

Abstract
Inflammation contributes to neurodegeneration in post-ischemic brain, diabetes, and Alzheimer’s disease. Participants to this inflammatory response include activation of microglia and astrocytes. We studied the role of microglia treated with amyloid-β peptide (Aβ) on hemichannel activity of astrocytes subjected to hypoxia in high glucose. Reoxygenation after 3 h hypoxia in high glucose induced transient astroglial permeabilization and reduction in intercellular communication via Cx43 based channels. Both responses were greater and longer lasting in astrocytes previously exposed for 24 h to conditioned medium from Aβ-treated microglia (CM-Aβ). The effects of CM-Aβ were mimicked by TNF-α and IL-1β and were abrogated by neutralizing TNF-α with soluble receptor and IL-1β with a receptor antagonist. Astrocytes under basal conditions protected neurons against hypoxia, but exposure to CM-Aβ made them toxic to neurons subjected to a sub-lethal hypoxia/reoxygenation episode, revealing the additive nature of the insults. Astrocytes exposed to CM-Aβ induce permeabilization of cortical neurons through activation of neuronal pannexin 1 (Panx1) hemichannels by ATP and glutamate released through astroglial Cx43 hemichannels. In agreement, inhibition of NMDA or P2X receptors only partially reduced the activation of neuronal Panx1 hemichannels and neuronal mortality but simultaneous inhibition of both receptors completely prevented the neurotoxic response. Therefore, we suggest that exitotoxicity triggered by ATP and glutamate converges in activation of neuronal Panx1 hemichannels. Thus, we propose that blocking hemichannels expressed by astrocytes and/or neurons in the inflamed nervous system could represent a novel and alternative strategy to reduce neuronal loss.
Keywords: pannexin, connexin, Alzheimer’s disease, diabetes mellitus, gap junctions, stroke, amyloid β-peptide, cytokines
Introduction
The most common acute brain insult is ischemic stroke, where transient or permanent reduction in cerebral blood flow deprives the tissue of oxygen and glucose and permits buildup of potentially toxic substances, effects that together lead to rapid or delayed cell death (Dirnagl et al. 1999). An association between Alzheimer’s disease (AD) and ischemic stroke has been established. Indeed, patients that on autopsy show cerebral infarcts and AD pathology are more cognitively impaired than patients with AD pathology alone (Snowdon et al. 1997). Moreover, the presence of high levels of a neurotoxic fragment of amyloid β-peptide (Aβ25-35) in a model of focal cerebral ischemia is associated with increased infarct size, greater inflammation, and more pronounced cognitive deficits (Whitehead et al. 2007).
It has long been known that hyperglycemia worsens the outcome of acute brain ischemia by increasing the extent of tissue injury in animals and in humans (Kagansky et al. 2001). Interestingly, both AD and hyperglycemic conditions developed during diabetes mellitus (DM) produce persistent inflammation that can cause neuronal death (LaFerla et al. 2007, Pasquier et al. 2006). More relevant to this point is that DM accelerates memory dysfunction via cerebrovascular inflammation and Aβ deposition in an AD transgenic mouse (Takeda et al. 2010). Thus, inflammation seems to be a common factor in the neuronal damage induced by stroke, AD and DM. However, the molecular and/or cellular targets involved in these processes remain to be elucidated.
Microglia are the most sensitive central nervous system (CNS) detectors of adverse conditions, including fibrillar Aβ depositions (Block et al. 2007). Interestingly, upon stimulation with LPS, microglial cells release TNF-α and IL-1β which reduce intercellular communication via gap junctions and increase hemichannel activity in astrocytes (Retamal et al. 2007). More prominent changes are observed in astrocytes exposed to hypoxia in high glucose (Orellana et al. 2010). Gap junctions are membrane specializations that provide a direct cytoplasmic pathway between contacting cells by aggregates that contain a few tens to thousands of cell-cell channels, termed gap junction channels (Sáez et al. 2003). They are formed by the docking of two hemichannels, contributed by each contacting cell (Sáez et al. 2003). Each hemichannel is formed by oligomerization of six protein subunits termed connexins (Cxs), which are expressed by astrocytes, microglia and neurons (Orellana et al. 2009). A more recently described three-member protein family, termed pannexins (Panxs), can also form hemichannels at the cell surface of diverse mammalian cells, including astrocytes and neurons (Iglesias et al. 2009, Thompson et al. 2008). They are permeable to ATP and are activated by intracellular Ca2+ and extracellular ATP acting on P2 receptors (Pelegrin & Surprenant 2006, Locovei et al. 2006).
