greater than previously observed (Mullen et al., 2002), which may be due to differences in the DR1-bound peptide, which could indirectly affect the gp42 binding affinity, or in the expression source of the HLA-DR1 protein.
We next combined gp42 with gHgL at a 1:1 molar ratio and incubated for 2hr at room temperature as before. We observed one elution peak at 166 kDa (Figure 7B and 7C, cyan line and Table 1), which is consistent with gp42 complexing with gHgL in a 1:1 molar ratio similar to previously published data (Kirschner et al., 2006). We confirmed that this eluted fraction contained gp42 and gH/gL by coomassie stained SDS-PAGE (data not shown).
Finally, gHgL, gp42, and HLA-DR1 proteins were combined at a 1:1:1 molar ratio, incubated at room temperature for 2hrs and loaded onto the S200 column. We observed one main elution peak corresponding to 217 kDa (Figure 7C, brown line and Table 1) which is consistent with gp42, gHgL, and HLA-DR1 complexing in a 1:1:1 molar ratio. Because gp42 complexed with HLA-DR1 as a dimer would be predicted to have an apparent molecular weight close to 217 kDa, we confirmed that the eluted fraction contained gp42, gHgL and HLA-DR1, by coomassie stained SDS-PAGE (data not shown). Our data shows the addition of HLA-DR1 protein does not alter the 1:1 stoichiometry of gHgL:gp42. Thus, we were unable to detect any gp42 dimers or multimers in any of the conditions tested using soluble forms of the relevant proteins, indicating that either dimerization or multimerization does not occur or it occurs only transiently and/or with low affinity such that they are not readily detected in solution by gel filtration.
Native gel analysis of CHO-KI cells transfected with gp42 expresssion vector (wild type gp42 and the gp42 mutants d36Flag, F88AFlag, d87-93Flag), with or without HLA class II, showed the mutations in gp42 at F88A and d87-93 did not affect HLA class II binding (data not shown). Wild type gp42 and each of the gp42 mutants bound HLA class II and produced a slower migrating complex. The complex was absent when HLA class II was co-expressed with a previously described gp42 point mutant (Y107A), that does not bind HLA class II (Silva et al., 2004). Thus, the slower migrating complex is consistent with a 1:1 association of gp42:HLA, as seen by gel filtration.
Discussion
EBV infects B cells via the fusion complex of gp42-gH/gL-gB. While it is clear that gH requires gL for export to the cellular membrane (Pulford et al., 1995; Yaswen et al., 1993), it is not clear if the other EBV glycoproteins are complexed together within the cell or alternatively if the fusion complex is formed at the cellular membrane prior to envelopment. It is known that gp42 complexes with gH/gL through its N-terminus and with its receptor, HLA class II, through its CTLD. However, whether the gH/gL binding site on gp42 changes after HLA class II binding, is not clear. In addition, X-ray crystallography data (Kirschner et al., 2009; Mullen et al., 2002) suggested that gp42 may form multimers in the absence of HLA class II or dimers after engagement with HLA class II. Crystal structures of gp42, both bound and unbound to HLA, indicated that residues 87–93 and F88, in particular, may be important contact residues for multimerization or dimerization, providing the basis for our studies (Fig. 1A, B).
Using tagged mutants of gp42, we show when gp42 is expressed endogenously or added to cells exogenously, it does not form dimers at the cell surface, in the absence of HLA. The inability to dimerize is not due to a loss of ability to bind gH/gL or HLA class II.
In the presence of HLA class II, a complex was detected for wild type gp42 as well as for the tagged mutants regardless of mutation of the putative multimerization or dimerization site. This complex likely represents a 1:1 complex of gp42 and the HLA class II heterodimer. The fact that the complex is detected for d87-93Flag, suggests gp42 does not dimerize, or, alternatively, that gp42 dimerization is not dependent on the predicted site. In addition, our data show the presence of gH/gL does not promote gp42 self-association formation.
