

Abstract
Autosomal dominant familial spastic paraplegia (AD-FSP) is a genetically heterogeneous neurodegenerative disorder characterized by progressive spasticity of the lower limbs. Three loci on chromosome 14q (SPG3), 2p (SPG4), and 15q (SPG6) were shown to be responsible for AD-FSP. Analysis of recombination events in three SPG3-linked families allowed us to narrow the critical interval from 9 to 5 cM. An ∼5-Mb YAC contig comprising 32 clones and 90 STSs was built from D14S301 to D14S991, encompassing this region of 14q21. Fifty-six ESTs assigned previously to this region with radiation hybrid (RH) panels Genebridge 4 and G3 were precisely localized on the YAC contig. The 90 STSs positioned on the contig were tested on the TNG RH panel to compare our YAC-based map with an RH map at a high level of resolution. Comparison between our map and the whole genome mapping data on this interval of chromosome 14q is discussed.
Familial spastic paraplegia (FSP) is a heterogeneous group of degenerative disorders of the central motor system characterized by progressive spasticity of the lower limbs. FSP has been classified (Sutherland 1975) according to the mode of inheritance and whether spasticity occurs in isolation (pure FSP) or with additional symptoms (complicated FSP). In pure FSP, inheritance is most commonly autosomal dominant (AD-FSP) although both pure and complicated FSP may be transmitted as autosomal dominant, autosomal recessive, or X chromosome-linked traits.
Pure AD-FSP is genetically heterogeneous and three AD-FSP-causing loci have been identified to date: SPG3 on chromosome 14q (Hazan et al. 1993; Gispert et al. 1995), SPG4 on chromosome 2p (Hazan et al. 1994; Hentati et al. 1994), and SPG6 on chromosome 15q (Fink et al. 1995). Known AD-FSP loci have been excluded in ∼50% of AD-FSP families (Hereditary Spastic Paraplegia Working Group 1996), providing evidence for the existence of at least a fourth locus. Linkage to chromosome 2p has been observed in the majority of the AD-FSP pedigrees analyzed so far (Hazan et al. 1994; Dubé et al. 1995; Lennon et al. 1995; Hereditary Spastic Paraplegia Working Group 1996), whereas linkage to chromosome 15q has only been detected in one large North American kindred (Fink et al. 1995). Linkage to chromosome 14q has been reported in five AD-FSP pedigrees, including one French (Hazan et al. 1993), one German (Gispert et al. 1995), one Tibetan (Huang et al. 1997), and two North American (Hentati et al. 1994; Lennon et al. 1995) families. Recombinant analysis and linkage data first placed SPG3 within a 15-cM interval between markers D14S266 and D14S66 (Hazan et al. 1993). The SPG3 candidate region was then reduced to a 7-cM interval flanked by loci D14S288 and D14S281 (Gispert et al. 1995). The distance between D14S288 and D14S281 was then re-evaluated at 9 cM in the last version of the Généthon map (Dib et al. 1996).
Here, we report linkage to chromosome 14q in an additional French family and present the refined genetic map of the SPG3 candidate interval using microsatellite markers from a published set (Dib et al. 1996). Subsequent studies of recombination events in two pedigrees from France and one from Germany allowed us to narrow the SPG3 region to 5 cM between markers D14S259 and D14S1018. As a first step toward the isolation of one of the genes responsible for AD-FSP, we constructed an ∼5-Mb YAC contig from D14S301 to D14S991 consisting of 32 clones. To evaluate the whole genome mapping data in this particular region of 14q21, our physical map was compared with the Whitehead/MIT contig WC14.1 (Hudson et al. 1995), which revealed some discrepancies in both STS order and YAC/STS content. To establish a transcript map, 104 ESTs that had been localized within or near the SPG3 interval (Schuler et al. 1996) with two radiation hybrid (RH) panels, Genebridge 4 (Gyapay et al. 1996) and G3 (Stewart et al. 1997), were tested by PCR on the 32 YAC clones of the contig: 56 ESTs including 10 known genes were mapped to the YAC contig between D14S301 and D14S991; 24 of them belong to the restricted SPG3 interval flanked by loci D14S259 and D14S1018. To compare the YAC physical map of the critical region with an RH map with a higher level of resolution, the 90 STSs positioned on the SPG3 YAC contig were tested on a third RH panel, the TNG panel (Lunetta et al. 1996) which could be used to order markers at 50-kb resolution. In this paper we integrate all of the RH and YAC data into an accurate high-resolution physical and transcript map of the SPG3 interval that represents a powerful tool for identification of the gene responsible for this form of FSP.
RESULTS
Narrowing the SPG3 Genetic Interval
Four microsatellite markers from the SPG3 locus (D14S1068, D14S269, D14S978, and D14S1018) were initially chosen from a published set (Dib et al. 1996) and tested for linkage to FSP in family 014. Maximum lod scores of 4.39 and 3.68 at Υ = 0 were obtained with locus D14S1068 in family 014, assuming complete penetrance and using liability classes, respectively. A total of 16 microsatellite markers spanning the SPG3 candidate interval were then analyzed to confirm the linkage to this region. As shown in Figure 1, we constructed extended haplotypes spanning a 9-cM interval using 10 microsatellites genotyped on the three SPG3-linked pedigrees. The recombination events visualized on the haplotypes define the narrowest interval containing the SPG3 gene. In family C, a crossover between D14S259 and D14S1055 in individual IV-12 delimited the SPG3 interval on the centromeric side with locus D14S259. Recombinant IV-12 in family C enabled us to order loci D14S259 and D14S1055 within the cluster of five nonrecombining markers described in Dib et al. (1996): the order 14qcen-D14S976/D14S259-D14S1055/D14S1068-14qtel is also consistent with the position of these markers on the YAC contig (see Fig. 2). On the telomeric side, the interval was restricted by two recombination events between D14S978 and D14S1018: Individual III-8 in family W and subject II-1 in family 014 defined the telomeric flanking locus at D14S1018 (Fig. 1). The genetic distance between D14S259 and D14S1018 was estimated at 5 cM from the Généthon map of human chromosome 14 (Dib et al. 1996).