It has been proposed that under pathological conditions the increased hemichannel opening and reduced gap junctional communication in astrocytes deprive neurons of glial protective functions, which could increase neuronal vulnerability and the incidence of neuronal death (Orellana et al. 2009). In agreement with this notion, it was recently demonstrated that astroglial hemichannel opening induced by pro-inflammatory cytokines potentiate glutamate-induced neurotoxicity (Froger et al. 2010). However, soluble factors released by activated astrocytes that enhance neuronal vulnerability to injuries remain to be identified. The aim of this study was to evaluate if changes in hemichannels and/or gap junction channels of cultured cortical astrocytes exposed to sub-threshold pro-inflammatory conditions in vitro are potentiated. In addition, the impact of changes in astroglial hemichannels/gap junction channels and neuronal hemichannels in the viability of cortical neurons was evaluated, while both glutamate and ATP released via astroglial hemichannels were identified as relevant mediators of the observed neuronal death.
Materials and methods
Reagents and antibodies
SuperSignal kit for enhanced chemiluminescence (ECL) detection, Sulfo-NHS-SS-biotin, and immobilized NeutrAvidin were purchased from Pierce. Gap26 (VCYDKSFPISHVR, first extracellular loop domain of Cx43, Cx32 and Cx26), Gap27 (SRPTEKTIFII, second extracellular loop domain of Cx43, Cx37, Cx32 and Cx26), 10panx1 (WRQAAFVDSY, extracellular loop domain of Panx1), and E1b (SSFSWRQAAFVDS, extracellular loop domain of Panx1) peptides were obtained from NeoMPS, SA. (Straousburg, France). Aβ25-35 and Aβ35-25 peptide were purchased from Bachem (King of Prussia, PA). HEPES, DMEM, H2O, LaCl3 (La3+), ethidium (Etd) bromide, Lucifer yellow (LY), cytosine arabinoside (Ara-C), glutamate, 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP), oATP, suramin, brillian blue G (BBG), apyrase, ATP, anti-MAP2 monoclonal antibody H-M2 and probenecid were purchased from Sigma-Aldrich (St. Louis, MO, USA). Penicillin, streptomycin, isolectin GS-IB4 and goat anti-mouse Alexa Fluor 488 were obtained from Invitrogen (Carlsbad, CA). TNF-α and IL-1β were obtained from Roche Diagnostics (Indianapolis, MI). Proteinase K was purchased from Promega (Madison, WI, USA). Anti-GFAP monoclonal antibody was purchased from ICN Chemicals, (Irvine, CA). Anti-Cx43 monoclonal antibody was obtained from BD Biosciences (Franklin Lakes, NJ). A soluble form of the TNF-α receptor (sTNF-aR1) and a recombinant receptor antagonist for IL-1β (IL-1ra) were from R&D Systems (Minneapolis, MN). Cx43E2 antibody specifically for blocking hemichannels was generated and affinity purified as previously described (Siller-Jackson et al. 2008).
Animals
Microglia, neuron and astrocyte cultures were prepared from OF1 mice (Charles River, L’Arbresle, France). In addition, Cx43-deficient astrocytes were obtained from neonatal mice born to Cx43+/− female mice (Reaume et al. 1995). Homozygous mutant (Cx43−/−) and their wild-type control (Cx43+/+) neonatal mice were the product of mating between heterozygous Cx43+/− mice. Genotyping was performed from a tissue sample, using PCR analysis, as previously described (Naus et al. 1997). All experiments were carried out in accordance with the European Community Council Directives of November 24, 1986 (86/609/EEC) and all efforts were made to minimize the number of animals used and their suffering.
Cell cultures
Astrocyte cultures
Primary astrocyte cultures were prepared from the cortex of newborn OF1 mice. Briefly, the brains were removed, and the cortices were dissected. Meninges were carefully peeled off and tissue was mechanically dissociated. Cells were seeded into 100-mm-diameter plastic dishes (Nunc, Roskilde, Denmark) at a density of 3 × 106 cells/dish or into 60 mm diameter plastic dishes at a density of 2 × 106 cells/dish in DMEM, supplemented with penicillin (5 U/ml), streptomycin (5 μg/ml), and 10% FCS. Alternatively, cells were seeded on glass coverslips (Gassalem, Limeil-Brevannes, France) placed inside 16 mm diameter 24-well plastic plates (NunClon) at the density of 1 × 105 cells/well in the same culture conditions. After 8–10 days, when cells had reached confluence, 1 μM of cytosine-arabinoside was added to the culture medium for 3 days to eliminate proliferating microglial cells. At that stage, these cultures contained >95% GFAP+ cells and >95% S100β+ cells. No neurons were detected as judged by MAP2 staining. At the end of these experiments cell cultures were stained with DAPI to quantify the total number of astrocytes per culture.
