

Abstract
The first highly enantioselective α-fluorination of ketones using organocatalysis has been accomplished. The long-standing problem of enantioselective ketone α-fluorination via enamine activation has been overcome via high-throughput evaluation of a new library of amine catalysts. The optimal system, a primary amine functionalized Cinchona alkaloid, allows the direct and asymmetric α-fluorination of a variety of carbo- and heterocyclic substrates. Furthermore, this protocol also provides diastereo-, regio- and chemoselective catalyst control in fluorinations involving complex carbonyl systems.
Stereodefined organofluorine compounds display a range of distinctive physical properties that often render them valuable to the pharmaceutical, agrochemical and polymer industries.1 In particular, fluorine atom incorporation has become an effective tool for medicinal chemists to tailor the physical and metabolic profiles of drug candidates.2 Despite the broad-spectrum utility of such C–F containing compounds, it is remarkable to consider that only a few catalytic methods exist for the asymmetric installation of fluorine onto carbogenic frameworks3 and that most of this research has focused on the generation of non-enolizable products such as α-alkyl-β-ketoesters. Given that α-fluoro carbonyl compounds have been identified as high value synthons for chemical synthesis, several laboratories (including our own) have established enamine catalysis4 as a viable strategy for the enantioselective α-fluorination of aldehydes, a protocol that is sufficiently mild to avoid post-reaction racemization (eq 1).5
 |
(Eq 1) |
 |
(Eq 2) |
Notably, this transformation has been utilized to build C–F stereocenters on a range of aromatic medicinal agents.6 However, this technology is not readily translated from aldehydic to ketonic substrates, a problem that is continually encountered throughout all facets of asymmetric catalysis (metal or organocatalysis). Indeed, despite the availability of a variety of organocatalysts (eq 2: catalysts 1-4), the utility of simple ketones in enamine catalyzed α-fluorination reactions has remained comprehensively elusive for more than six years.
Design Plan
The field of organocatalysis has recognized two critical concerns that have thus far hindered a solution to the “ketone fluorination problem.” First, in comparison to aldehydic substrates, ketones slowly condense with secondary amine catalysts to form low equilibrium quantities of enamine, a limitation that dramatically impacts overall reaction efficiency.
Second, enamines undergo electrophilic substitution from one of the two N–olefin rotational isomers that allow maximal overlap between the nitrogen lone pair and π* orbital of the adjacent C=C system (180° rotation apart). While the relative populations of these two rotational isomers are often dramatically distinct in aldehyde-based enamines (controlled by the steric differentiation of C–H versus C=C in relation to the catalyst framework), ketone-derived enamines often exist as approximately equimolar mixtures of both systems. Unfortunately, this inherent lack of enamine organizational control typically leads to diminished enantiocontrol. In the face of these challenges, the best result to date has been demonstrated by Enders and coworkers using 4-hydroxyproline (4) to achieve a notable 56% yield and 34% ee in the α-fluorination of cyclohexanone.7
In an effort to overcome the “ketone fluorination problem,” we undertook the evaluation of a large and diverse set of catalyst structures, including primary and secondary amines. Specifically, using cyclohexanone as a prototypical substrate, we developed a robotic platform to automate the parallel execution of ∼400 small-scale reactions to determine the utility of a library of 250 novel and known organocatalysts in this important fluorination reaction. An illuminating selection of results from our catalyst evaluation is shown in Figure 1.
Figure 1.
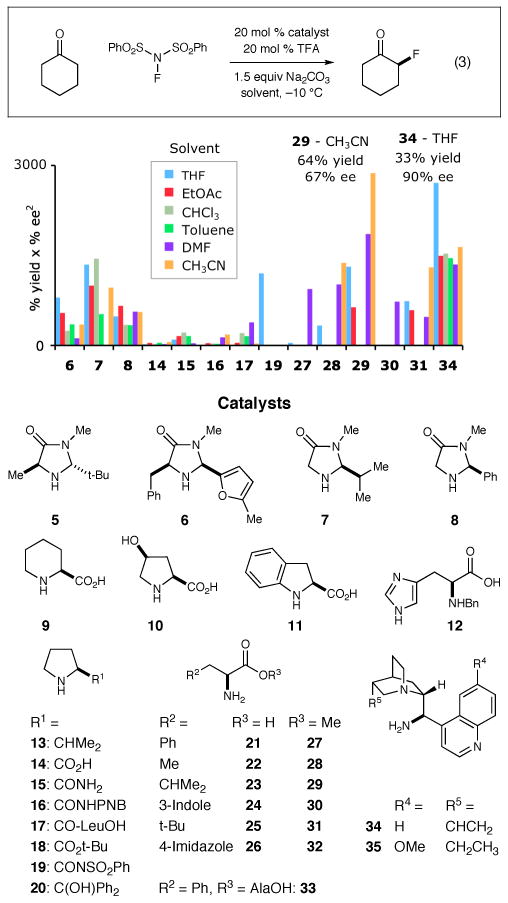
Simultaneous high-throughput variation of catalyst architecture and solvent. Catalysts with [% yield × (% ee)2] < 5 omitted for clarity, see Supporting Information. Catalysts evaluated for the α-fluorination of cyclohexanone.