Figure 1.



Pedigrees of families C, W, and 014. Affected individuals are represented by black symbols and unaffected family members by open symbols. Boxes indicate disease chromosomes. Critical recombination events in patients are marked with an arrow. (A) Family C; (B) family W; (C) family 014.
Figure 2.
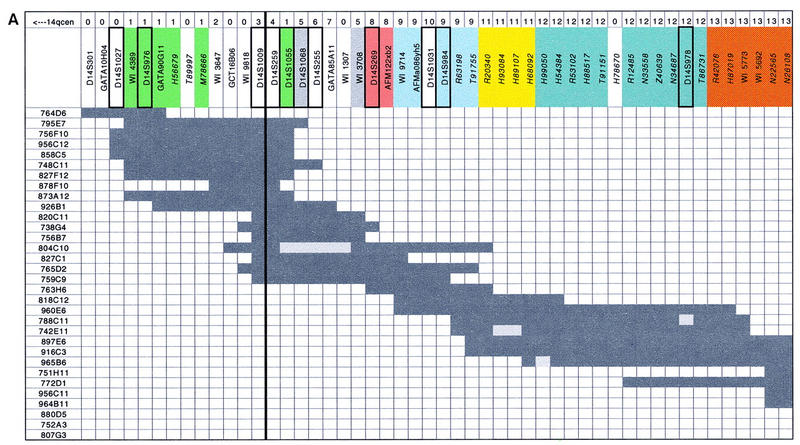
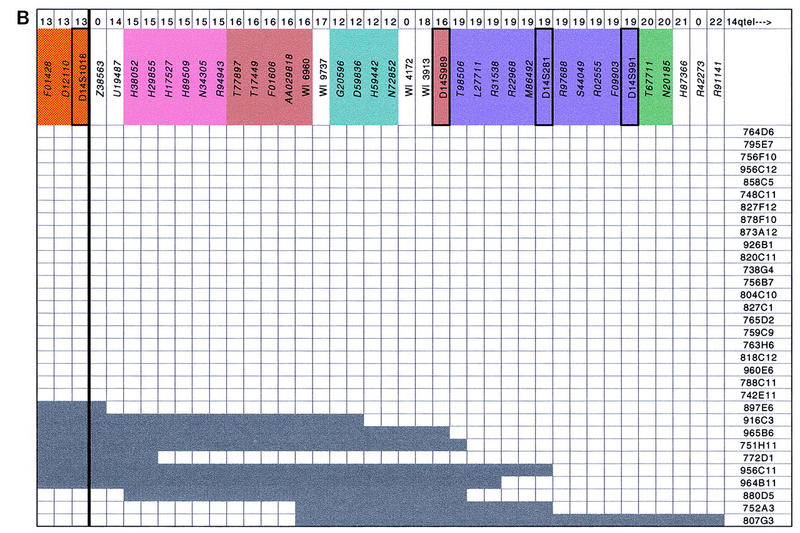
YAC contig map of the SPG3 region. The 90 STSs, including polymorphic markers and ESTs for which GenBank accession numbers are indicated in italics, are listed along the second line at top. The two thick black lines surrounding loci D14S259 and D14S1018 represent the proximal and distal borders of the SPG3 interval, respectively. Polymorphic markers appearing on the last version of Généthon genetic map are framed with a thicker line. Each of the 32 YAC clones is depicted by dark grey rectangles with its name given to the left and to the right. Presence or absence of an STS in the corresponding YAC clone is indicated by dark or light grey squares. RH mapping data using the TNG panel are presented at top: The 22 linkage groups defined by a pairwise lod score above 4 are indicated on the first line, a 0 is used to describe STSs that did not amplify on TNG, and the colors along the second line correspond to the 11 linkage groups containing more than one STS. Positions of ESTs H68092 and H99050, which generate an internal deletion in YAC 965B6, were those defined by TNG RH mapping data.
YAC Contig
The CEPH mega-YAC library was initially screened by PCR with the STSs D14S1055, D14S269, D14S255, D14S978, and D14S281. A total of 25 YAC clones were selected by this method. Additional YACs that had more than one verified STS hit were chosen from the publicly available Whitehead Institute/MIT chromosome 14 integrated map (Hudson et al. 1995). Ten clones that were identified by the screening but were absent from contig WC14.1 (Hudson et al. 1995) were not studied further. The STS content of each YAC clone was then determined by PCR with 34 STSs generated by Généthon, Cooperative Human Linkage Center (CHLC), and the Whitehead Institute/MIT. To confirm each YAC/STS hit, all the PCR products were electrophoresed on agarose gels, transferred onto membranes, and hybridized with the corresponding primer. A total of 32 YAC clones were used to establish a contig of overlapping YACs spanning an ∼5-Mb region from D14S301 to D14S991 as shown in Figure 2. A minimum of five YACs is required to form a continuous clone path that links D14S301 with D14S991. The depth of clone coverage is variable along the YAC contig, from a single clone at both ends to a maximum of 16 clones at D14S259 and D14S1009. Four YACs (804C10, 788C11, 742E11, and 965B6) appear to have undergone internal deletions.
