

Abstract
Background
The skeleton is the most common site of prostate cancer metastasis, which often results in osteoblastic lesions. The role of transforming growth factor-beta (TGFβ) signaling in prostate cancer-induced osteoblastic metastasis is not clear. We investigated the role of TGFβ signaling in prostate cancer-induced bone metastasis using a novel human prostate cancer cell line, PacMetUT1.
Methods
We injected PacMetUT1/Luc-GFP cells in male nude mice by intracardiac and intratibia injections and then investigated the effect of TGFβ signaling abrogation on osteoblastic tumor growth and incidence in vivo by using fluorescence and bioluminescence imaging analysis and quantifying bone and tumor volume by histomorphometry analysis. Osteoclasts were counted using TRAP assay.
Results
Osteoblastic bone metastasis in skull, rib and femur was detected after 10 to 16 weeks of intracardiac injection of the PacMetUT1 cells. Stable knockdown of TGFβ1 with an shRNA resulted in decreased tumor incidence and bone formation when the cells were directly injected into the tibiae. Systemic administration of either a small inhibitor of TGFβ type I receptor kinase or a pan TGFβ binding protein (BGERII) also decreased bone tumor growth and osteoblastic bone formation in vivo after seven weeks of treatment.
Conclusions
Our results for the first time indicate that blockade of TGFβ signaling in the PacMetUT1 model significantly inhibits osteoblastic bone formation and tumor incidence. Thus, TGFβ signaling pathway may be a viable target for the prevention and treatment of prostate cancer-induced bone metastasis.
Keywords: metastasis, TβRI kinase inhibitor, BGERII, osteoblastic lesion, osteoclasts
Introduction
Prostate cancer is the second leading cause of cancer death in men in the US with an estimated 200,000 new cases and approximately 28,000 deaths occurring in 2009 (www.cancer.gov). More than 80% of prostate cancer patients develop bone metastasis during the advanced stages of cancer [1, 2]. Bone metastatic lesions of prostate cancer are predominantly osteoblastic in nature, whereas most other bone metastatic cancers such as breast, lung and myeloma generate osteolytic lesions [3–5].
Transforming growth factor-beta (TGFβ) signaling has been shown to developmentally regulate bone mass and bone matrix properties [6]. TGFβ released from bone matrix by bone resorbing osteoclasts has been shown to promote osteolytic metastases induced by breast cancer, by stimulating the secretion of bone active cytokines such as parathyroid hormone protein (PTHrP), which enhances bone resorption and further tumor growth through a vicious cycle [3, 5, 7–9]. TGFβ signaling in osteoblasts is required for normal osteoblast differentiation in vivo [10]. The expression of PTHrP in prostate cancer cells can also be induced by TGFβ, resulting in PTHrP-induced bone resorption [10, 11]. Contrary to this, TGFβ1 has also been shown to enhance the expression of osteoprotegerin (OPG), which inhibits osteoclasts, thereby regulating bone turnover [12, 13]. Thus, the role of TGFβ signaling in prostate cancer induced bone metastasis appears ill defined. This is in part due to a lack of suitable models, both in vitro and in vivo, that mimics bone metastasis for biological investigation and pre-clinical research. Prostate cancer xenograft models in mice rarely metastasize spontaneously to bone following orthotopic tumor growth, unlike human prostate cancer. Bone metastasis xenograft models using immunocompromised mice have been developed using prostate cancer cell lines, PC-3 [14], LNCaP [14], LuCaP 35 and LuCaP 23.1 [15]. However, PC-3 and LuCaP 35 induced osteolytic lesions while LNCaP induced mixed lesions. Thus, development of osteoblastic metastasis model of prostate cancer that closely mimics human disease should aid in defining the role of TGFβ signaling in prostate cancer-induced osteoblastic metastasis.
TGFβ transduces its signal through two transmembrane serine/threonine kinase receptors called type I (TβRI) and type II (TβRII) receptors [16]. After the formation of a ligand-receptor complex, TβRII phosphorylates TβRI at the serine/threonine residues in the GS region of TβRI. Activated TβRI then phosphorylates Smad2 and Smad3 proteins, which upon interaction with Smad4, translocates to nucleus, and regulates transcription of target genes. Several potential therapeutic interventions targeting various components of TGFβ signaling pathway are currently in the process of development [17, 18]. Due to its central role in TGFβ signaling, TβRI is being targeted for the blockade of the tumor promoting activity of TGFβ pathway [19]. In the present study, we have used a small TβRI kinase inhibitor (TβRI-KI) and a pan-TGFβ binding protein to effectively block the TGFβ signaling pathway in a novel human prostate cancer cell line, PacMetUT1. PacMetUT1 was shown to induce osteoblastic bone metastasis in nude mice and was used for the investigation of the role of TGFβ in osteoblastic metastasis. Our results, for the first time, show that the treatment with TGFβ inhibitors significantly inhibited osteoblastic bone formation and tumor incidence in the lung and bone, suggesting that blockade of TGFβ pathway may be a novel therapeutic strategy for osteoblastic bone metastases from prostate cancer.
Materials and Methods
Cell cultures
PacMetUT1 was isolated from the lymph node of a 57-year-old male with prostate cancer in our institution [20] and used in this study. DU-145, MDA-PCa-2b, PC-3 and LNCaP were originally from the American Type Culture Collection (ATCC). The culture was maintained in McCoy’s 5A medium supplemented with pyruvate, L-serine, L-asparagine, 100X nonessential amino acids for MEM, MEM amino acids without L-Glutamine, MEM vitamins, penicillin, streptomycin, gentamycin, sodium bicarbonate and 10% fetal bovine serum (FBS). MDA-PCa-2b was maintained in F-12k medium supplemented with cholera toxin, EGF, hydrocortisone, selenious acid, insulin, phosphoethanolamine, penicillin, streptomycin, and 10% FBS. Cells were maintained at 37 C in a 5% CO2 humidified incubator.
Generation of stable TGFβ1 knockdown cells
Lentiviral shRNA constitutive expression vectors against TGFβ1 were purchased from Sigma. Several TGFβ1 shRNA expression vectors were tested and two were found to significantly knockdown TGFβ1 expression. They are named as TGFβ1 shRNA1: (5′-CCGGGCCTCTAAGTATCTGTACCATCTCGAGATGGTACAGATACTTAGAGGCTTT-3′) and TGFβ1 shRNA 2: (5′-CCGGAGAAGGTTATTGGCACTAATACTCGAGTATTAGTGCCAATAACCTTCTTTTT-3′). The control vector, pLKO.1, only contains puromycin resistant gene, but no shRNA sequence. These vectors were transfected into HEK 293FT cells together with two other plasmids, which were purchased from Invitrogen and express necessary proteins for lentivirus packaging. Viral supernatants were harvested for infection into PacMetUT1 cells. Positive cells were selected with puromycin (2 μg/ml).
Generation of GFP- and luciferase-expressing cell lines
Parental PacMetUT1 cells were stably transfected with the enhanced green fluorescent protein (GFP) in the retroviral vector, pLXSN (Clontech Laboratories, Inc.), to aid in the identification of these cells in animal tissues with green fluorescence imaging. For bioluminescence imaging, the control and TGFβ1-knockdown cells were tranduced with pLV411G effLuc-flag-IRES-hrGFP (Luc-GFP), a lentiviral expression vector, kindly provided by Dr. Brian Rabinovich at MD Anderson Cancer Center.