Previously, purified soluble forms of gHgL and gp42 were found to associate in a 1:1 stochiometry (Kirschner et al., 2006). We sought to determine if binding to HLA class II would change this stoichiometry. The size of the gHgL-gp42-HLA-DR1 elution complex determined by gel filtration of purified soluble proteins was 217.08, consistent with a 1:1:1 ratio of glycoproteins to receptor. We were unable to detect gp42 dimers or multimers in solution regardless of the presence of HLA. Thus, our data strongly suggest that the dimers or multimers observed in the gp42 crystal structures are likely the result of crystal packing. However, we have not ruled out the possibility that gp42 may dimerize with low affinity or transiently upon HLA binding and therefore is undetectable by our methods.
Interestingly, herpes simplex virus (HSV) gD, a functional homolog of gp42, masks it own receptor binding site as a part of its fusion triggering mechanism. Like gp42, gD binds receptor and acts to trigger fusion mediated by HSV gH/gL and gB. Crosslinking studies with HSV virions have suggested gD is oligomeric in the absence of its receptor (Handler et al., 1996). In addition, gel filtration chromatography data have suggested gD may form dimers in the presence of receptor (Whitbeck et al., 1997). In contrast, gD crystallized as a monomer, both on its own and when bound to HVEM, however the C-terminus was truncated to produce the purified protein (Carfi et al., 2001). The introduction of a disulfide bond at the C-terminus of the gD ectodomain stabilized a dimer form of gD. The crystal structure shows dimerization contacts within the C-terminal region, proximal to the membrane (Krummenacher et al., 2005). Comparison of the structures of gD alone and in complex with its receptor HVEM demonstrate that prior to receptor binding, the C-terminus of gD masks its receptor binding site (Krummenacher et al., 2005). Upon receptor binding, this C-terminus, termed the pro-fusion domain, is displaced and upon its release, it interacts with gH/gL and/or gB to trigger fusion.
gp42 is a type II protein and thus its N-terminus (membrane proximal prior to cleavage) is positionally analogous to the C-terminal region of type I protein gD (although gD is not cleaved). The crystallographic multimerization of unbound gp42 is asymmetric with residues 87–93 from one gp42 contacting residues 104–111, 154 and 155 in another gp42, which coincides with the HLA binding site (Fig. 1A, B). One might postulate from these crystal data that the HLA class II receptor binding site is masked by the flexible N-terminal region of the neighboring gp42. Binding of HLA class II to gp42 could be postulated to displace the gp42 N-terminal extension of the neighboring gp42, thereby dissolving the gp42 self-association and releasing the gp42 N-terminal extension. This freshly released gp42 N-terminal extension, including residue F88, could then serve as a signal to trigger fusion by binding symmetrically to another gp42 as found in the gp42 crystal structure when bound to HLA class II.
Other than the crystallographic data, however, we have not been able to show gp42 dimer formation. Therefore, it is unlikely that gp42 binding to HLA dissociates gp42 dimers. Further evidence that gp42 does not mask its own receptor binding site is that the F88A mutant doesn’t show increased binding to HLA. For HSV, a mutation gD-W294A reduces the binding of the gD C-terminus to the gD receptor binding site and results in increased receptor binding affinity. Since the unbound gp42 dimer structure shows F88 interacting with the HLA site, the fact that F88A does not alter HLA binding is further evidence to dispel the presence of these dimeric contacts in solution.
The lack of evidence for gp42 dimerization suggests that the reduced level of fusion seen for the gp42 F88A and d87-93 mutants is due to another defect in the proteins. Both gp42 mutants retain their ability to bind to gH/gL and HLA, however they are unable to trigger fusion. Perhaps gp42 residues 87–93 are responsible for transmitting an activation signal to the gH/gL complex. These residues lie downstream of the defined gH/gL interaction site, but they could function to activate gH/gL after binding. Alternatively, these residues could be involved in triggering a conformational change in gB to drive fusion. Future experiments are needed to further evaluate these possibilities.