We then compared the STS content data obtained by the Whitehead Institute for the YAC clones of the SPG3 interval with our data set. Of 31 STSs tested in common on the 32 YAC clones, 118 YAC/STS hits shown in contig WC14.1 were confirmed by our analysis, whereas a total of 98 YAC/STS hits that do not appear in the MIT/Whitehead database were detected and verified, which is consistent with the approximate rate of false-negatives (at most 20%) estimated in Hudson et al. (1995); four false-positive hits were identified, which is also in accordance with the false-positive estimate of at most 5% (Hudson et al. 1995). All of the discrepancies revealed by the comparison between the Whitehead Institute/MIT and our STS content data are listed in Table 1. The number of false-negatives is highly variable from one locus to another, or from one YAC clone to another: For example, in the Whitehead Institute database marker, D14S259 was found negative for the 16 YAC clones that were shown positive for this locus in our experiments, which caused its misplacement in contig WC14.1 (see below); on the other hand, only one discrepancy of ten was identified with STSs WI4389, D14S976, and GATA90G11. Likewise, YACs 765D2 and 759C9 were incorrectly scored negative for 10 loci in contig WC14.1, whereas none, or only one, false-negative was detected in YACs 763H6, 818C12, 772D1, 756F10, and 751H11.
Table 1.
Discrepancies Between the Whitehead Institute/MIT Contig WC14.1 and our Data
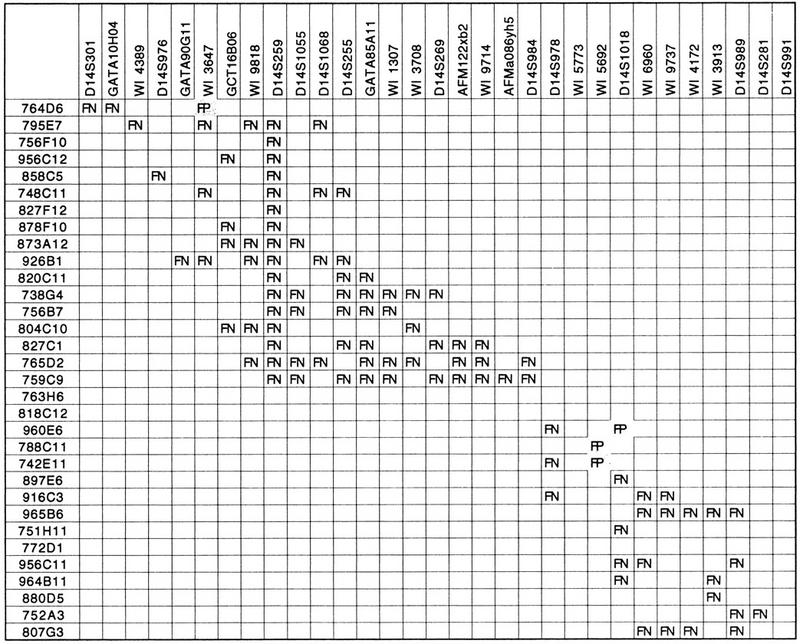
YAC clone names are at left; STS names are on top. (FN) False negative; (FP) False positive.
The main discordance revealed by the comparison between contig WC14.1 (Hudson et al. 1995) and our physical map concerns the STS order that we confirmed with RH and genetic mapping. According to the Whitehead Institute map of the region, the STS order on the proximal side of the SPG3 interval is the following: 14qcen_D14S288_S14S259_ WI3647_ WI4389_D14S976_GATA90G11_ D14S1055_GCT16B06_WI9818_WI3708_D14S1068_ WI1307_GATA85A11_D14S255_D14S269_ AFMa086yh5_WI9714_D14S984_AFM122xb2_ 14qtel, whereas the order we have established is 14qcen_D14S288_WI4389_D14S976_GATA90G11_ WI3647_GCT16B06_WI9818_D14S259_D14S1055_ D14S1068_GATA85A11_WI1307_WI3708_ D14S269_AFM122xb2_WI9714_AFMa086yh5_ D14S984_14qtel.
Transcript Map
To establish a gene map of the SPG3 interval and to compare the positioning of ESTs between the human genome RH map (Schuler et al. 1996) and our YAC contig, 104 ESTs assigned previously to this region of 14q21 (Schuler et al. 1996; G. Gyapay, unpubl.) using RH panels Genebridge 4 (Gyapay et al. 1996) and G3 (Stewart et al. 1997) were selected for precise localization on the YAC contig. Of these ESTs, six were exclusively typed on the G3 panel and assigned to the SPG3 region in the National Center for Biotechnology Information (NCBI) Gene Map database (Schuler et al. 1996). A total of 98 ESTs were tested on at least the Genebridge 4 panel: To be selected, these ESTs had to be localized with a Log10-likelihood difference from the best location (Δlog10L) ranging from 0 to 3 in one of the two RH framework intervals, [D14S259;D14S978] and [D14S978;D14S276], which encompass the entire SPG3 interval. ESTs mapping to these intervals with a lower likelihood were not considered. The 32 YAC clones were tested twice by PCR for the presence or absence of these 104 ESTs. A total of 56 ESTs were precisely positioned on the YAC contig (see Fig. 2). Of these, 24 belong to the restricted SPG3 interval. Among the 56 ESTs localized on the contig, 10 transcripts correspond to known genes including the genes encoding β-1,2-N-acetyl glucosaminyl transferase II (MGAT2, Tan et al. 1995; EST T91755 on the YAC contig), a guanine nucleotide exchange factor (hSos2, Chardin et al. 1993; EST H93084), an STE20-homologous protein (KHS, Tung and Blenis 1997; EST H99050), osteonidogen (GenBank Accession no. D86425, Ohno et al. unpubl.; EST H87019), GTP-binding regulatory protein γ6 subunit (Robishaw et al. 1989; EST N26108), prostaglandin E2 receptor (Regan et al. 1994; EST U19487), a phosphatase (KAP1, Hannon et al. 1994; EST L27711), glia maturation factor β (Kaplan et al. 1991; EST M86492), GTP cyclohydrolase I (Togari et al. 1992; EST S44049), and kinectin (Futterer et al. 1995; EST H87366). Of these 10 genes, 5 (MGAT2, hSos2, KHS, osteonidogen, and GTP-binding regulatory protein γ6 subunit) are in the narrowed SPG3 interval. These 10 transcripts were either identified directly in the NCBI Gene Map database (Schuler et al. 1996) and assigned to the contig, or by full sequencing of the corresponding IMAGE cDNA clones and comparison of the query sequence in the GenBank database by use of the BLASTN and BLASTX programs (Altschul et al. 1990).