A critical analytical component of high-throughput asymmetric catalysis is developing a mathematical formula that allows both reaction efficiency and enantiocontrol to be appropriately weighted when identifying potentially useful catalysts. For example, we generally view enantiocontrol as a more difficult parameter to optimize in comparison to yield. On this basis, we interpret the output from such catalyst library screens as a function of the reaction efficiency and square of the observed enantiocontrol (as displayed in Figure 1). For this study, such a mathematical treatment quickly enabled our catalyst leads to be narrowed to only two candidates: the higher yielding leucine methyl ester 29 in acetonitrile (64% yield, 67% ee) and lower yielding but highly enantioselective cinchonidine-derived 34 in tetrahydrofuran (33% yield, 90% ee).8,9 Systematic optimization of both lead series quickly demonstrated that superior results were available with cinchona-based alkaloid catalysts. Indeed, use of the dihydroquinidine catalyst 35 with trichloroacetic acid (TCA) as the co-catalyst10 at −20 °C provided α-fluorocyclohexanone in 88% yield and 99% ee (Table 1, entry 1), and as such was established as the optimal protocol for this α-fluorination study.11
Table 1.
 |
Isolated yield.
Determined by chiral GC-FID, absolute configuration determined by chemical correlation, X-ray analysis or by analogy.
GC yield.
Regioselectivity.
Using 20 mol % 35·TCA.
Using 10 mol % epi-34·TCA.
The difluorination product was obtained in 34% yield.
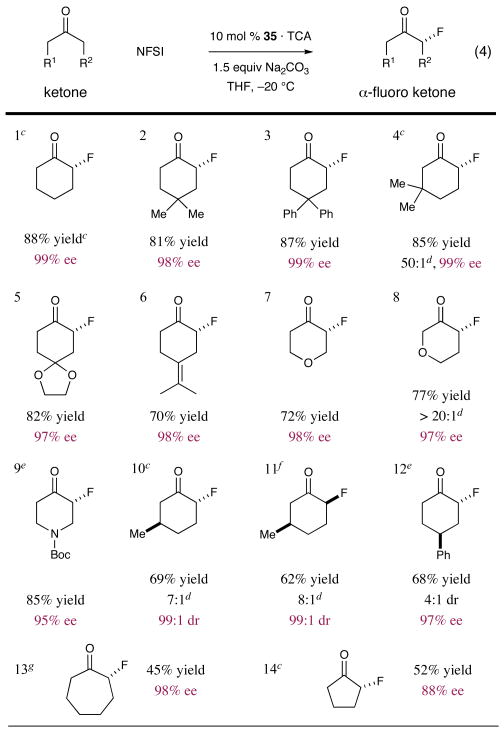
The scope and limitations of this new fluorination reaction have been extensively investigated (Table 1). Notably, a wide array of carbocyclic and heterocyclic ring systems (five-, six-, and seven-membered) are amenable to this enamine activation approach (entries 1-14). Moreover, the introduction of geminal disubstitution at the cyclohexanone 4-position can be tolerated without loss in yield or selectivity (entry 2: dimethyl, 81%, 98% ee; entry 3: diphenyl, 87%, 99% ee). Remarkably, and of high synthetic value, the introduction of substituents at the cyclohexanone 3-position engenders enantioselective fluorination with regioselectivity away from the more substituted site (entries 4, 10 and 11: 62-85%, 99% ee, ≥7:1 regiocontrol). While such selectivities presumably arise from steric control, it should be noted that the oxacyclic analog tetrahydropyran-3-one also enjoys the same positional selectivity, presumably due to electronic factors in the enamine formation step (entry 8, 77% yield, 97% ee, >20:1 regiocontrol). With respect to functional group tolerance, olefins, carbamates and ketals are readily tolerated using these mild reaction conditions (entries 5, 6 and 9: 70-85%, ≥95% ee). Moreover a wide array of heterocycles such as tetrahydropyran-4-one, tetrahydropyran-3-one and N-Boc-piperidin-4-one were successfully fluorinated (entries 7-9: 72-85%, 95-98% ee).
Importantly, we have found that this new protocol can be readily employed with five- and seven-membered cyclic ketones. Despite competing difluorination, 2-fluoro-cycloheptanone was obtained with excellent enantioselectivity and reasonable yield (entry 13: 45%, 98% ee), while fluorination of cyclopentanone proved to be more efficient (entry 14: 52%, 88% ee). It should be noted that this enantioselective fluorination has yet to be successfully implemented with acyclic ketones (diminished yields and enantioselectivities are observed for this subclass).