Preparation of TGFβ Inhibitors
The TβRI-KI used in our study was reported previously to be an ATP-competitive inhibitor of the TβRI kinase domain [21, 22]. The chemical name of the compound is [3-(pyridine-2yl)-4-(4-quinonyl)]-1H pyrazole, which was synthesized according to the procedure described by Sawyer et al. [21]. BGERII, a pan-TGFβ antagonist, was made in our laboratory as previously described [23]. Briefly, a recombinant fusion protein containing the endoglin domain of rat betaglycan (BGE) and the extracellular domain of human TβRII was constructed and expressed in E. coli. The fusion protein (BGERII) was purified from bacterial inclusion bodies by immobilized metal ion chromatography (IMAC) and refolded by controlled oxidation. The refolded BGERII was further purified with gel filtration chromatography.
Animal experiments
Four- to five-week-old male athymic nude mice (Harlan Sprague-Dawley, Inc.) were used for this study. Animals were maintained under the care and supervision of the Laboratory Animal Research facility at the University of Texas Health Science Center, San Antonio, Texas. The animal protocol was approved and monitored by the Institutional Animal Care and Use Committee.
Intracardiac injections
Intracardiac injections of PacMetUT1/GFP cells was performed as described previously [24, 25]. Briefly, PacMetUT1/GFP cells were harvested from subconfluent, exponentially growing cultures. The cells were injected into the left cardiac ventricle of anesthesized male nude mice with a 27-gauge needle attached to a 1-ml syringe using a micromanipulator. Each mouse was injected with 1×105 cells in 0.1 ml of PBS, and successful injection was characterized by pumping of arterial blood into the syringe. Development of bone metastasis was monitored at regular intervals for up to 24 weeks using whole body animal imaging for the detection of GFP-expressing tumor cells growing in skull, leg and ribs using a Nikon SMZ1500 fluorescence stereoscope attached to a coolSNAP CCD camera (Photometrics, Tucson, AZ).
Intratibia injections
Mice were anesthesized by i.p. injections with a mouse cocktail containing 3 ml ketamine (100 mg/ml), 2 ml Rompun (20 mg/ml) and 5 ml saline and were also given buprenorpine-HCl (0.3 mg/ml) as an analgesic. An incision of about 5 mm was made on the skin above the knee joint. A 30-gauge, 1/2 inch tuberculin needle was inserted about 5 mm into the proximal end of tibia with drilling motion to make a hole. PacMetUT1 cells expressing Luc-GFP (1×105 in 10ul of PBS) were inoculated into the bone marrow area of right tibias through the pre-made hole by a Hamilton syringe fitted with a 27-gauge needle. PBS was injected into left tibias as control. The TGFβ inhibitor, TβRI-KI at 20μg/100 μl PBS or BGERII at 50 μg/100 μl PBS, was administered i.p every other day for 7 weeks. Intratibia tumor growth was monitored with bioluminescence imaging with a Xenogen IVIS-Spectrum imaging system (Xenogen Corporation) every other week starting from three weeks after tumor cell inoculation. X-ray and whole animal GFP images were also taken every other week starting from five weeks after tumor cell inoculation. At the termination of the experiment in the seventh week after tumor cell inoculation, lungs were harvested and green metastatic cancer cell colonies were visually observed and counted in the whole lungs with an inverted fluorescence microscope.
Bone histomorphometry analysis
Bone tissues were fixed in 10% neutral-buffered formalin (Fisher Scientific) for 24 hours at room temperature, decalcified in 10% EDTA, and embedded in paraffin. Sections were stained with H & E, orange G, and phloxine. The tumor burden and trabecular bone volume in tibia were examined under a Nikon Eclipse E800 microscope equipped with a QImaging QICAM-F fast color digital camera and quantified with a Bioquant Osteo System (Bioquant Image Analysis Corporation, Nashville, TN). Briefly, bone volume, tissue volume and tumor volume were measured by taking a defined region of 2.87 mm2 area from 100 microns beneath the growth plate in the stained tibia sections. The tumor burden is expressed as the ratio of tumor volume to total tissue volume. The osteoblastic bone formation is measured by taking the ratio of trabecular bone volume to total tissue volume.
Bioluminescence imaging analysis
Mice were anesthetized and D-luciferin (Xenogen) was injected i.p at 75 mg/kg in PBS. Xenogen IVIS-Spectrum Imaging system was used to acquire bioluminescence images at 10 min after injection. Acquisition time was set at 60 seconds at the beginning and reduced later on in accordance with signal strength to avoid saturation. Analysis was performed using LivingImage software (Xenogen) by measurement of photon flux (measured in photons/s/cm2/steradian) with a region of interest (ROI) drawn around the bioluminescence signal to be measured. Tumor burden was taken by drawing an ROI around the major bioluminescence signal from the hind limb.
Radiographic analysis of bone metastasis
Mice were exposed with an X-ray at 35 KVP for 5 sec by using a Faxitron Digital Radiographic Inspection unit against the detector as described previously [7].
Cell proliferation assay
Cells were plated in a 96-well plate at 1,000 cells/well in triplicates. After every consecutive day for 8 days, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) solution (2mg/ml) was added and incubated for 2 hours in a tissue incubator. The blue colored formazon product was dissolved in DMSO and quantified at 595 nm wavelength on a Biotek plate reader.
ELISA assay
Various human prostate cancer cell lines were grown to confluence in the complete medium. The cells were washed twice with PBS and then serum-free medium was added. After 48 h, the conditioned medium was collected, filtered using a 0.45-μm syringe filter, and frozen at −80°C until ready for use. TGFβ1 and TGFβ2 secreted in conditioned medium were quantified using an Emax Immunoassay System (Promega) following the manufacturer’s protocol.
Western Blot Analysis
Cells were harvested and lysed in Laemmli buffer with a cocktail of protease inhibitors. The total protein concentrations were quantified by the BCA protein assay (Thermo Scientific, Rockford, IL). An equal amount of total protein was resolved by SDS-PAGE, transferred to a nitrocellulose membrane under constant voltage and blocked with 5% non-fat dried milk in TBST (10mM Tris pH 7.5, 150mM NaCl and 0.05% Tween-20) followed by washing with TBST. Primary antibodies and secondary antibodies were diluted in TBST and applied with a washing step in between. Proteins were detected using the Amersham ECL Western blotting detection kit (GE Healthcare, Piscataway, NJ, USA). Antibody to p-Smad2 was from Cell Signaling), to Actin from Sigma, and to GAPDH from CalBioChem.
Soft Agarose assay
Cells (2×103) in the complete medium were mixed with 0.8% agarose in one to one ratio. The mixture was poured on top of 1 ml of 0.8% hardened agar layer in 6-well plates and allowed to solidify. The cells were grown at 37°C in a 5% CO2 humidified incubator for 10 days. The colonies were then stained with 0.5 ml/well of 1 mg/ml INT (p-Iodonitrotetra zolium violet, Sigma) staining solution overnight in an incubator. The plates were scanned for colony number.
In vitro luciferase assay
Cells were seeded in triplicates in a 12-well plate at a density of 1.8×105 cells/well. When cultures were about 80% confluent, they were co-transfected with 1.0 μg of a β-galactosidase expression plasmid and a TGFβ responsive promoter-luciferase construct (pSBE4-Luc) using 2.0 μl of Lipofectamine 2000 (Invitrogen) in a serum-free medium following the manufacturer’s protocol. After 5 h, the medium was replaced with the serum-containing medium. After overnight incubation, the cells were lysed in a buffer (100 mM K2HPO4, 1 mM DTT, and 1% Triton X-100) and the luciferase activity in the cell lysate was measured as previously described [24]. Luciferase activity was normalized for transfection efficiency with β-galactosidase activity.