Methods
Cells and Antibodies
Mammalian cells were grown in 25-cm2 or 75-cm2 cell culture flasks (Corning) in medium supplemented with 10% FetalPlex animal serum complex (Gemini Bio-Products) and 1% penicillin-streptomycin (BioWhittaker). Mammalian B cells were Daudi lymphocytes that are EBV positive, express HLA class II and CD21 (American Type Culture Collection), and are modified to stably express T7 RNA polymerase under selection of G418 (700 ug/ml) in RPMI 1640 medium (BioWhittaker) (Silva et al, 2004). Chinese hamster ovary K1 (CHO-K1) cells were grown in Ham’s F-12 medium (BioWhitaker). Trypsin-Versene (BioWhittaker) was used to detach adherent cells. Polyclonal anti-gp42 antibody serum (PB114) and polyclonal anti-HLA-DRβ were used as previously described (McShane et al., 2003). Monoclonal antibody 3H3 (anti-gp42) was obtained as previously described (Kirschner et al., 2006). Monoclonal anti-Flag M2 antibody (F1804) and polyclonal anti-Flag antibody (F7425) were obtained from Sigma-Aldrich Chemical Company.
Plasmids
Gp42 mutant d37-41 was described previously (Kirschner et al., 2007; Sorem et al., 2009). Flag tagged gp42 (designated d36Flag) was generated by PCR amplifying from wild type gp42 (cloned into PCAGGS), using primer 5′-GGAATTCCGTGGCAGCCGCGGCC-3′ and primer 5′-CAGATCTGTTAGCTATTTGATCTTTGACTGAC-3′ resulting in deletion of the first 36 a.a. of gp42. The PCR product was purified by Qiagen PCR purification kit, digested with EcoRI and Bgl II, purified by Qiagen gel extraction kit and ligated into the gel purified EcoRI- and Bgl II-digested pFLAG-myc-CMV-21 expression vector (E5776; Sigma-Aldrich). Flag tagged point mutant F88A (designated F88AFlag) was generated by PCR amplifying from a gp42 F88A mutant that has been previously described (Kirschner et al., 2009) using the same primers as above, resulting in deletion of the first 36 a.a. of gp42. D87-93F was generated by four primer PCR using the primers 5′-GGAATTCCGTGGCAGCCGCGGCC-3′ and 5′-CAGTTAGCTTTGGTATAGTGCAGGGTGGGTGTCC-3′ in one PCR reaction and primers 5′-GGACACCCACCCTGCACTATACCAAAGCTAACTG-3′ and 5′-CAGATCTGTTAGCTATTTGATCTTTGACTGAC-3′ in another PCR reaction in the first round of PCR. Outside primers 5′-GGAATTCCGTGGCAGCCGCGGCC-3′ and 5′-CAGATCTGTTAGCTATTTGATCTTTGACTGAC-3′ were used in the second round of PCR to generate the deletion of amino acids 87–93 which was then cloned into the EcoR I and Bgl II- digested pFLAG-myc-CMV-21 expression vector, as described above.
PCR products were ligated overnight at 14°C with pFlag-Myc-CMV-21 digested vector, transformed into DH5α, and selected on ampicillin plates. Minipreparations were prepared from overnight cultures using Qiagen miniprep kit, digested to confirm the presence of insert and sequenced by the Northwestern Genomic Core Facility. Large scale preparations were isolated using Qiagen Endo-Free Plasmid maxiprep kit and used in subsequent experiments.
HLA-DRα and HLA-DRβ008 were cloned into pSG5 (Stratagene) as previously described (Haan et al., 2000).
Transfection
CHO-KI cells were transfected in Opti-Mem (Gibco) medium using Lipofectamine 2000 (Invitrogen) according the manufactures directions. Briefly, 24 hrs after plating in a six well dish, various combinations of expression vectors were transfected with 6 ul of lipofectamine in Opti Mem.
CELISA and Fusion (Fig. 4): pFLAG-myc-CMV-21 (vector) 2μg, wild type gp42 2μg, d37-41 2μg, d36Flag 2μg, F88AFlag 2μg, d87-93Flag 2μg, and gH 0.5μg, gL 0.5μg, gB 0.5μg, T7 luciferase 0.8μg
Expression Analysis (Fig. 3): pCAGGS 4μg, Wild type gp42 4μg, d37-41 4μg, pFLAG-myc-CMV-21 (vector) 4μg, d36Flag 4μg, F88AFlag 4μg, d87-93Flag 4μg.