Of the six ESTs typed on the G3 panel, only four were located in the same interval on the YAC contig and on the RH map. No conclusion can be drawn about the other two, which were approximately localized on the RH map: One is defined as linked to D14S269 in the gene map database (Schuler et al. 1996) and the other is positioned between loci D14S269 and D14S276 according to the RH map, whereas neither locus D14S276 nor this EST belong to the YAC contig. The fact that RH framework interval [D14S978;D14S276] is bigger than the region covered by our YAC contig precluded us from comparing the localization of some ESTs between the RH map and the YAC contig. The comparison between RH and YAC contig mapping with respect to the 98 other ESTs is summarized in Table 2. For each of these 98 ESTs, we determined, in addition to the best location on the RH map, the second and third most likely locations and their associated Log10-likelihood differences from the best location (Δlog10L) using the RHMAP package (Boehnke et al. 1991; Lange et al. 1995). Fifteen of the 98 ESTs appear in both intervals [D14S259;D14S978] and [D14S978;D14S276] on Table 2: An EST may, for instance, be localized within intervals [D14S978;D14S276] and [D14S259;D14S978] with Δlog10L of 0 (i.e., best location) and 1.4 (i.e., second most likely interval), respectively. Of the 98 ESTs, 36 were mapped to the same intervals on the Genebridge 4 panel and on the YAC contig (∼37% concordant assignments), 25 were misplaced in the RH map of the region (∼26% discrepant assignments) and no conclusion could be drawn from the results obtained for the 37 remaining ESTs (∼37% not determined), which is mainly due to the absence of RH framework marker D14S276 and of several ESTs from the YAC contig. The 25 wrongly assigned ESTs are mainly divided into two groups: (1) 14 that were localized between loci D14S259 and D14S978 with a Δlog10L of 0 according to the Genebridge 4 data, do not appear in this interval on the YAC contig. These 14 ESTs were placed in a second-most likely interval with Δlog10L ranging from 0.22 to >3. Two ESTs are actually localized on the YAC contig in interval [D14S978;D14S276], which also corresponds to their second most likely RH location with Δlog10L of 0.4 and 0.98. Among the remaining twelve, seven (R22818, H56679, H89672, D59836, M78666, U15128, and T89997) are positioned in their second, third, or fourth most likely location, which is unexpectedly at least 103 times less probable than their best location: whereas three of them are physically localized in their second most likely location with Δlog10L of 3.12, 3.72, and 3.77 respectively, the other four are positioned in their third or fourth most likely interval with Δlog10L of 4.86, 5.41, 5.9, and 6.19. (2) Twelve ESTs for which the second most likely location is interval [D14S978;D14S276] with Δlog10L ranging from 0.17 to 0.98 are actually placed between these two flanking markers on the YAC contig.
Table 2.
Comparison of EST Localization between RH Genebridge 4 Panel and YAC Contig
| ESTs mapped with RH Genebridge 4 panel | ||||
|---|---|---|---|---|
| Δlog10L = 0 | 0 < Δlog10L < 1 | 1 < Δlog10L < 2 | 2 < Δlog10L < 3 | |
| A. Interval (D14S259; D14S978) | ||||
| Assigned ESTs (RH) | 30 | 3 | 8 | 14 |
| Concordant mappinga (RH/YAC contig) | 16 | 2 | 4 | 2 |
| Discordant mappinga (RH/YAC contig) | 14 | 0 | 0 | 1 |
| RH mapping vs YAC contig undetermined | 0 | 1 | 4 | 11 |
| B. Interval (D14S978; D14S276) | ||||
| Assigned ESTs (RH) | 26 | 22 | 3 | 7 |
| Concordant mappinga (RH/YAC contig) | 20 | 1 | 2 | 0 |
| Discordant mappinga (RH/YAC contig) | 0 | 12 | 0 | 0 |
| RH mapping vs YAC contig undetermined | 6 | 9 | 1 | 7 |
Δlog10L = log10. Likelihood difference from the best location.
The position found on the YAC contig is identical to the most likely position obtained on the RH map.
Although the EST localization according to the RH map of the SPG3 region appears to be correct in most cases, the distance between an EST and the flanking markers estimated with the RHMAP package (Boehnke et al. 1991; Lange et al. 1995) is generally inaccurate and should not be considered in an exhaustive mapping analysis of ESTs positioned near a disease locus. For instance, EST H38052, which was localized between loci D14S978 and D14S276 at respective distances of 18.9 and 6.2 centirays (cR) according to the RHMAP calculations was positioned close to markers D14S978 and D14S1018 on the YAC contig (see Fig. 2). Another example of erroneous RH distances affects EST Z40639 which was placed at 26.6 cR from locus D14S259 and 47 cR from locus D14S978, although it is actually located on the same YAC clones as marker D14S978 on the physical map of the region.
Fine RH Mapping
To compare our YAC-based physical map with an RH map at a higher level of resolution and to evaluate the resolving power of the TNG RH panel generated at Stanford (Lunetta et al. 1996) with a radiation dose of 50,000 rads, whereas Genebridge 4 and G3 panels were created with 3000 and 10,000 rads of irradiation, respectively (Gyapay et al. 1996; Stewart et al. 1997), the 90 STSs positioned on the SPG3 YAC contig were tested on the 90 RHs of the TNG panel. Eleven STSs that were coded 0 on top of Figure 2 could not be amplified on the TNG panel. Three different sets of linkage groups were made up assuming minimal pairwise lod scores between STSs of 3, 4, and 6. Although minimal pairwise lod scores of 3 or 6 appear to be too loose or too stringent criteria, respectively, the linkage groups obtained with two-point lodscores > 4 seem to be significant for this RH panel. The 79 markers showing a mean retention frequency of 0.22 with a range varying from 0.08 to 0.46 fell into 22 linkage groups (see Fig. 2) including 11 singletons. These 11 singletons consist of three ESTs including two localized at the very end of the YAC contig, three STSs developed by the Whitehead Institute/MIT (Hudson et al. 1995), and five microsatellites. Five of these 11 singletons show abnormally elevated or reduced retention frequencies, which may result in overestimation in the number of breaks between markers and sometimes in the number of linkage groups. However, abnormally elevated or reduced retention frequencies were also observed for some STSs that belong to linkage groups containing more than two markers.