TRAP (tartarate resistant acid phosphatase) assay
TRAP assay was used for the identification of osteoclasts as previously described [26]. Briefly, formalin fixed, EDTA decalcified, paraffin-embedded bone specimens were deparaffinized in xylene for 2 min, hydrated in 100%, 95% and 80% ethanol sequentially for 1 min each and finally in water. Slides were incubated at 42°C for 30 min in a substrate solution containing tartaric acid and naphthol AS-BI phosphate. Slides were then put directly into the color reaction solution containing sodium nitrite and pararosaniline dye. For nuclear staining, slides were incubated with Harris’s acid hematoxylin (20 sec), followed with water rinsing and 10 sec incubation in ammonia water. Osteoclasts were stained bright red. The slides were scanned using a Nikon Eclipse E400 microscope equipped with a CalComp (Scottsdale, AZ) digitizing tablet and a Sony (Japan) color video camera using OsteoMetrics (Decatur,GA) computer software. The osteoclasts were counted by taking a 3 mm2 defined area from 500 microns below the growth plate in the scanned images of tibia sections.
Statistical analysis
Results are expressed as mean ± SEM. Two-tailed Student’s t-tests were used to compare two groups. One-way analysis of variance was used for the examination of differences among more-than-two groups followed by Tukey-Kramer post-hoc test. P < 0.05 was considered as statistically significant.
Results
In this study, we have used a novel human prostate cancer cell line, PacMetUT1, to evaluate its metastatic potential to bone and its effect on osteoblastic bone remodeling. We have also used this unique model for the determination of the role of TGFβ pathway in the regulation of bone metastasis.
PacMetUT1 induces bone metastasis and formation of osteoblastic lesions
To determine the effect of PacMetUT1 on skeletal metastasis, we injected PacMetUT1/GFP cells into the left cardiac ventricle of male nude mice at 1×105 cells/mouse. Because the cells were labeled with GFP, we were able to observe green fluorescent metastatic tumors in femur/tibia area by whole-body green fluorescence imaging (Fig.1A). Green fluorescence imaging also revealed the presence of metastatic tumors in skull, rib, and femur when these bones were excised at the termination of the experiment (Fig.1B). Bone metastasis was generally detected between 10 and 16 weeks after tumor cell inoculation. Histologic staining revealed extensive formation of new bone tissue inside tumor areas in the sections of calvaria and femur containing metastatic tumors as compared to the control calvaria and femur sections (Fig.1C). These results show that PacMetUT1 cells can induce bone metastasis and formation of osteoblastic lesions after intracardiac inoculation in male nude mice. To determine whether bone metastatic PacMetUT1 cells have higher metastatic potential than their parental counterparts, we isolated and cultured PacMetUT1 cells from a tumor in the femur of a male nude mouse and then re-inoculated the cells through intracardiac injection. However, the cells were not more bone metastatic than the parental cells, which induced detectable bone metastasis in 10 to 15 weeks at a low frequency of 10%. Because of the weak bone metastatic potential after intracardiac inoculation, we directly injected PacMetUT1 cells in the tibia of male nude mice for subsequent studies.
Figure 1. Induction of bone metastasis and osteoblastic lesion formation by PacMetUT1 cells in an intracardiac tumor injection model.
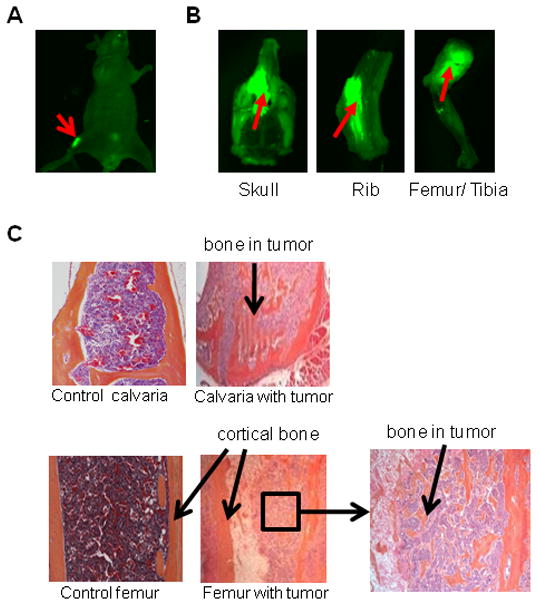
(A) PacMetUT/GFP cells were injected into the left cardiac ventricle at 1×105 cells/mouse. Whole-body green fluorescence imaging shows the presence of metastatic tumors in femur/tibia at 10th week of injection. (B) Metastasis to skull, ribs and femur/tibia was detected at 20th week of injection. (C) H& E stained bone sections at the termination of experiment. Tumor cells are stained light blue in color with a larger nucleus as compared to normal brain and bone marrow cells. Control calvaria and femur sections were obtained from a mouse without bone metastases whereas sections of calvaria and femur with metastases show bone formation inside metastatic tumors. An inset of femur showing the osteoblastic bone formation in an area of tumor cells is also presented.
PacMetUT1 produces large amounts of TGFβ1
TGFβ has been shown to be a key mediator of metastasis to bone, lung and other organs [20, 27, 28]. TGFβ1 was found to be overexpressed in prostate cancer [29]. We compared secreted levels of TGFβ1 and TGFβ2 by PacMetUT1 cells with those by other prostate cancer cell lines using a sandwich ELISA. As shown in Fig.2A, PacMetUT1 cell produced copious amounts of active TGFβ1 as compared to other prostate cancer cell lines. Interestingly, either active or total TGFβ2 was not detected in the medium conditioned by PacMetUT1 cells. As such, PacMetUT1 is an appropriate prostate cancer model allowing us to investigate the role of TGFβ1 in prostate cancer-induced osteoblastic metastasis. Therefore, we examined the effect of knocking down TGFβ1 in the PacMetUT1 cell on its ability to form tumors and induce osteoblastic lesions in tibia. Stable knockdown of TGFβ1 in PacMetUT1 cells was achieved with lentiviral expression vectors containing two different TGFβ1 shRNA sequences. To confirm the knockdown of TGFβ1 expression and signaling in the transduced cells, two assays were performed. The secreted active and total TGFβ1 were found to be significantly reduced in the knockdown cells with either of the two TGFβ1 shRNA sequences as compared to the PacMetUT1 control cells (Fig.2B). The knockdown of TGFβ1 also significantly reduced autocrine TGFβ1 signaling as reflected by a significant reduction of a TGFβ-responsive promoter activity (Fig.2C).
Figure 2. Stable knockdown of TGFβ1 in PacMetUT1 cells decreases TGFβ1 secretion and autocrine TGFβ signaling activity.