Last, we turned our attention to the diastereoselective fluorination of cyclic ketones that incorporate pre-existing stereogenicity. Using simple six-membered carbocycles such as (R)-3-methyl cyclohexanone, nearly complete diastereocontrol (trans) was observed (entry 10: 69%, 99:1 dr, 99% ee). Moreover, using the pseudo-enantiomeric cinchonidine catalyst (epi-34), the cis fluorination product was readily obtained with similar levels of reaction efficiency (entry 11: 62%, 99:1 dr, 99%ee). In a similar fashion, α-fluorination of the meso substrate 4- phenyl-cyclohexanone (entry 12) proceeded with high enantioselectivity in the desymmetrization step (97% ee).
Having successfully examined a series of prototypical cyclohexanone systems, we next directed our standard fluorination conditions to more complex substrates (Figure 2). With the hydrogenated Hajos–Parrish ketone, issues of both carbonyl chemoselectivity (cyclopentyl versus cyclohexyl) as well as α-carbonyl positional selectivity (C(3) versus C(5)) were encountered. Remarkably, this new enamine activation reaction enabled chemo-, regio- and diastereoselective fluorination of the C(5) cyclohexyl ring (74%, 98:2 dr, >99:1 regiocontrol, >99:1 carbonyl selectivity, 99% ee). The generality of such an approach was again demonstrated with allo-preganedione wherein two ketones and three α-methylene sites are effectively partitioned with high levels of catalyst controlled selectivities (91% yield, 95:5 dr, >99:1 regiocontrol, >99:1 carbonyl selectivity, 99% ee). Finally, in the case of the steroid cholestanone, fluorination occurs with complete regiocontrol and again with excellent efficiency (85% yield, 97:3 dr, 99% ee, >99:1 regiocontrol).12
Figure 2.

Chemo- and regioselective α-fluorination of polycycles.
In conclusion, a highly enantioselective ketone α-fluorination reaction utilizing an electrophilic fluorine reagent (NFSI) and a primary amine organocatalyst has been accomplished. The primary amine catalyst enables high levels of regio-, chemo-, enantio- and diastereoselectivity for a variety of ketone substrates. The resultant methodology serves as a direct entry into very useful stereogenic carbon-fluorine synthons for chemical synthesis.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM078201-01-01) and kind gifts from Merck. PK thanks the Foundation for Polish Science and JCC thanks NSERC for postdoctoral fellowships.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided (44 pages, pdf)
References
- 1.For reviews on fluorination in medicinal chemistry, see: Kirk KL. Org Process Res Dev. 2008;12:305.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c.Hagmann WK. J Med Chem. 2008;51:887. doi: 10.1021/jm800219f.Ojima I. Chem Bio Chem. 2004;5:628. doi: 10.1002/cbic.200300844. Crop protection: Jeschke P. Chem Bio Chem. 2004;5:570.
- 2.(a) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (b) Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. Chem Bio Chem. 2004;5:637. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 3.Pioneering work: Hintermann L, Togno A. Angew Chem Int Ed. 2000;39:4359. doi: 10.1002/1521-3773(20001201)39:23<4359::AID-ANIE4359>3.0.CO;2-P.Hamashima Y, Yagi K, Takano H, Tamás L, Sodeoka M. J Am Chem Soc. 2002;124:14530. doi: 10.1021/ja028464f.Suzuki T, Hamashima Y, Sodeoka M. Angew Chem Int Ed. 2007;46:5435. doi: 10.1002/anie.200701071.Paull DH, Scerba MT, Alden-Danforth E, Widger LR, Lectka T. J Am Chem Soc. 2008;130:17260. doi: 10.1021/ja807792c. Reviews: Lectard S, Hamashima Y, Sodeoka M. Adv Syn Catal. 2010;352:2708.Brunet VA, O'Hagan D. Angew Chem Int Ed. 2008;47:1179. doi: 10.1002/anie.200704700.Ma JA, Cahard D. Chem Rev. 2008;108:PR1. doi: 10.1021/cr800221v.Prakash GKS, Beirer P. Angew Chem Int Ed. 2006;45:2172. doi: 10.1002/anie.200503783.Pihko PM. Angew Chem Int Ed. 2006;45:544. doi: 10.1002/anie.200502425.Brunet VA, O'Hagan D. Angew Chem Int Ed. 2007;46:2. Recent examples: Kalow JA, Doyle AG. J Am Chem Soc. 2010;132:3268. doi: 10.1021/ja100161d.Katcher MH, Doyle AG. J Am Chem Soc. 2010;132:17402. doi: 10.1021/ja109120n.