Eleven linkage groups composed of at least two markers were identified with pairwise lod scores above four and are represented by the different colors at the top of Figure 2. Except for the four ESTs belonging to linkage group 12 and embedded in linkage group 16, these linkage groups are perfectly concordant with the positioning of STSs on the YAC contig, which confirms the physical map of the SPG3 region and illustrates the high resolving power of the TNG RH panel. The presence of the 4 ESTs from group 12 within linkage group 16, which is still observed with pairwise lod scores above 6, might be explained by genotyping errors, although the PCR reactions were repeated twice on both the TNG panel and the YAC clones, or by a possible rearrangement, either in the donor DNA used to construct the RH TNG panel or in the donor DNA used to build the CEPH YAC library.
DISCUSSION
AD-FSP is a genetically heterogeneous group of neurodegenerative diseases characterized by progressive spasticity of the lower limbs. Three loci on chromosomes 14q (SPG3; Hazan et al. 1993; Gispert et al. 1995), 2p (SPG4; Hazan et al. 1994; Hentati et al. 1994) and 15q (SPG6; Fink et al. 1995) are involved in AD-FSP and the existence of at least a fourth locus was clearly demonstrated (Hereditary Spastic Paraplegia Working Group 1996; Kobayashi et al. 1996; Bruyn et al. 1997), because this form of FSP was not linked to any of the known loci in ∼50% of the kindreds analyzed so far.
Locus SPG3 on chromosome 14q was shown to be responsible for AD-FSP in five large families (Hazan et al. 1993; Hentati et al. 1994; Gispert et al. 1995; Lennon et al. 1995; Huang et al. 1997). In this study we have demonstrated that AD-FSP is linked to the genetic markers spanning the SPG3 region in a sixth kindred of French descent. Analysis of the recombination events in three of these SPG3-linked families enabled us to narrow the genetic interval from 9 to 5 cM. We established a YAC contig composed of 32 clones that span an ∼5Mb region encompassing the entire SPG3 interval located at 14q21. This YAC-based physical map comprises a total of 90 different STSs with an average spacing of 50 kb. These 90 loci consist of 22 polymorphic microsatellite markers generated either by Généthon or CHLC, 56 ESTs from various cDNA libraries, and 12 STSs isolated by the Whitehead Institute/ MIT. A minimum of two mega-YAC clones, 765D2 and 916C3, are required to generate a continuous tiling path covering the restricted SPG3 interval for which the physical size was approximately estimated at 2.5 Mb.
Comparison of our YAC-based map with contig WC14.1 obtained from the Whitehead Institute/ MIT database (Hudson et al. 1995) revealed several discrepancies with respect to the YAC/STS hits as well as the STS order in this region of chromosome 14q. A total of 31 STSs were tested in common on the 32 YAC clones of the contig: Of these 992 PCR amplifications, 118 YAC/STS hits were identified in both analyses, whereas 98 hits that were found negative in contig WC14.1 were detected and confirmed by the present study. This false-negative rate of ∼10% is consistent with the rate of at most 20% predicted by Hudson et al. (1995). Similarly, our analysis revealed a false-positive rate of ∼0.4% in this part of contig WC14.1, which is in accordance with the rate of at most 5% estimated in the whole genome physical mapping project (Hudson et al. 1995). The presence of false-negative hits resulted in the establishment of a wrong STS order in the Whitehead Institute/ MIT map of the region. Our STS order, which deviates more on the proximal side of the map from contig WC14.1 was confirmed by RH mapping. It should be noted, however, that the overlap relationships among the 32 YAC clones are globally the same in the two contigs, which leads to the conclusion that the whole genome physical map (Hudson et al. 1995) represents a good starting point for subsequent fine mapping efforts.
To establish a gene map of the region, 104 ESTs that had been localized within the SPG3 restricted interval or in adjacent intervals (Schuler et al. 1996) by use of the RH panels Genebridge 4 (Gyapay et al. 1996) and G3 (Stewart et al. 1997), were tested by PCR on the YAC clones of the contig. A total of 56 ESTs were positioned on the contig and 24 of them were located within the narrowed SPG3 interval. Most of these ESTs (98 of 104) were selected for this study on the basis that they had been mapped, with a Log10-likelihood difference from the best location (Δlog10L) ranging from 0 to 3, to one of the two RH framework intervals, [D14S259;D14S978] and [D14S978;D14S276], with the Genebridge 4 panel: The best location as well as the second and third most likely locations were then determined for each of these ESTs. The comparison between the YAC-based physical map and the RH map showed that 37% of the ESTs were localized in the same interval on the Genebridge 4 panel and on the YAC contig, 26% were misplaced on the RH map of the region, and no conclusion could be drawn for the remaining 37% because of the absence of framework RH marker D14S276 and of several ESTs from the YAC contig. Odds ratios between the best and the second most likely locations obtained for the majority of wrongly assigned ESTs are smaller than 10:1, which indicates that definite assignments could not be resolved with this RH panel. The fact that these ESTs were actually positioned in their second most likely interval on the YAC contig, tends to demonstrate the relatively high reliability of the RH map. However, the most likely position was supported at >1000:1 odds (Δlog10L ranging from 3.12 to 6.19) for seven ESTs that were placed in their second, third, or fourth most likely location according to the YAC contig. This confirms the necessity of examining all ESTs that were localized on the RH maps, not only within a genetic interval containing a disease-causing gene, but also in the adjacent intervals. Nevertheless, the existence of these seven misplaced ESTs shows that the limits we had fixed for selecting the ESTs to be tested (i.e., ESTs mapped to an RH interval of interest with a Δlog10L < 3) were not sufficient to permit an exhaustive search for transcripts.