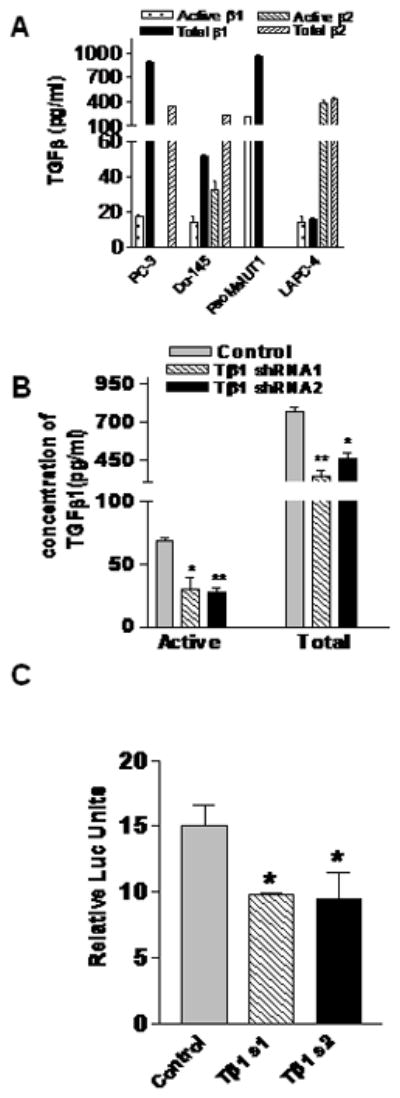
(A) Secretion of TGFβ1 & β2 was measured by different prostate cancer cells using an ELISA assay. Data presented are mean ± SEM from triplicate measurements. (B) PacMetUT1 cells were stably transfected with lentiviral expression vector plasmids, pLKO.1, containing TGFβ1-specific short hairpin RNA sequence 1 or 2. Knockdown of TGFβ1 in these cells was quantified using an ELISA assay. Data presented are mean ± SEM from triplicate measurements. Statistically significant difference between the control and TGFβ1 knockdown groups with a student’s t-test is indicated with asterisk “*” at P<0.002 and asterisk “**” at P<0.0006. (C) TGFβ1 knockdown in control and TGFβ1 knock down PacMetUT1 cells were assessed using pSBE4-Luc reporter assay. Cells were transiently transfected with pSBE4-Luc and a β-gal expression construct for 16 hours. β-gal normalized luciferase activity is presented as mean ± SEM from triplicate measurements. Asterisk “*” indicates statistically significant difference between the control and TGFβ1 knockdown groups with a student’s t-test at P<0.01.
TGFβ inhibition decreases Smad2 activation in PacMetUT1 cells
TGFβ action is mediated through the phosphorylation of intracellular Smad2 and Smad3 proteins thereby affecting gene transcription. To confirm that the knockdown or blockade of TGFβ signaling results in decreased Smad activation, we examined the level of Smad2 phosphorylation after TGFβ1 stable knockdown and also by using specific TGFβ inhibitors. The basal level of phosphorylated Smad2 protein was reduced to an undetectable level in the two TGFβ1 knockdown cells when compared to the control PacMetUT1 cells (Fig.3A) suggesting that TGFβ1 knockdown resulted in an inhibition of Smad2 protein activation in the knockdown tumor cells. Treatment with exogenous TGFβ1 restored phosphorylated Smad2 level in the two knockdown cells (Fig. 3A). But, its action was blocked by a TGFβ type I receptor kinase inhibitor, TβRI-KI (Fig. 3B), or a soluble chimeric TGFβ receptor, BGERII (Fig. 3C) demonstrating that the inhibitors can block TGFβ/Smad signaling.
Figure 3. Effect of TGFβ1 blockade on Smad activation in PacMetUT1 cells.
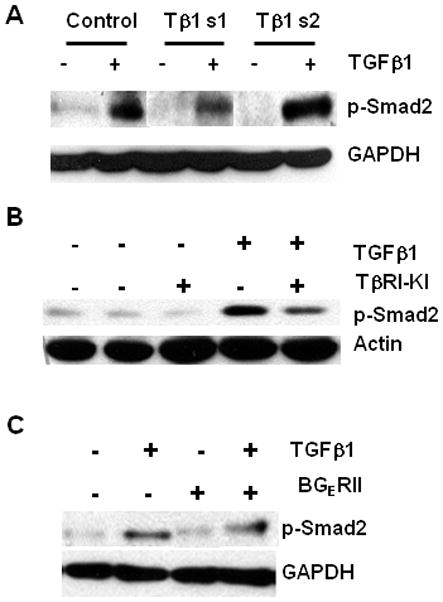
(A) Control and TGFβ1 knockdown PacMetUT1 cells were treated with or without TGFβ1 (5 ng/ml) for 45min. The media was changed to serum free a day before treatment. The cell lysates were used for Western analysis to measure the level of phosphorylated Smad2 (p-Smad2). GAPDH was used as a loading control. (B) PacMetUT1 cells were treated with TβRI-KI (100 nM) for 24h followed by treatment with or without TGFβ1 (2 ng/ml) for 45min. The cells were harvested and probed for p-Smad2 and actin using respective antibodies. The left two lanes refer to serum-free and vehicle-treated cells respectively. (C) PacMetUT1 cells were treated with BGERII (3 μg/ml) in presence or absence of TGFβ1 (2 ng/ml) for 45min. The cell lysates were probed for p-Smad2 and GAPDH with their respective antibodies.
Effect of TGFβ1 knockdown on anchorage-dependent and -independent growth of PacMetUT1 cells
TGFβ is known to inhibit proliferation of epithelial cells. However, many aggressive carcinoma cells are resistant to its growth inhibitory activity, resulting in a gain of proliferative advantage [30]. We examined the effect of knockdown of endogenous TGFβ1 in PacMetUT1 cells on the growth of cells in vitro. TGFβ1 knockdown cells showed a moderately lower growth rate on plastic during the early exponential growth phase compared to the control cells, but attained similar cell numbers when they reached saturation phase (Fig.4A). To compare the in vitro tumorigenic property of the control and knockdown cells, we also examined their ability to form colonies when suspended in soft agar. As shown in Fig.4B, knockdown of TGFβ1 in PacMetUT1 cells did not show significant effect on the colony formation in soft agar.
Figure 4. Effect of TGFβ1 knock down on the growth and in vitro tumorigenicity of PacMetUT1 cells.
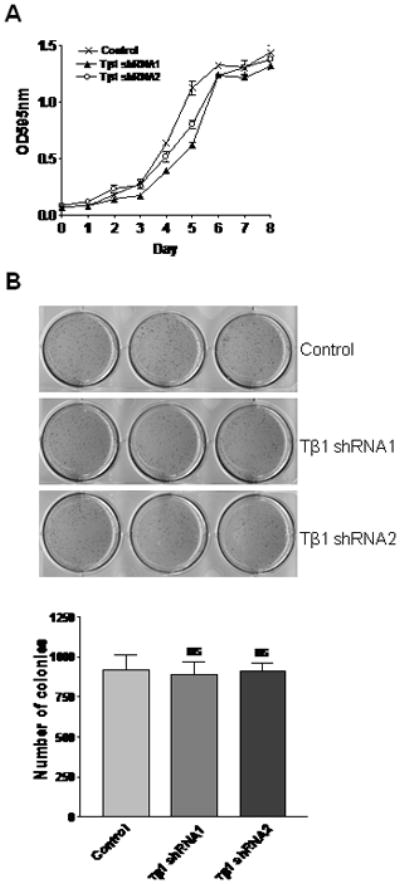
(A) Growth of control and TGFβ1 knock down PacMetUT1 cells was measured with MTT assay at an absorbance of 595 nm after every consecutive day for 8 days. Data presented are mean ± SEM from triplicate measurements. (B) Anchorage-independent growth by TGFβ1 knockdown PacMetUT1 cells was compared to control cells after 10 days of growth in soft agar. Figures presented are triplicates for each cell line. The number of colonies were counted and is presented as mean ± SEM from triplicate wells. “ns” denotes no significant difference with a one-way ANOVA analysis.