Cx43−/− and Cx43+/+ astrocyte cultures
Cx43−/− and Cx43+/+ astrocyte cultures were prepared from the cortex of Cx43−/− and WT mice, as described for OF1 mice. The mouse genotype was determined by PCR analysis as described previously (Naus et al. 1997).
Microglial cultures, astrocyte-microglia co-cultures, and conditioned media
After dissociation, astroglial cells were seeded into 100-mm-diameter culture dishes (NunClon) at 3 × 106 cells/10 ml/dish in DMEM, containing 10% heat-inactivated FCS (Abcys, Paris, France). The medium was changed at 1 and 3 DIV, and microglia were collected at 10 DIV by shaking the culture dishes to detach cells adherent to the astrocyte monolayer. The collected population resulted in > 98% of cells bearing the Mac-1 antigen, a specific marker of macrophage cells. Freshly collected microglia were either seeded on confluent astrocytes (astrocyte-microglia co-cultures, 3 × 104 cells/16mm wells) or cultured to generate conditioned medium (CM). Co-cultures were maintained for 24 h in DMEM containing 5% FCS and then treated (or not for control) for another 24 h. Immunostaining with astrocyte and microglia markers (GFAP and isolectin B4, respectively) indicated that astroglial cultures contained 98.9 ± 0.2 astrocytes (GFAP-positive) and 1.1 ± 0.1% microglia (isolectin B4-positive) (n = 3), whereas astrocyte-microglia co-cultures contained 81.5 ± 0.1% astrocytes and 18.5 ± 0.5% microglia (n=3).
To obtain CM from microglia, freshly collected microglia were seeded in DMEM containing 5% FCS (1.7 × 106 cells/ml/dish in 35 mm dishes) and treated with 10 μM Aβ25-35 (CM-Aβ25-35) for 24 h. CM of non-activated microglia was obtained from sister cultures, and effects of activated and non-activated CM on astrocyte cultures were compared. In addition, to obtain CM from astrocytes (CM-Ast), astrocytes were treated for 24 h with CM-Aβ25-35 and then exposed to 3 h of hypoxia in fresh medium containing 5 mM glucose followed by 1 h of reoxygenation in fresh medium. The final supernatants from treated microglia and astrocytes were collected, filtered (0.22 μm), and stored at −20°C before use.
Neurons and astrocyte-neuron co-cultures
Neuron-astrocyte co-cultures were obtained by plating cell suspensions dissociated from E16 mice cerebral cortex (5 × 104 cells/coverslip) on 3 week old astrocyte monolayer in MEM containing 5% horse serum and 5% FCS. After 24 h, the medium was replaced by one containing 2 × 10−5 M 5′-fluoro-2-deoxyuridine + uridine (10−5 M), insulin (5 μg/mL), pyruvate (110 μg/mL), 5% horse serum and 1% Ultroser-G, a serum substitute (Pall-Biosepra). Partial medium changes (1/4) were done twice a week. In this medium, astrocytes are healthy, neuronal cells differentiate, and potentially dividing cells (neural progenitors and/or microglial cells) are killed due to the continuous presence of an anti-mitotic agent. Enriched neuronal cultures were switched to the co-culture medium and submitted to the same medium changes as their sister co-cultures.
Cell treatments
Some astrocyte-microglia co-cultures were treated for 24 h with 10 μM Aβ25-35 and then used for experiments. Astrocyte or neuron cultures and astrocyte-neuron co-cultures were treated with either CM-Aβ25-35 (diluted four times at the final concentration) or the mixture of cytokines TNF-α plus IL-1β (10 pg/ml of each) for 24 h and then exposed to an in vitro hypoxia model in the presence of normal or high glucose. Briefly, astrocyte-microglia and astrocyte-neuron co-cultures or highly enriched astrocytes and neuron cultures were subjected to 3 h of hypoxia in ischemic brain solution (in mM: 51 NaCl, 65 K-gluconate, 0.13 CaCl2, 1.5 MgCl2, 10 HEPES, and pH 6.8) (Orellana et al. 2010), containing normal (5 mM) or high (27 mM) glucose concentrations. Hypoxia was induced as described (Orellana et al. 2010). In brief, cell cultures were kept inside a chamber with the air removed by a CO2/N2 flow for 7 min and maintaining the chamber closed for 3 h. Then, oxygenation was restored, and the medium replaced with normal medium. In hypoxic protocols in 5 mM glucose, 22 mM sucrose was added to achieve the same osmolarity as the high, 27 mM glucose medium. Connexin hemichannel blockers, La3+ (200 μM) and synthetic peptides, Gap26 and Gap27 (200 μM), were co-applied with Etd for uptake measurements. In other experiments, Panx1 hemichannel blockers, 10panx1 (200 μM), E1b (200 μM) and probenecid (200 μM), were applied similarly. In some experiments, CM-Aβ was preincubated (2 h) with 100 ng/ml sTNF-aR1, a soluble form of the receptor that binds TNF-α, and/or 100 ng/ml IL-1ra, an IL-1β receptor blocker were applied before the addition of CM to astrocyte cultures.