Monolayer Binding assay (Fig. 5): pCAGGS 4μg, wild type gp42 4μg, d37-41 4μg, gH 2μg, gL 2μg
CELISA assay (Fig. 6): pCAGGS 2ug, pFLAG-myc-CMV-21 (vector) 2μg, wild type gp42 2μg, d37-41 2μg (Fig 6 only), HLA-DRα 1μg, HLA-DRβ 1μg, gH 1μg, gL 1μg, d36Flag 2μg, F88AFlag 2μg, d87-93Flag 2μg
Western Blotting
CHO-K1 cells were transfected as stated above. The medium was changed 5–6 hrs post transfection to complete Ham’s F12 medium and cells and culture supernatants were collected at 48 h post transfection. Cells were detached with Versene, washed with PBS, and lysed using a 1% Triton x-100 buffer containing protease inhibitors (One milliliter of lysis buffer/10 million cells). Culture supernatants were collected prior to cell detachment and spun down to pellet detached cells. Supernatants and lysates were run on 12% Bio-Rad Criterion gels in sodium dodecyl sulphate (SDS) sample buffer at 90V for l.5 hrs. Proteins were transferred to Whatman Optitran 0.45um nitrocellulose membrane in transfer buffer at 100V for 90 min. Blots were blocked in Tris-buffered saline with 5% milk for 1 hr at room temperature or overnight at 4°C and then incubated for 2 hrs at room temperature with polyclonal anti-flag antibody (Sigma, F7425) 1:1000 or polyclonal anti-gp42 antibody serum (PB114) 1:2000 in blocking solution, as previously described (Fan and Longnecker, 2010; McShane et al., 2003). Blots were washed and horseradish peroxidise (HRP) conjugated anti-rabbit immunoglobulin G (Cell Signaling) was applied for 1.5 hrs at room temperature. The blots were washed and overlaid with equal volumes of ECL Western blotting detection reagents (GE Healthcare) and exposed to hyperfilm (Amersham Biosciences).
Cell enzyme-linked immunosorbent Assay (CELISA)
CELISA was used to test the cell surface expression. Briefly, CHO-K1 cells were co-transfected in a 6 well dish with each of the Flag tagged mutants and either wild-type gp42, d37-41, HLA or empty vector. The medium was changed 5–6 hours post transfection. Twenty four hours post transfection the cells were detached with Versene, counted using a Beckman Coulter Z1 particle counter, 37,500 cells were transferred to a 96 well plate and the total volume was adjusted to 150 ul with complete Ham’s F12 medium. Twenty-four hours later cells were washed once with phosphate-buffered saline (PBS), and CELISA was performed using the monoclonal antibody anti-FLAG-M2 (F1804; Sigma). After incubation with antibody, the cells were washed, fixed, and incubated with biotinylated goat anti-mouseIgG (Sigma), followed by streptavidin-horseradish peroxidase (HRP) (GE Healthcare) and TMB one component HRP substrate (BioFX). Absorbance readings were taken at 380 nm using a Wallac-Victor luminometer (Perkin-Elmer).
CELISA and Fusion Assay
CHO-K1 cells were transiently transfected as described above. The medium was changed 5–6 hrs post transfection and 24 hours post transfection the cells were detached with Versene and 37,500 cells were transferred to duplicate 96-well plates, one plate was used for CELISA with monoclonal anti-gp42 antibody (3H3) and the other plate was overlaid with equal numbers of Daudi B-target cells. The total volume was adjusted to 150 μl with complete Ham’s F12 medium. Eighteen to twenty hours after overlay, cells were washed with PBS and lysed for 10 min with 50 μl passive lysis buffer (Promega) per well. Luciferase activity was measured with a Perkin-Elmer Victor plate reader immediately after addition of 50 μl/well of luciferase reagent (Promega).
Monolayer Binding Assay
A “monolayer binding assay” was performed as previously described (Fan and Longnecker, 2010) to assess the abilities of the secreted gp42 flag mutants to bind wild type gp42 or transmembrane bound gp42. CHO-K1 cells seeded in six well plates for 24 hrs were transfected with wild type gp42, d37-41, gH/gL or HLA using 6ul of Lipofectamine 2000. After 24 h of incubation, the cells were washed twice with cold PBS and incubated with each of the flag mutant protein (isolated following 48 hr transfection and normalized to each other by Western blot, data not shown) for 1 h at 4°C. The cells were then washed with cold PBS four times and lysed with 200 ul of lysis buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM Na3VO3, 1% Nonidet P-40) containing a protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN). Proteins were separated on Bio-Rad Criterion 12% SDS-PAGE gels after boiling for 10 min under reducing conditions. Western blot analyses were performed using the polyclonal anti-Flag antibody (F7425) at 1:1000 and polyclonal anti-gp42 antibody serum (PB114) at 1:2000.