TGFβ1 knockdown decreases tumor incidence and osteoblastic lesions in vivo
To determine the effect of TGFβ1 on the tumorigenicity and bone remodeling, we injected the control and TGFβ1 knockdown PacMetUT1 cells into right tibias of five-week old male nude mice at 1×105 cells/mouse. Both TGFβ knockdown cells showed decreased ability to form tumors in the tibias of the mice with TGFβ1 shRNA2 group showing a statistically significant difference at P<0.05 as compared to the control group as detected by whole-body GFP imaging, x-ray radiography, and histology analysis (Table I). Thus, TGFβ1 signaling appears to be necessary for the tumorigenicity of PacMetUT1 cells in bone. Another observation we made was that the tumors formed by TGFβ1 knockdown cells induced less bone formation as detected by histologic staining (Fig.5A) and radiographic analysis (Fig.5B) of the right tibias. Our results show that TGFβ1 leads to osteoblastic lesion formation which is in contrast to what has been reported on the role of TGFβ on bone and TGFβ mediated osteolysis in breast carcinomas [26, 31]. Next, we quantitated the bone volume to total tissue volume in the H & E stained sections of tibias with tumor. The result also showed an inhibition of bone formation in the TGFβ1 knockdown cells as compared to the PacMetUT1 control cells (Fig.5C). Because TGFβ1 shRNA2 PacMetUT1 cells didn’t generate any tumors in mice (Table I), we didn’t include this group in the analyses. Therefore, our results indicate that TGFβ1 signaling in PacMetUT1 cells contributes to both tumorigenesis and osteoblastic lesion formation in the bone of nude mice.
Table I.
GFP imaging, radiographic and histologic analyses of tumor and bone formation incidence in vivo.
| Group | # GFP Imaging (11 wks) | §X-ray (11 wks) | % H&E staining (12 wks) |
|---|---|---|---|
| Control | 4/8 | 4/8 | 5/8 |
| Tβ1 shRNA1 | 2/8 | 2/8 | 2/8 |
| Tβ1 shRNA2 | *0/8 | *0/8 | *0/8 |
denotes engraftment of tumor cells to femur/tibia in mouse
denotes bone formation as detected by X-ray imaging in right tibias
denotes bone formation inside tumor area in stained tibia sections
denotes significantly different from the control at P<0.05 with a Fisher exact probability test
Figure 5. Decrease in tumor incidence and osteoblastic bone lesions by TGFβ1 knockdown in PacMetUT1 cells in vivo.
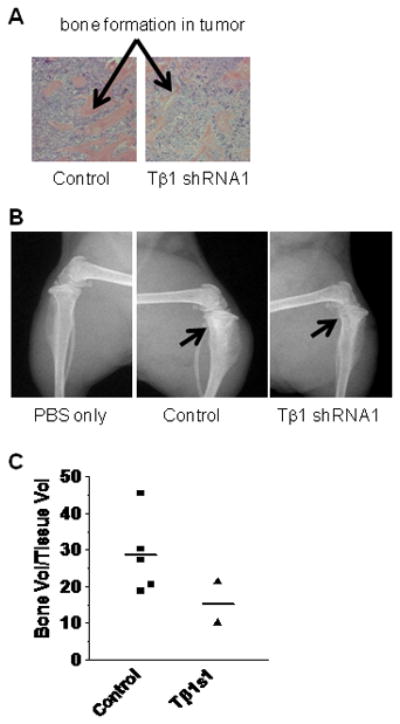
(A) Representative sections of H & E stained tibia are presented from mice inoculated with the control or TGFβ1 knockdown tumor cells. Arrow shows new bone formation inside tumor area. (B) Representative radiographs of tibia injected with PBS only, or control, or TGFβ1 knockdown PacMetUT1 cells. The arrow indicates where tumor cells were injected and new bone formation occurred. (C) Bone volume to total tissue volume was quantified in control and TGFβ1 knockdown groups using BIOQUANT analysis software. The line represents average value of bone volume to total tissue volume for each group.
Systemic administration of TGFβ inhibitors inhibits bone tumor growth and lung metastasis
As TGFβ1 produced by the PacMetUT1 cells contributes to osteoblastic tumor growth, we next investigated the effect of systemic treatment with TGFβ inhibitors on the ability of the tumor cells to induce osteoblastic tumor formation. The male nude mice were inoculated with PacMetUT1/Luc-GFP cells in the right tibias and treated with one of two TGFβ inhibitors, TβRI-KI or BGERII, for seven weeks. The TβRI-KI used in the current study was initially described as a highly specific inhibitor of TβRI [21, 22]. A dose of 20 μg/mouse of TβRI-KI was chosen for administration as it was shown to be effective, safe and non-toxic in our previous study [32]. We also used another potent pan-TGFβ inhibitor, BGERII, which we have previously shown to inhibit TGFβ signaling extracellularly and blocked the activity of all three TGFβ isoforms [23]. Our previous studies showed that systemic treatment with a soluble TβRIII (sRIII), which binds all three TGFβ isoforms, at 100 μg/mouse every other day was effective in inhibiting tumor progression in various models [33, 34]. Since the molecular weight of sRIII is approximately twice that of BGERII, we decided to use 50 μg/mouse of BGERII for the animal studies. TβRI-I (20 μg/mouse) and BGERII (50 μg/mouse) were administered i.p. every other day for seven weeks. The control mice were injected i.p. with PBS. Because the PacMetUT1 cells used in this study was labeled with luciferase and GFP, we were able to assess bone tumor burden and the number of lung metastatic colonies with bioluminescence and fluorescence imaging analyses respectively. Treatment with TβRI-KI or BGERII initially significantly inhibited tumor growth in the tibias as reflected by total photon flux (Fig. 6A and 6B). The inhibition eventually became statistically insignificant at week 7 when the experiment was terminated. In agreement with the terminal tibia tumor burden obtained with bioluminescence imaging analysis, histomorphometric analysis also revealed a moderately lower tumor burden, though not statistically significant, in the groups treated with TGFβ inhibitors in comparison with the control group (Fig.6C). Interestingly, we observed lung metastasis with bioluminescence imaging and green micrometastases in the excised lungs with green fluorescence imaging suggesting that the intratibia tumors led to lung metastasis. Although the tumor burden in the tibias of the TGFβ inhibitor-treated groups was only moderately lower than that of the control group at the termination of the experiment (Fig.6C), the treatment with TGFβ inhibitors markedly reduced the number of lung metastatic colonies as shown in Fig.6D. The treatment with BGERII prevented lung metastasis in seven out of nine mice, which had tibia tumors whereas all eight control mice that had tibia tumors developed lung metastasis (Fig.6D). The difference between the control and RI-KI groups was not statistically significant because of one mouse in the RI-KI treatment group that showed a very large number of lung colonies. Thus, our results indicate that systemic administration of TGFβ inhibitors can inhibit bone tumor growth and reduce lung metastasis of PacMetUT1 cells.
Figure 6. Decrease of bone tumor growth and lung metastasis with the systemic administration of TGFβ inhibitors in vivo.