Ten of the 56 ESTs that were mapped to the YAC contig correspond to known genes. Five genes encoding β-1,2-N-acetyl glucosaminyl transferase II (MGAT2, Tan et al. 1995), a guanine nucleotide exchange factor (hSos2, Chardin et al. 1993), an STE20-homologous protein (KHS, Tung and Blenis 1997), GTP-binding regulatory protein γ6 subunit (Robishaw et al. 1989) and osteonidogen are localized within the restricted SPG3 interval. On the other hand, the genes encoding the prostaglandin E2 receptor (Regan et al. 1994), KAP1 phosphatase (Hannon et al. 1994), glia maturation factor β (Kaplan et al. 1991), GTP cyclohydrolase I (Togari et al. 1992) and kinectin (Futterer et al. 1995) were definitely excluded. Interestingly, the exclusion of GTP cyclohydrolase I gene, which was shown to be responsible for dopa responsive dystonia (Ichinose et al. 1994), clearly demontrates that SPG3-linked FSP and dopa responsive dystonia (DRD) are genetically distinct disorders. Allelic heterogeneity had been suggested for these two neurological disorders because elements suggesting spasticity could be part of DRD and the SPG3 and DRD genetic intervals were initially overlapping (Nygaard et al. 1993).
To compare our YAC-based map with an RH map with a high level of resolution, we tested the 90 STSs localized in the SPG3 interval on the TNG RH panel (Lunetta et al. 1996). Twenty-two linkage groups with pairwise lod scores above four were obtained for the 79 ESTs that could be amplified on this RH panel. Except for the presence of four ESTs from linkage group 12 within linkage group 16, which could not be explained so far, the positioning of STSs on our YAC contig are concordant with the linkage groups defined on the TNG panel. The similarity between these two maps ascertains the reliability of our YAC contig and confirms the high resolving power of this RH panel which, combined with the Whitehead Institute/MIT YAC maps, could be used to generate high-confidence maps. In conclusion, the integration of all the data produced by the whole genome mapping projects (Hudson et al. 1995; Dib et al. 1996; Schuler et al. 1996) at the SPG3 locus enabled us to establish a robust and accurate map encompassing this region of chromosome 14q21, which should lead to the identification of the gene responsible for this form of FSP.
METHODS
Clinical Evaluation
All the patients from the three families presented in Figure 1 fulfill the diagnostic criteria of pure and progressive spastic paraplegia with pyramidal signs in the lower limbs as defined by Harding (1981). The two pedigrees of German (family W) and French (family C) descent were reported in Gispert et al. (1995) and Hazan et al. (1993), respectively. Eleven patients of family 014 were examined at 26 ± 21 years by one of the authors (A. Dürr) by a standardized clinical protocol. Age of onset could be determined in 10 patients and ranged from 16 months up to age 50, mean 9 ± 15 years, median 3 years. In two patients, the functional handicap was moderate, five patients could not run, and three patients required help to walk. Spasticity in lower limbs was marked in six, moderate in three, and slight or absent in two subjects. Reflexes in lower limbs were hyperactive in 10 subjects and they were abolished in one patient after a disease duration of 50 years. Reflexes in upper limbs were hyperactive in three subjects and normal or slightly increased in eight patients. In all subjects, the plantar response was extensor. Vibration sense was decreased in four, abolished in one, and normal in six. Five patients complained of urinary urgency. In one patient, the vertical eye gaze was limited and two had nystagmus. All DNAs were extracted from whole blood by standard procedures.
Genotyping and Linkage Analysis
PCRs were carried out as described previously (Hazan et al. 1993). Four amplification products, generated with separate primer sets on identical DNA samples, were coprecipitated and comigrated in a single lane of a 6% polyacrylamide denaturing gel. Separated products were then transferred to Pall membranes and hybridized successively with nonradiolabeled (ECL, Amersham) PCR primers as reported in Vignal et al. (1993). Two-point lod scores were calculated by MLINK of the LINKAGE package (version 5.1; Lathrop et al. 1985) under the assumption of an AD-FSP gene with a frequency of 10−4 and equal female and male recombination rates. On the basis of the clinical evaluation, we chose to estimate the pairwise lod scores using penetrance values of 100% and liability classes. Five liability classes were obtained from the cumulative age of onset curve designed previously (Hazan et al. 1994): 0.17 for individuals from 0 to 19 years old, 0.32 for 20–24 years, 0.55 for 25–30 years, 0.92 for 31–51 years and 0.99 for individuals over 51 years. Marker allele frequencies were assumed to be equal. Simulation analyses with extensive alterations of the allele frequencies did not modify the conclusions of the linkage analyses (Freimer et al. 1993).
Construction of a YAC Contig
YAC DNA pools from the CEPH mega-YAC library (Cohen et al. 1993) were screened by PCR with polymorphic STSs D14S1055, D14S269, D14S255, D14S978, and D14S281. The PCR conditions, as well as the sequences of these STS primers have been published previously (Weissenbach et al. 1992; Dib et al. 1996). Amplification products were analyzed on 2% NuSieve (FMC) agarose gels. The 32 YAC clones identified were subsequently tested by PCR with a set of STSs generated by Généthon, CHLC, and the Whitehead Institute/MIT. Amplifications were carried out in a total volume of 50 μl containing 20 ng of YAC DNA, 50 pmole of each primer, 125 μm dNTPs, 50 mm KCl, 10 mm Tris (pH 9), 1.5 mm MgCl2, 0.1% Triton X-100, 0.01% gelatin, and 0.5 Units of Taq polymerase (Cetus). The reactions were performed with a hot-start procedure: Taq polymerase was added after a denaturation step of 5 min at 96°C. Samples were then processed through 35 cycles of denaturation (94°C for 40 sec), annealing (55°C for 30 sec) and elongation (72°C for 30sec), followed by one last step of elongation (5 min at 72°C). The PCR products were electrophoresed on 2% agarose gels, transferred by Southern blotting to charged nylon membranes (PALL) and hybridized with one of the corresponding nonradiolabelled (ECL, Amersham) primers.