- 4.(a) Mukherjee S, yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]; (b) Pihko PM, Majander I, Erkkilä Asymmetric Organocatalysis [Google Scholar]; List B, editor. Top Curr Chem. Vol. 291. Springer-Verlag; Berlin Heidelberg: 2009. p. 29. [Google Scholar]
- 5.(a) Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]; (b) Marigo M, Fielenbach D, Braunton A, Kjærsgaard A, Jørgensen KA. Angew Chem Int Ed. 2005;44:3703. doi: 10.1002/anie.200500395. [DOI] [PubMed] [Google Scholar]; (c) Steiner D, Mase N, Barbas CF., III Angew Chem Int Ed. 2005;44:3706. doi: 10.1002/anie.200500571. [DOI] [PubMed] [Google Scholar]
- 6.For cascade reactions employing aldehyde α-fluorination, see: Huang Y, Walji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2005;127:15051. doi: 10.1021/ja055545d.Fadeyi OO, Lindsley CW. Org Lett. 2009;11:943. doi: 10.1021/ol802930q.Appayee C, Brenner-Moyer SE. Org Lett. 2010;12:3356. doi: 10.1021/ol101167z.
- 7.Enders D, Hüttl MRM. Synlett. 2005:991. [Google Scholar]
- 8.Alkaloids have been used as stochiometric fluorinating agents and it is possible that fluorination proceeds in this case via dual activation of the ketone and fluorine source: Shibata N, Ishimaru T, Suzuki E, Kirk K. J Org Chem. 2003;68:2494. doi: 10.1021/jo026792s.
- 9.Review of primary amine catalysts used for organocatalysis: Peng F, Shao Z. J Mol Catal A: Chem. 2008;285:1. Alkaloid-derived catalysts used for enamine activation: McCooey SH, Connon SJ. Org Lett. 2007;9:599. doi: 10.1021/ol0628006.Bencivenni G, Galzerano P, Mazzanti A, Bartoli G, Melchiorre P. Proc Nat Acad Sci USA. 2010:20642. doi: 10.1073/pnas.1001150107.Bergonzini G, Vera S, Melchiorre P. Angew Chem Int Ed. 2010;49:9685. doi: 10.1002/anie.201004761. Alkaloid-derived catalysts used for iminium activation: Bartoli G, Bosco M, Carlone A, Pesciaioli F, Sambri L, Melchiorre P. Org Lett. 2007;9:1403. doi: 10.1021/ol070309o.Carlone A, Bartoli G, Bosco M, Pesciaioli F, Ricci P, Sambri L, Melchiorre P. Eur J Org Chem. 2007:5492. doi: 10.1021/ol070309o.Xie JW, Chen W, Li R, Zeng M, Du W, Yue L, Chen YC, Wu Y, Zhu J, Deng JG. Angew Chem Int Ed. 2007;46:389. doi: 10.1002/anie.200603612.Xie JW, Yue L, Chen W, Du W, Zhu J, Deng JG, Chen YC. Org Lett. 2007;9:413. doi: 10.1021/ol062718a.Chen W, Du W, Yue L, Li R, Wu Y, Ding LS, Chen YC. Org Biomol Chem. 2007;5:816. doi: 10.1039/b616504d.Li X, Cun L, Lian C, Zhong L, Chen Y, Liao J, Zhu J, Deng J. Org Biomol Chem. 2008;6:349. doi: 10.1039/b713129a.Singh RP, Bartelson K, Wang Y, Su H, Lu X, Deng L. J Am Chem Soc. 2008;130:2422. doi: 10.1021/ja078251w.Wang X, Reisinger CM, List B. J Am Chem Soc. 2008;130:6070. doi: 10.1021/ja801181u.Lu X, Liu Y, Sun B, Cindric B, Deng L. J Am Chem Soc. 2008;130:8134. doi: 10.1021/ja802982h.Ricci P, Carlone A, Bartoli G, Bosco M, Sambri L, Melchiorre P. Adv Synth Catal. 2008;350:49. Thiourea-alkaloid catalysts: Ye J, Dixon DJ, Hynes P. Chem Commun. 2005:4481. doi: 10.1039/b508833j.Vakulya B, Varga S, Csámpai A, Soós T. Org Lett. 2005;7:1967. doi: 10.1021/ol050431s.
- 10.See Supporting Information for optimization details; lowering the catalyst loading to 2.5 mol % resulted in a 71% yield.
- 11.The opposite enantiomer (R, 78%, 99% ee) can be obtained from the catalyst diastereomer derived from dihydrocinchonidine epi-35.
- 12.The reaction does not proceed in the absence of catalyst. Diastereoselective two-step fluorination of cholestanone is known: Nakanishi S, Morita KI, Jensen EV. J Am Chem Soc. 1959;81:5259.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.