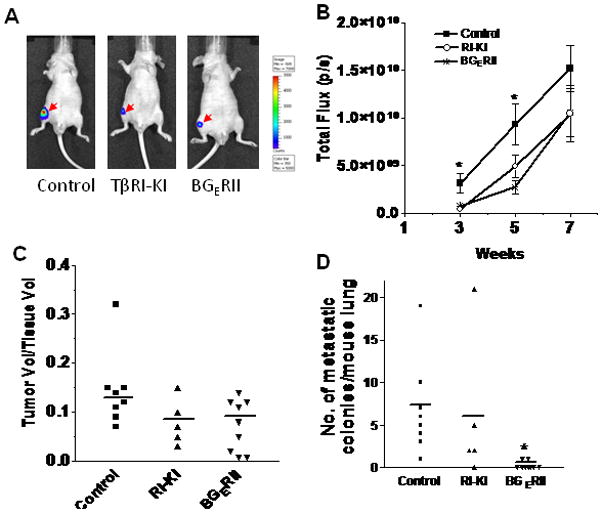
Exponentially growing, PacMetUT1/Luc-GFP cells were injected into the right tibias at 1×105 cells per mouse. The mice were treated on the same day with TβRI-KI at 20 μg/mouse and with BGERII at 50 μg/mouse every other day for seven weeks. (A) Representative whole-body bioluminescence imaging for control and treatment groups shows a decreased tumor growth in tibias for the TβRI-KI and BGERII treated mouse after three weeks of treatment. (B) Total photon flux for each of the three groups was taken at 3, 5 and 7 weeks after tumor cell inoculation. Data presented are mean ± SEM from each group. Asterisk “*” indicates statistically significant difference between the control and TβRI-KI and BGERII inhibitor treatment groups with one-way ANOVA followed with Tukey’s post-hoc test at P<0.05. (C) Tumor volume to tissue volume ratio was quantified in control, TβRI-KI, and BGERII treatment groups using BIOQUANT analysis software. The line represents average value of tumor volume to total tissue volume for each group. (D) After the termination of experiment at seven weeks, lungs were excised and metastatic cancer cell colonies were visually counted in the whole lungs of mice from each of the three groups. The line represents average number of colonies in each group. Asterisk “*” indicates statistically significant difference between the control and BGERII treatment groups with a student’s t-test at P<0.05.
Systemic administration of TGFβ inhibitors inhibits osteoblastic bone lesions
In addition to the inhibition of bone tumor growth, the systemic treatment with TGFβ inhibitors also significantly inhibited PacMetUT1-induced osteoblastic bone formation as detected with histomorphometry analysis (Fig.7A) confirming that TGFβ contributes to the bone formation in this model system. TGFβ is an important functional modulator of osteoclasts and attenuation of TGFβ activity can reduce mammary tumor-induced osteolysis [9]. As such, the inhibition of bone formation by TGFβ inhibitors is not likely due to an increase in osteoclasts. To rule out this possibility, TRAP (tartarate resistant acid phosphatase) staining for osteoclasts in the tibia sections was performed. Osteoclasts were mostly observed below the growth plate in the non tumor bearing left tibias that were only injected with PBS. However, they were found near the area where new trabecular bones were forming in the tibias with tumor (Fig. 7B). The treatment with TGFβ inhibitors led to a decrease in osteoclast counts when they were compared with tumor bearing tibias treated with PBS only (Fig.7C). These results rules out the involvement of osteoclasts in the inhibition of PacMetUT1-induced bone formation by TGFβ inhibitors.
Figure 7. Decrease in osteoblastic bone lesions with the systemic administration of TGFβ inhibitors in vivo.
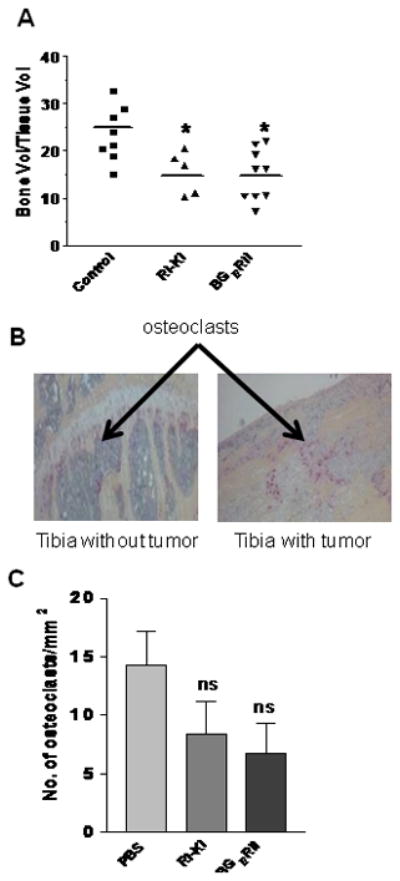
(A) Bone volume to total tissue volume in tibia sections was quantified in control, TβRI-KI, and BGERII treatment groups using BIOQUANT analysis software. The line represents average value of bone volume to total tissue volume in the each group. Asterisk “*” indicates statistically significant difference among the control, TβRI-KI, and BGERII inhibitor treatment groups with one-way ANOVA followed with Tukey’s post-hoc test at P<0.01. (B) After the termination of experiment at seven weeks, bone sections from tibias without (injected with PBS) or with PacMetUT1 cell-formed tumors were stained for osteoclasts using TRAP assay. Representative TRAP stained bone sections showing presence of the stained osteoclasts are presented. (C) The TRAP-stained osteoclasts below the growth plate were counted in the sections of the tibias containing tumors from the three groups of mice treated with PBS, TβRI- KI, or BGERII. Data presented are mean ± SEM from all tibias with tumors in each group (8 from control, 5 from RI-KI group, and 9 from BGERII group). “ns” denotes no significant difference.
Discussion
Prostate cancer metastasizes to bone in more than 80% of patients suffering from advanced disease [2]. Unlike breast and lung cancer, the nature of bone lesions is predominantly osteoblastic resulting in hypocalcaemia, pain, bone fractures and nerve compression syndromes. We have used a novel human prostate cancer cell line, PacMetUT1, in our study. Our results indicate that the PacMetUT1 cells have the potential for skeletal metastasis and bone formation in a nude mice model after intracardiac inoculation. To elucidate the molecular mechanism that mediates PacMetUT1-induced bone formation, we investigated the role of TGFβ signaling because PacMetUT1 secretes large amounts of TGFβ1. TGFβ regulates a broad range of biological processes, including cell proliferation, survival, differentiation, migration, and production of extracellular matrix (ECM). However, TGFβ acts as a double-edged sword during tumor progression by inhibiting cell proliferation during early stage and stimulating metastasis in the later stages [35]. TGFβ suppresses cancer progression by stimulating cyclin-dependent kinase inhibitors through its canonical signaling pathway involving Smad-2/3 proteins. We observed a diminished Smad2 protein activation after TGFβ1 knockdown suggesting that the downstream Smad signaling pathway is getting altered after TGFβ blockade in PacMetUT1 cells. However, we observed a moderate decrease in cell proliferation after knocking down TGFβ1 in PacMetUT1 cells in vitro. On the other hand, TGFβ1 knockdown did not affect the tumorigenicity of PacMetUT1 cells in vitro as shown in our soft agar clonogenic assay. As such, its signaling in the cells does not appear to confer tumor suppression.