EST Assignment
A total of 104 ESTs that were localized previously in the SPG3 region on the chromosome 14 RH map were tested on the 32 YAC clones by the same PCR conditions as those used for the STS analysis described above. The primers defining these 104 ESTs were those reported in the NCBI Gene Map Database (Schuler et al. 1996). The PCR products were separated on 2% agarose gels.
RH Mapping
STSs were tested by PCR on the TNG RH panel by the hot-start and touchdown procedures described in Gyapay et al. (1996). The PCR products were electrophoresed on agarose gels containing 1% SeaKem and 3% NuSieve agarose (FMC) and images of the gels were recorded with a high resolution CCD camera. Linkage groups were obtained with the RHMAP package (v. 3.0; Boehnke et al. 1991; Lange et al. 1995).
Acknowledgments
We are grateful to the family members for their participation in this study. We thank Susan Cure for critical reading of the manuscript, Corinne Cruaud, Christophe Caloustian, and Françoise Gary for sequencing facilities, Marie-Pascale Pankowiak for technical assistance at the beginning of the project, and Denis Le Paslier for providing the YAC clones. This work was supported by the Association Française contre les Myopathies (AFM).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jamile@genethon.fr; FAX 33-1-60-770951.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Boehnke M, Lange K, Cox DR. Statistical methods for multipoint radiation hybrid mapping. Am J Hum Genet. 1991;49:1174–1188. [PMC free article] [PubMed] [Google Scholar]
- Bruyn RPM, van Veen MMM, Kremer H, Scheltens PH, Padberg GW. Familial spastic paraplegia: Evidence for a fourth locus. Clin Neurol Neurosurg. 1997;99:87–90. doi: 10.1016/s0303-8467(97)00602-1. [DOI] [PubMed] [Google Scholar]
- Chardin P, Camonis JH, Gale NW, Van Aelst L, Schlessinger J, Wigler M H, Bar-Sagi D. Human Sos1: A guanine nucleotide exchange factor for Ras that binds to GRB2. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- Cohen D, Chumakov I, Weissenbach J. A first-generation physical map of the human genome. Nature. 1993;366:698–701. doi: 10.1038/366698a0. [DOI] [PubMed] [Google Scholar]
- Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Dubé M-P, Rouleau GA, Kibar Z, Farlow MR, Ebers G, Harper P, Kolodny EH, Baumbach L, Figlewicz DA. Autosomal dominant hereditary spastic paraplegia: Linkage analysis of a heterogeneous trait. Am J Hum Genet. 1995;57:A190. [PMC free article] [PubMed] [Google Scholar]
- Fink JK, Wu CB, Jones SM, Sharp GB, Lange BM, Lesicki A, Reinglass T, Varvil T, Otterud B, Leppert M. Autosomal dominant familial spastic paraplegia: Tight linkage to chromosome 15q. Am J Hum Genet. 1995;56:188–192. [PMC free article] [PubMed] [Google Scholar]
- Freimer NB, Sandkuijl LA, Blower SM. Incorrect specification of marker allele frequencies: Effects on linkage analysis. Am J Hum Genet. 1993;52:1102–1110. [PMC free article] [PubMed] [Google Scholar]
- Futterer A, Kruppa G, Kramer B, Lemke H, Kronke M. Molecular cloning and characterization of human kinectin. Mol Biol Cell. 1995;6:161–170. doi: 10.1091/mbc.6.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gispert S, Santos N, Dahmen R, Voit T, Schulz J, Klockgether T, Orozco G, Kreuz F, Weissenbach J, Auburger G. Autosomal dominant familial spastic paraplegia: reduction of the FSP1 candidate region on chromosome 14q to 7 centimorgans and locus heterogeneity. Am J Hum Genet. 1995;56:183–187. [PMC free article] [PubMed] [Google Scholar]
- Gyapay G, Schmitt K, Fizames C, Jones H, Vega-Czarny N, Spillett D, Muselet D, Prud’Homme J-F, Dib C, Auffray C, et al. A radiation hybrid map of the human genome. Hum Mol Genet. 1996;5:339–346. doi: 10.1093/hmg/5.3.339. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Casso D, Beach D. KAP: a dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc Natl Acad Sci. 1994;91:1731–1735. doi: 10.1073/pnas.91.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AE. Hereditary “pure” spastic paraplegia: A clinical and genetic study of 22 families. J Neurol Neurosurg Psychiatry. 1981;44:871–883. doi: 10.1136/jnnp.44.10.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan J, Lamy C, Melki J, Munnich A, de Recondo J, Weissenbach J. Autosomal dominant familial spastic paraplegia is genetically heterogeneous and one locus maps to chromosome 14q. Nature Genet. 1993;5:163–167. doi: 10.1038/ng1093-163. [DOI] [PubMed] [Google Scholar]
- Hazan J, Fontaine B, Bruyn RPM, Lamy C, van Deutekom JCT, Rime C-S, Dürr A, Melki J, Lyon-Caen O, Agid Y, Munnich A, et al. Linkage of a new locus for autosomal dominant familial spastic paraplegia to chromosome 2p. Hum Mol Genet. 1994;3:1569–1573. doi: 10.1093/hmg/3.9.1569. [DOI] [PubMed] [Google Scholar]