Expression and purification of soluble proteins
HLA-DR1
Soluble HLA-DR1 from DRA*0101 and DRB1*0101 genes (Sloan et al., 1995) were produced as empty αβ heterodimers secreted from stably transfected S2 Schneider cells. Protein expression was induced with 1mM CuSO4 and the cell supernatant was collected after 5–6 days. The supernatant was centrifuged, filtered through 0.22 μm membrane and loaded onto L243 antibody column. Purification was done by washing the column with PBS and eluting with CAPS pH 11.5 buffer, following with immediate neutralization with 1 M sodium phosphate pH 6.0. Peptide A2 derived from HLA-A2 (residue 103–117) was bound to the purified HLADR1 protein with 2–10 fold molar excess peptide. The HLA-DR1 and A2 peptide mixture was incubated at 37°C for 72 hrs in the presence of 1 mM EDTA, 1 mM PMSF, 0.1 mM iodoacetamide, 3 mM NaN3 in PBS. Excess peptide was removed by using a 10 kDa centrifugal filter unit (Amicon Ultra-15, Millipore) and the peptide loaded HLA-DR1 was concentrated to 10 mg/mL and stored at 4°C
gp42 and gHgL
Soluble gHgL and gp42 was expressed and purified as previously described (Kirschner et al., 2006) (Mullen et al., 2002). Briefly, residues 33–223 of gp42 containing a 6His-tag, S-tag, and an enterokinase cleavage site at the N-terminus was expressed in Hi-5 insect cells infected with amplified recombinant baculovirus. Cell supernatant containing gp42 protein was collected by centrifugation after 72 hrs of incubation and sterile filtered. Initial purification was done using a Talon metal affinity resin (Clontech) following the manufacturer’s manual for batch purification. Enterokinase was used to cleave the tags. Concentrated gp42 sample was further purified by S75 gel filtration column (GE Healthcare) in 25 mM Tris pH 7.4, 150 mM NaCl (TBS), buffer.
Residues 18–279 of gH and residues 24–137 of gL were expressed simultaneously as secreted proteins using pBACgus4x-1 (Novagen) baculovirus transfer plasmid containing two promoters. Sf+ insect cells were used for protein expression. Cell supernatant was collected by centrifugation at 72 hrs post infection with amiplified baculovirus stock. Sterile filtered supernatant containing gHgL was purified using E1D1 antibody affinity column and eluted with 0.1 M Sodium Citrate pH2.5, immediately neutralized by adding 1M Sodium Citrate Tribasic salt and 5M NaCl to be final pH 5.5. E1D1 antibody hybridoma was provided to us by L. Hutt-Fletcher. Concentrated gHgL was applied to S200 gel filtration column (GE Healthcare). 50 mM sodium citrate pH5.5, 50mM NaCl was used for the final purification step.
Gel Filtration Chromatography
Purified HLADR1 loaded with A2 peptide, gp42, and gHgL proteins were each concentrated and buffer was exchanged into TBS. S200 gel filtration column was connected to an AKTA purifier (GE Healthcare) and absorbance of 280 nm was monitored. To create a calibration curve to estimate the relation between the elution volume and molecular weight, the S200 column was equilibrated with TBS buffer and molecular weight markers Thyroglobulin (669 kDa), Apoferritin (443 kDa), beta-Amylase (200 kDa), Alcohol Dehydrogenase (150 kDa), Albumin (66 kDa), Ovalbumin (43 kDa), Carbonic Anhydrase (29 kDa), Ribonuclease A (13.7 kDa) (Sigma) were combined into two separate injections and loaded onto the column. The elution volume of each protein sample was monitored at a flow rate of 0.6 ml/min. HLA-DR1, gp42, and gHgL proteins were injected individually and as complexes at 0.5 ml/min or 0.6 ml/min in TBS. Eluted samples were collected as 0.5 ml fractions.