TGFβ is secreted in the bone matrix by osteoblasts and is a critical regulator of osteogenesis. TGFβ plays a key role in osteoblast differentiation, bone development and remodeling. It has been shown to stimulate proliferation and early osteoblast differentiation, while inhibiting terminal differentiation [6]. TβR1 inhibitors were shown to increase osteoblast differentiation and bone formation while reducing osteoclast differentiation and bone resorption in the post-natal skeleton [26]. TGFβ released during osteolytic resorption from bone matrix has been shown to stimulate osteolytic metastasis and osteoclast activation in breast cancer by activating bone resorbing cytokines [36]. But the role of TGFβ signaling in prostate cancer-induced bone metastatic lesions is not very clear in part due to a lack of model system which can mimic osteoblastic bone metastasis process. In this study, we have evaluated the role of TGFβ pathway in osteoblastic bone tumor formation by PacMetUT1 cells. Because of the low incidence of bone metastasis in intracardiac injection, we used direct intra-tibia injection route for studying the effect of TGFβ signaling abrogation on tumor growth in the tibias of nude mice. We have observed a decrease in tumor incidence and osteoblastic lesions in vivo in the TGFβ1 knockdown group as compared to the control group. Systemic administration of the TβRI-KI and a pan-TGFβ inhibitor BGERII also resulted in a decreased bone tumor growth. This result is in contrast to what has previously been observed with TGFβ mediated osteolysis in breast cancer model system. However, a recent report shows that TGFβ1 induces osteogenic differentiation of murine bone marrow stromal cells by increasing osteoblast differentiation markers (Runx-2, osteopontin, collagen-1) as well as alkaline phosphatase activity as compared to control cells [37]. The decreased osteoblastic bone formation by inhibition of TGFβ signaling in tumor cells may be because of reduced expression of osteoblast specific factors like alkaline phosphatase, collagen, osteonectin and osteopontin [38, 39]. TGFβ1 after being activated by prostate specific antigen (PSA), which is an osteoblast stimulating factor, can result in an increase in mature osteoblasts and a decrease in osteoclast population [40, 41]. However, PacMetUT1 cells do not express PSA, suggesting that the increase in bone formation was mediated by a PSA-independent mechanism. Bone cells such as osteoblasts express aromatase, which is the enzyme required for estrogen biosynthesis in humans [42, 43]. TGFβ1 was shown to stimulate expression of the aromatase gene in osteoblast-like cells [44]. The autocrine or paracrine secreted TGFβ1 from PacMetUT1 cells might result in an increased aromatase gene expression and activity leading to an enhanced estradiol synthesis which would in turn lead to an increase in bone formation as estrogen is a known osteogenic factor.
TGFβ has been shown to be a potential modifier of receptor activator of NF-kappaB ligand (RANK)-dependent osteoclast activation and osteolysis in breast cancer [36]. TGFβ can also activate osteoclastic differentiation and activity [26]. In our study, we observed a decrease in osteoblastic bone lesions with a moderate decrease in osteoclast number by the systemic administration of TGFβ signaling inhibitors in the nude mice. This rules out the possibility that the decreased bone formation is due to an increase in osteoclast number. We also observed that in the tumor-containing tibias, osteoclasts migrated to the area where new bone formation was taking place, which is consistent with the dogma that new bone formation occurs in the area of osteolytic resorption [45]. Further studies are required to reveal the mechanism of TGFβ mediated increase in bone formation in PacMetUT1 cells.
Pulmonary metastasis commonly develops after bone metastasis of prostate cancer. Few patients with prostate cancer present initially with symptomatic metastatic lung lesions without any other concomitant distant dissemination [46]. TGFβ has been shown to cause lung metastasis in several animal models of breast cancer [34, 47]. In our study, we have observed that systemic administration of TGFβ inhibitors, TβRI-KI and BGERII, reduced the number of lung metastatic colonies from intratibia PacMetUT1 tumors. One possible mechanism for the reduced incidence of bone tumor-derived lung metastasis by TGFβ inhibitors may be because of the inhibition of TGFβ-induced migration of tumor cells in the bone marrow, similar to the TGFβ-induced extravasation in the pulmonary metastasis model of breast cancer [48].
In summary, our study revealed the predominant osteoblastic nature of a novel human prostate cancer cell line, PacMetUT1. Our results indicated for the first time that abrogation of TGFβ signaling by the systemic administration of TβRI-KI and BGERII or by knockdown of TGFβ1 with shRNA’s can effectively inhibit the tumor incidence and osteoblastic lesion formation by this novel prostate cancer model in the bone microenvironment. Our long-term goal is to determine whether TGFβ antagonists alone or in combinatorial therapy will be suitable as therapeutic agents for the treatment and prevention of prostate cancer-induced bone metastasis.
Acknowledgments
Financial Support: This work was supported in part by NIH Grants R01CA079683 and R01CA075253 to LZS, R01CA104505 to JAC, and the Cancer Therapy and Research Center at the University of Texas Health Science Center at San Antonio through the NCI Cancer Center Support Grant 2 P30 CA054174-17.
The authors thank the institutional cores of FACS, Optical Imaging, and Pathology for their assistance in obtaining part of the presented data. The authors also thank Dr. Brian Rabinovich at MD Anderson Cancer Center for the pLV411G effLuc-flag-IRES-hrGFP vector.
Reference List
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 3.Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–784. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 4.Roudier MP, Morrissey C, True LD, Higano CS, Vessella RL, Ott SM. Histopathological assessment of prostate cancer bone osteoblastic metastases. J Urol. 2008;180:1154–1160. doi: 10.1016/j.juro.2008.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye L, Kynaston HG, Jiang WG. Bone metastasis in prostate cancer: molecular and cellular mechanisms (Review) Int J Mol Med. 2007;20:103–111. [PubMed] [Google Scholar]
- 6.Mundy GR, Bonewald LF. Role of TGF beta in bone remodeling. Ann N Y Acad Sci. 1990;593:91–97. doi: 10.1111/j.1749-6632.1990.tb16102.x. [DOI] [PubMed] [Google Scholar]
- 7.Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, Yoneda T, Mundy GR. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 9.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massague J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filvaroff E, Erlebacher A, Ye J, Gitelman SE, Lotz J, Heillman M, Derynck R. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development. 1999;126:4267–4279. doi: 10.1242/dev.126.19.4267. [DOI] [PubMed] [Google Scholar]
- 11.Sanders JL, Chattopadhyay N, Kifor O, Yamaguchi T, Brown EM. Ca(2+)-sensing receptor expression and PTHrP secretion in PC-3 human prostate cancer cells. Am J Physiol Endocrinol Metab. 2001;281:E1267–E1274. doi: 10.1152/ajpendo.2001.281.6.E1267. [DOI] [PubMed] [Google Scholar]
- 12.Brown JM, Vessella RL, Kostenuik PJ, Dunstan CR, Lange PH, Corey E. Serum osteoprotegerin levels are increased in patients with advanced prostate cancer. Clin Cancer Res. 2001;7:2977–2983. [PubMed] [Google Scholar]