Scrape loading/dye transfer technique
Gap junction permeability was evaluated at room temperature using the scrape-loading/dye transfer (SL/DT) technique, on either astrocyte cultures or microglia-astrocyte co-cultures. Briefly, cultures were washed for 10 min in HEPES-buffered salt solution containing the following (in mM): 140 NaCl, 5.5 KCl, 1.8 CaCl2, 1 MgCl2, 5 glucose, 10 HEPES, pH 7.4 followed by washing in a Ca2+-free HEPES solution for 1 min. Then, a razor blade cut was made in the monolayer in a HEPES-buffered salt solution with normal Ca2+ concentration containing the fluorescent dye LY. After 1 min, LY (100 μM) was washed out several times with HEPES-buffered salt solution. Eight minutes after scraping, fluorescent images were captured using an inverted fluorescent microscope equipped for epifluorescence (Diaphot-Nikon, Tokyo, Japan). For each trial, data were quantified by measuring fluorescence areas in five representative fields using an image analyzer system (Lucia-Nikon, Tokyo, Japan). Quantification of changes in gap junctional communication induced by different treatments was performed by measuring the fluorescence area, expressed as arbitrary units (AU).
Dye uptake
For single image visualization of dye uptake, astrocytes were exposed to 5 μM Etd for 10 min at 37°C with HBSS (in mM: 137 NaCl, 5.4 KCl, 0.34 Na2HPO4, 0.44 KH2PO4, pH 7.4) with 1.2 mM CaCl2 (HBSS-Ca2+), mounted in Fluoromount, and examined by epifluorescence (518 nm excitation and 605 nm emission) using an inverted microscope (Diaphot-Nikon) equipped with a CCD camera (Nikon). Captured images were analyzed with image analyzer software (Lucia-Nikon) and the NIH ImageJ program.
For time lapse fluorescence imaging, fluorescence of cells bathed with HBSS-Ca2+ containing 5 μM Etd was recorded every 30 s using a Olympus BX 51W1I microscope. To test for changes in slope, regression lines were fitted to points before and after various treatments using Microsoft (Seattle, WA) Excel, and mean values of slopes were compared using Graphpad Software (San Diego, CA).
Biotinylization
Confluent cultures in 100 mm diameter dishes were washed three times with HBSS-Ca2+. Three ml of Sulfo-NHS-SS-biotin (0.5 mg/ml dissolved in HBSS-Ca2+) was added to cultures, which were then incubated for 30 min at 4°C. Cells were washed three times with HBSS-Ca2+ solution plus 15 mM glycine (pH 8.0), to quench unreacted biotin, and harvested by scraping with a rubber policeman in the presence of protease inhibitors (200 μg/ml soybean trypsin inhibitor, 1 mg/ml benzamidine, 1 mg/ml ε-aminocaproic acid, and 2 mM PMSF) and phosphatase inhibitors (see below, Western blot analysis). Then, cells were pelleted and lysed by sonication in 50 μl of ice cold solution containing proteases and phosphatases inhibitors. NeutrAvidin was added to the samples (1 μl of NeutrAvidin solution per 3 μg of biotinylated protein, based on the assumption that 40% of total membrane protein was biotinylated), and the mixture was maintained for 1 h at 4°C. One milliliter of binding buffer (HBSS, pH7.2, plus 0.1% SDS and1% NP-40) was added, mixed by vortexing, and centrifuged for 2 min at 14.000 rpm at 4°C, and the supernatant was removed. This wash procedure was repeated three times. After the final wash, 40 μl of HBSS, pH 2.8 (to release the protein from the avidin) plus 0.1 M glycine was added to the pellet, which was resuspended and centrifuged at 14.000 rpm for 2 min at 4°C. The supernatant was removed and placed in a 1.5 ml Eppendorf (Westbury, NY) tube, and pH was adjusted to 7.4 immediately by adding 10 μl of 1 M Tris, pH 7.4. Relative Cx43 levels present in each sample were measured by Western blot analysis (see below).