Acknowledgments
We thank the members of the Longnecker and Jardetzky laboratories for their help and support. We thank Sarah Connolly for critical reading of this manuscript. This research was supported by AI076183 (R.L. and T.J.) and AI067048 (R.L.) from National Institute of Allergy and Infectious Diseases by CA117794 (R.L. and T.J.) and CA133063 (R.L. and C.L.R) from the National Cancer Institute.
Abbreviations
- EBV
Epstein Barr Virus
- CELISA
cell enzyme-linked immunosorbent assay
- a.a
amino acid
- PCR
polymerase chain reaction
- ug
microgram
- V
volts
- hr
hour
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CLR and HM were responsible for the experimental work, intellectual design, and drafting the manuscript. RJ and TSJ participated in the intellectual design of this study and the final manuscript. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Cynthia L. Rowe, Email: c-rowe@northwestern.edu.
Hisae Matsuura, Email: hisaem@stanford.edu.
Theodore S. Jardetzky, Email: tjardetz@stanford.edu.
References
- Bieging KT, Swanson-Mungerson M, Amick AC, Longnecker R. Epstein-Barr virus in Burkitt’s lymphoma: a role for latent membrane protein 2A. Cell Cycle. 2010;9:901–908. doi: 10.4161/cc.9.5.10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borza CM, Hutt-Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med. 2002;8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8:169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A. 2009;106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q, Longnecker R. The Ig-like v-type domain of paired Ig-like type 2 receptor alpha is critical for herpes simplex virus type 1-mediated membrane fusion. J Virol. 2010;84:8664–8672. doi: 10.1128/JVI.01039-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A. 1984;81:4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan KM, Kwok WW, Longnecker R, Speck P. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J Virol. 2000;74:2451–2454. doi: 10.1128/jvi.74.5.2451-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler CG, Eisenberg RJ, Cohen GH. Oligomeric structure of glycoproteins in herpes simplex virus type 1. J Virol. 1996;70:6067–6070. doi: 10.1128/jvi.70.9.6067-6070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennecke J, Carfi A, Wiley DC. Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 2000;19:5611–5624. doi: 10.1093/emboj/19.21.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennecke J, Wiley DC. Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J Exp Med. 2002;195:571–581. doi: 10.1084/jem.20011194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Gu X, Nathan CO, Hutt-Fletcher L. Laser-capture microdissection of oropharyngeal epithelium indicates restriction of Epstein-Barr virus receptor/CD21 mRNA to tonsil epithelial cells. J Oral Pathol Med. 2008;37:626–633. doi: 10.1111/j.1600-0714.2008.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner AN, Lowrey AS, Longnecker R, Jardetzky TS. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J Virol. 2007;81:9216–9229. doi: 10.1128/JVI.00575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J Virol. 2006;80:9444–9454. doi: 10.1128/JVI.00572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner AN, Sorem J, Longnecker R, Jardetzky TS. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure. 2009;17:223–233. doi: 10.1016/j.str.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. Embo J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J Virol. 1997;71:4657–4662. doi: 10.1128/jvi.71.6.4657-4662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Marquardt G, Kirschner AN, Longnecker R, Jardetzky TS. Mapping the N-terminal Residues of EBV gp42 that Bind gH/gL Using Fluorescence Polarization and Cell-based Fusion Assays. J Virol. 2010 doi: 10.1128/JVI.00381-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longnecker R. Molecular biology of Epstein-Barr virus. In: McCance DJ, editor. Human tumor viruses. American Society for Virology; Washington, D.C: 1998. pp. 133–174. [Google Scholar]