- 13.Thirunavukkarasu K, Miles RR, Halladay DL, Yang X, Galvin RJ, Chandrasekhar S, Martin TJ, Onyia JE. Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-beta (TGF-beta). Mapping of the OPG promoter region that mediates TGF-beta effects. J Biol Chem. 2001;276:36241–36250. doi: 10.1074/jbc.M104319200. [DOI] [PubMed] [Google Scholar]
- 14.Wu TT, Sikes RA, Cui Q, Thalmann GN, Kao C, Murphy CF, Yang H, Zhau HE, Balian G, Chung LW. Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer. 1998;77:887–894. doi: 10.1002/(sici)1097-0215(19980911)77:6<887::aid-ijc15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 15.Corey E, Quinn JE, Bladou F, Brown LG, Roudier MP, Brown JM, Buhler KR, Vessella RL. Establishment and characterization of osseous prostate cancer models: intra-tibial injection of human prostate cancer cells. Prostate. 2002;52:20–33. doi: 10.1002/pros.10091. [DOI] [PubMed] [Google Scholar]
- 16.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 17.Lahn M, Kloeker S, Berry BS. TGF-beta inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2005;14:629–643. doi: 10.1517/13543784.14.6.629. [DOI] [PubMed] [Google Scholar]
- 18.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 19.Singh J, Ling LE, Sawyer JS, Lee WC, Zhang F, Yingling JM. Transforming the TGFbeta pathway: convergence of distinct lead generation strategies on a novel kinase pharmacophore for TbetaRI (ALK5) Curr Opin Drug Discov Devel. 2004;7:437–445. [PubMed] [Google Scholar]
- 20.Troyer DA, Tang Y, Bedolla R, Adhvaryu SG, Thompson IM, bboud-Werner S, Sun LZ, Friedrichs WE, deGraffenried LA. Characterization of PacMetUT1, a recently isolated human prostate cancer cell line. Prostate. 2008;68:883–892. doi: 10.1002/pros.20758. [DOI] [PubMed] [Google Scholar]
- 21.Sawyer JS, Anderson BD, Beight DW, Campbell RM, Jones ML, Herron DK, Lampe JW, McCowan JR, McMillen WT, Mort N, Parsons S, Smith EC, Vieth M, Weir LC, Yan L, Zhang F, Yingling JM. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46:3953–3956. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- 22.Singh J, Chuaqui CE, Boriack-Sjodin PA, Lee WC, Pontz T, Corbley MJ, Cheung HK, Arduini RM, Mead JN, Newman MN, Papadatos JL, Bowes S, Josiah S, Ling LE. Successful shape-based virtual screening: the discovery of a potent inhibitor of the type I TGFbeta receptor kinase (TbetaRI) Bioorg Med Chem Lett. 2003;13:4355–4359. doi: 10.1016/j.bmcl.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 23.Verona EV, Tang Y, Millstead TK, Hinck AP, Agyin JK, Sun LZ. Expression, purification and characterization of BG(E)RII: a novel pan-TGFbeta inhibitor. Protein Eng Des Sel. 2008;21:463–473. doi: 10.1093/protein/gzn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, Cornell JE, Pollock BH, Mundy GR, Sun LZ. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714–6721. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 25.Arguello F, Baggs RB, Frantz CN. A murine model of experimental metastasis to bone and bone marrow. Cancer Res. 1988;48:6876–6881. [PubMed] [Google Scholar]
- 26.Mohammad KS, Chen CG, Balooch G, Stebbins E, McKenna CR, Davis H, Niewolna M, Peng XH, Nguyen DH, Ionova-Martin SS, Bracey JW, Hogue WR, Wong DH, Ritchie RO, Suva LJ, Derynck R, Guise TA, Alliston T. Pharmacologic inhibition of the TGF-beta type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS One. 2009;4:e5275. doi: 10.1371/journal.pone.0005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 28.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 29.Steiner MS. Transforming growth factor-beta and prostate cancer. World J Urol. 1995;13:329–336. doi: 10.1007/BF00191213. [DOI] [PubMed] [Google Scholar]
- 30.Sun L. Tumor-suppressive and promoting function of transforming growth factor beta. Front Biosci. 2004;9:1925–1935. doi: 10.2741/1382. [DOI] [PubMed] [Google Scholar]
- 31.Guise TA. Molecular mechanisms of osteolytic bone metastases. Cancer. 2000;88:2892–2898. doi: 10.1002/1097-0142(20000615)88:12+<2892::aid-cncr2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 32.Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, Cornell JE, Pollock BH, Mundy GR, Sun LZ. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714–6721. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 33.Bandyopadhyay A, Wang L, Lopez-Casillas F, Mendoza V, Yeh IT, Sun L. Systemic administration of a soluble betaglycan suppresses tumor growth, angiogenesis, and matrix metalloproteinase-9 expression in a human xenograft model of prostate cancer. Prostate. 2005;63:81–90. doi: 10.1002/pros.20166. [DOI] [PubMed] [Google Scholar]
- 34.Bandyopadhyay A, Lopez-Casillas F, Malik SN, Montiel JL, Mendoza V, Yang J, Sun LZ. Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res. 2002;62:4690–4695. [PubMed] [Google Scholar]
- 35.Bachman KE, Park BH. Duel nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- 36.Futakuchi M, Nannuru KC, Varney ML, Sadanandam A, Nakao K, Asai K, Shirai T, Sato SY, Singh RK. Transforming growth factor-beta signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci. 2009;100:71–81. doi: 10.1111/j.1349-7006.2008.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L, Jiang S, Hantash BM. TGF-beta1 Induces Osteogenic Differentiation of Murine Bone Marrow Stromal Cells. Tissue Eng Part A. 2009 doi: 10.1089/ten.TEA.2009.0495. [DOI] [PubMed] [Google Scholar]
- 38.Noda M, Yoon K, Prince CW, Butler WT, Rodan GA. Transcriptional regulation of osteopontin production in rat osteosarcoma cells by type beta transforming growth factor. J Biol Chem. 1988;263:13916–13921. [PubMed] [Google Scholar]
- 39.Pfeilschifter J, D’Souza SM, Mundy GR. Effects of transforming growth factor-beta on osteoblastic osteosarcoma cells. Endocrinology. 1987;121:212–218. doi: 10.1210/endo-121-1-212. [DOI] [PubMed] [Google Scholar]
- 40.Goya M, Ishii G, Miyamoto S, Hasebe T, Nagai K, Yonou H, Hatano T, Ogawa Y, Ochiai A. Prostate-specific antigen induces apoptosis of osteoclast precursors: potential role in osteoblastic bone metastases of prostate cancer. Prostate. 2006;66:1573–1584. doi: 10.1002/pros.20375. [DOI] [PubMed] [Google Scholar]
- 41.Yonou H, Ogawa Y, Ochiai A. Mechanism of osteoblastic bone metastasis of prostate cancer. Clin Calcium. 2006;16:557–564. [PubMed] [Google Scholar]
- 42.Nawata H, Tanaka S, Tanaka S, Takayanagi R, Sakai Y, Yanase T, Ikuyama S, Haji M. Aromatase in bone cell: association with osteoporosis in postmenopausal women. J Steroid Biochem Mol Biol. 1995;53:165–174. doi: 10.1016/0960-0760(95)00031-t. [DOI] [PubMed] [Google Scholar]
- 43.Sasano H, Uzuki M, Sawai T, Nagura H, Matsunaga G, Kashimoto O, Harada N. Aromatase in human bone tissue. J Bone Miner Res. 1997;12:1416–1423. doi: 10.1359/jbmr.1997.12.9.1416. [DOI] [PubMed] [Google Scholar]
- 44.Shozu M, Zhao Y, Simpson ER. TGF-beta1 stimulates expression of the aromatase (CYP19) gene in human osteoblast-like cells and THP-1 cells. Mol Cell Endocrinol. 2000;160:123–133. doi: 10.1016/s0303-7207(99)00233-6. [DOI] [PubMed] [Google Scholar]
- 45.Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005;15:57–62. doi: 10.1038/sj.cr.7290266. [DOI] [PubMed] [Google Scholar]
- 46.Tohfe M, Baki SA, Saliba W, Ghandour F, Ashou R, Ghazal G, Bahous J, Chamseddine N. Metastatic prostate adenocarcinoma presenting with pulmonary symptoms: a case report and review of the literature. Cases J. 2008;1:316. doi: 10.1186/1757-1626-1-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tobin SW, Douville K, Benbow U, Brinckerhoff CE, Memoli VA, Arrick BA. Consequences of altered TGF-beta expression and responsiveness in breast cancer: evidence for autocrine and paracrine effects. Oncogene. 2002;21:108–118. doi: 10.1038/sj.onc.1205026. [DOI] [PubMed] [Google Scholar]
- 48.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]