- Hentati A, Pericak-Vance MA, Lennon F, Wasserman B, Hentati F, Juneja T, Angrist MH, Hung W-Y, Boustany R-M, Bohlega S, et al. Linkage of a locus for autosomal dominant familial spastic paraplegia to chromosome 2p markers. Hum Mol Genet. 1994;3:1867–1871. doi: 10.1093/hmg/3.10.1867. [DOI] [PubMed] [Google Scholar]
- Hereditary Spastic Paraplegia Working Group. Hereditary spastic paraplegia: Advances in genetic research. Neurology. 1996;46:1507–1514. doi: 10.1212/wnl.46.6.1507. [DOI] [PubMed] [Google Scholar]
- Huang S, Zhuyu Li H, Labu B, Lo WH, Fischer C, Vogel F. Another pedigree with pure autosomal dominant spastic paraplegia (AD-FSP) from Tibet mapping to 14q11.2-q24.3. Hum Genet. 1997;100:620–623. doi: 10.1007/s004390050563. [DOI] [PubMed] [Google Scholar]
- Hudson TJ, Stein LD, Gerety SS, Ma J, Castle AB, Silva J, Slonim DK, Baptista R, Kruglyak L, Xu SH, et al. An STS-based map of the human genome. Science. 1995;270:1945–1954. doi: 10.1126/science.270.5244.1945. [DOI] [PubMed] [Google Scholar]
- Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nature Genet. 1994;8:236–242. doi: 10.1038/ng1194-236. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Zaheer A, Jaye M, Lim R. Molecular cloning and expression of biologically active human glia maturation factor-β. J Neurochem. 1991;57:483–490. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Garcia CA, Alfonso G, Marks HG, Hoffman EP. Molecular genetics of familial spastic paraplegia: A multitude of responsible genes. J Neurol Sci. 1996;137:131–138. doi: 10.1016/0022-510x(95)00349-7. [DOI] [PubMed] [Google Scholar]
- Lange K, Boehnke M, Cox DR, Lunetta KL. Statistical methods for polyploid radiation hybrid mapping. Genome Res. 1995;5:136–150. doi: 10.1101/gr.5.2.136. [DOI] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM, Julier C, Ott J. Multilocus linkage analysis in humans: Detection of linkage and estimation of recombination. Am J Hum Genet. 1985;37:482–498. [PMC free article] [PubMed] [Google Scholar]
- Lennon F, Gaskell PC, Wolpert C, Aylsworth AS, Malin D, Warner C, Farrell CD, Boustany R-M, Albright SG, Kingston HM, et al. Linkage and heterogeneity in hereditary spastic paraparesis. Am J Hum Genet. 1995;57:A217. [Google Scholar]
- Lunetta KL. Selected locus and multiple panel models for radiation hybrid mapping. Am J Hum Genet. 1996;59:717–725. [PMC free article] [PubMed] [Google Scholar]
- Nygaard TG, Wilhelmsen KC, Risch NJ, Brown DL, Trugman JM, Gilliam TC, Fahn S, Weeks DE. Linkage mapping of dopa-responsive dystonia (DRD) to chromosome 14q. Nature Genet. 1993;5:386–391. doi: 10.1038/ng1293-386. [DOI] [PubMed] [Google Scholar]
- Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46:213–220. [PubMed] [Google Scholar]
- Robishaw JD, Kalman VK, Moomaw CR, Slaughter CA. Existence of two γ subunits of the G proteins in brain. J Biol Chem. 1989;264:15758–15761. [PubMed] [Google Scholar]
- Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, Rice K, White RE, Rodriguez-Tome P, Aggarwal A, Bajorek E, et al. A gene map of the human genome. Science. 1996;274:540–546. [PubMed] [Google Scholar]
- Stewart EA, McKusick KB, Aggarwal A, Bajorek E, Brady S, Chu A, Fang N, Hadley D, Harris M, Hussain S, et al. An STS-based radiation hybrid map of the human genome. Genome Res. 1997;7:422–433. doi: 10.1101/gr.7.5.422. [DOI] [PubMed] [Google Scholar]
- Sutherland JM. Familial spastic paraplegia. In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology. Vol. 22. Amsterdam, Netherlands: North Holland Publishing Company; 1975. pp. 421–431. [Google Scholar]
- Tan J, D’Agostaro GAF, Bendiak B, Reck F, Sarkar M, Squire JA, Leong P, Schachter H. The human UDP-N-acetylglucosaminyltransferase II gene (MGAT2). Cloning of genomic DNA, localization to chromosome 14q21, expression in insect cells and purification of the recombinant protein. Eur J Biochem. 1995;231:317–328. doi: 10.1111/j.1432-1033.1995.tb20703.x. [DOI] [PubMed] [Google Scholar]
- Togari A, Ichinose H, Matsumoto S, Fujita K, Nagatsu T. Multiple mRNA forms of human GTP cyclohydrolase I. Biochem Biophys Res Commun. 1992;187:359–365. doi: 10.1016/s0006-291x(05)81501-3. [DOI] [PubMed] [Google Scholar]
- Tung RM, Blenis J. A novel human SPS1/STE20 homologue, KHS, activates Jun N-terminal kinase. Oncogene. 1997;14:653–659. doi: 10.1038/sj.onc.1200877. [DOI] [PubMed] [Google Scholar]
- Vignal A, Gyapay G, Hazan J, Nguyen S, Dupraz C, Cheron N, Becuwe N, Tranchant M, Weissenbach J. Nonradioactive multiplex procedure for genotyping of microsatellite markers. In: Adolph KW, editor. Methods in molecular genetics. Vol. 1. San Diego, CA: Academic Press; 1993. pp. 211–221. [Google Scholar]
- Weissenbach J, Gyapay G, Dib C, Vignal A, Morissette J, Millasseau P, Vaysseix G, Lathrop M. A second-generation linkage map of the human genome. Nature. 1992;359:794–801. doi: 10.1038/359794a0. [DOI] [PubMed] [Google Scholar]