- McShane MP, Mullen MM, Haan KM, Jardetzky TS, Longnecker R. Mutational analysis of the HLA class II interaction with Epstein-Barr virus glycoprotein 42. J Virol. 2003;77:7655–7662. doi: 10.1128/JVI.77.13.7655-7662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen MM, Haan KM, Longnecker R, Jardetzky TS. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol Cell. 2002;9:375–385. doi: 10.1016/s1097-2765(02)00465-3. [DOI] [PubMed] [Google Scholar]
- Murthy VL, Stern LJ. The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure. 1997;5:1385–1396. doi: 10.1016/s0969-2126(97)00288-8. [DOI] [PubMed] [Google Scholar]
- Nemerow GR, Mold C, Schwend VK, Tollefson V, Cooper NR. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J Virol. 1987;61:1416–1420. doi: 10.1128/jvi.61.5.1416-1420.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulford DJ, Lowrey P, Morgan AJ. Co-expression of the Epstein-Barr virus BXLF2 and BKRF2 genes with a recombinant baculovirus produces gp85 on the cell surface with antigenic similarity to the native protein. J Gen Virol. 1995;76 (Pt 12):3145–3152. doi: 10.1099/0022-1317-76-12-3145. [DOI] [PubMed] [Google Scholar]
- Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, Hussey RE, Smolyar A, Hare B, Zhang R, Joachimiak A, Chang HC, Wagner G, Wang J. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- Ressing ME, van Leeuwen D, Verreck FA, Gomez R, Heemskerk B, Toebes M, Mullen MM, Jardetzky TS, Longnecker R, Schilham MW, Ottenhoff TH, Neefjes J, Schumacher TN, Hutt-Fletcher LM, Wiertz EJ. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U S A. 2003;100:11583–11588. doi: 10.1073/pnas.2034960100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressing ME, van Leeuwen D, Verreck FA, Keating S, Gomez R, Franken KL, Ottenhoff TH, Spriggs M, Schumacher TN, Hutt-Fletcher LM, Rowe M, Wiertz EJ. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J Virol. 2005;79:841–852. doi: 10.1128/JVI.79.2.841-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson A, Kieff E. Epstein-Barr virus. In: Fields BN, Howley DMKPM, editors. Fields’ Virology. Lippincott Williams& Wilkins; Philadelphia: 2007. pp. 2656–2700. [Google Scholar]
- Shaw PL, Kirschner AN, Jardetzky TS, Longnecker R. Characteristics of Epstein-Barr virus envelope protein gp42. Virus Genes. 2010;40:307–319. doi: 10.1007/s11262-010-0455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AL, Omerovic J, Jardetzky TS, Longnecker R. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J Virol. 2004;78:5946–5956. doi: 10.1128/JVI.78.11.5946-5956.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- Sorem J, Jardetzky TS, Longnecker R. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J Virol. 2009;83:6664–6672. doi: 10.1128/JVI.00195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs MK, Armitage RJ, Comeau MR, Strockbine L, Farrah T, Macduff B, Ulrich D, Alderson MR, Mullberg J, Cohen JI. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J Virol. 1996;70:5557–5563. doi: 10.1128/jvi.70.8.5557-5563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern LJ, Wiley DC. The human class II MHC protein HLA-DR1 assembles as empty alpha beta heterodimers in the absence of antigenic peptide. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
- Takada K. Role of Epstein-Barr virus in Burkitt’s lymphoma. Curr Top Microbiol Immunol. 2001;258:141–151. doi: 10.1007/978-3-642-56515-1_9. [DOI] [PubMed] [Google Scholar]
- Tanner J, Weis J, Fearon D, Whang Y, Kieff E. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell. 1987;50:203–213. doi: 10.1016/0092-8674(87)90216-9. [DOI] [PubMed] [Google Scholar]
- Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clin Cancer Res. 2004;10:803–821. doi: 10.1158/1078-0432.ccr-0670-3. [DOI] [PubMed] [Google Scholar]
- Wei WI, Sham JS. Nasopharyngeal carcinoma. Lancet. 2005;365:2041–2054. doi: 10.1016/S0140-6736(05)66698-6. [DOI] [PubMed] [Google Scholar]
- Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, Peng T, Nicola AV, Montgomery RI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J Virol. 1997;71:6083–6093. doi: 10.1128/jvi.71.8.6083-6093.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen LR, Stephens EB, Davenport LC, Hutt-Fletcher LM. Epstein-Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. Virology. 1993;195:387–396. doi: 10.1006/viro.1993.1388. [DOI] [PubMed] [Google Scholar]