Western blot analysis
Cultures were rinsed twice with PBS, pH 7.4, and harvested by scraping with a rubber policeman in ice solution containing protease and phosphatase inhibitors (1 mM orthovanadate, 10 mM α-glycerophosphate) and complete miniprotease inhibitor (Roche Diagnostics). Pelleted cells were resuspended in 40 μl of the protease and phosphatase inhibitor solution, placed on ice, and lysed by sonication (Ultrasonic cell disrupter, Microson, Ultrasons, Annemasse, France). Proteins were measured in aliquots of cell lysates with the Bio-Rad protein assay (Bio- Rad, Richmond, CA). Samples were stored at −80°C or analyzed by immunoblotting. Aliquots of cell lysates (50 μg of protein) or biotinylated surface membrane proteins were resuspended in a final concentration of 1 × Laemli’s sample buffer, boiled for 5 min, separated on 8% SDS-PAGE and electro-transferred to nitrocellulose sheets as described previously (Orellana et al. 2010). Nonspecific protein binding was blocked by incubation of nitrocellulose sheets in PBS-BLOTTO (5% nonfat milk in PBS) for 30 min, and then blots were incubated with primary monoclonal antibody for 1 h at room temperature or overnight at 4°C, followed by four 15 min PBS washes. Blots were incubated with goat anti-mouse antibody conjugated to horseradish peroxidase. Immunoreactivity was detected by ECL detection using the SuperSignal kit (Pierce, Rockford, IL) according to the provider instructions.
Immunofluorescence and confocal microscopy
For all immunostaining experiments, cells grown on coverslips were fixed at room temperature with 2% paraformaldehyde for 30 min and then washed three times with PBS. Then, they were sequentially incubated in 0.1 M PBS-glycine, three times for 5 min each, and then in PBS-0.1% Triton X-100 containing 10% NGS for 30 min. To identify astrocytes and microglia, we used a specific molecular marker for each one (GFAP antibody and isolectin B4, respectively). We first incubated cells for 2 h at room temperature with anti-GFAP monoclonal antibody (IgG1, 1:500) diluted in 0.1% PBS-Triton X-100 with 2% NGS. After three rinses in 0.1% PBS-Triton X-100, cells were then incubated for 50 min at room temperature with both goat anti-mouse Alexa Fluor 355 (1:1500) and isolectin GS-IB4 (1:100), diluted in the same solution as the first antibody. To identify neurons an anti-MAP2 monoclonal antibody (1/500) was used following the same protocols mentioned above. After several washes, coverslips were mounted in Fluoromount and examined with an upright microscope equipped with epifluorescence (Eclipse E800, Nikon). To visualize double immunostaining, a confocal laser-scanning microscope (TBCS SP2; Leica, Wetzlar, Germany) was used. Stacks of consecutive confocal images taken with a 63 × objective at 500 nm intervals were acquired sequentially with two lasers (argon 488 nm and helium/neon 543 nm), and Z projections were reconstructed using the Leica confocal software.
Measurement of ATP and glutamate release
Astrocytes were plated in multi well culture trays (106 cells/well/0.5 ml) and 48 h after later were used for experiments. Extracellular ATP was measured by luciferin/luciferase bioluminescence assay kit (Sigma-Aldrich, St. Louis, MO, USA). Levels of extracellular glutamate were determined using an enzyme-linked fluorimetric assay as described by Genever et al. (Genever & Skerry 2001). In the presence of glutamate dehydrogenase (GDH) and β-nicotinamide adenine dinucleotide phosphate (NADP+), glutamate is oxidized to α-ketoglutarate, yielding NADPH, which can be determined fluorimetrically (excitation and emission wavelengths of 355 nm and 460 nm) to provide an indirect quantification of glutamate concentration.
For each assay, standard curves were constructed by using known concentrations of ATP or glutamate. The concentration of ATP and glutamate in samples of extracellular medium were calculated from standard curves and referred to 106 cells). The fraction of ATP or glutamate released by cells to the extracellular milieu was estimated by the difference between the concentration detected in the medium of cells under resting conditions and the concentration measured after stimulation in the presence or absence of hemichannel inhibitors.
Neuronal death quantification
Neuronal death was measured as fraction of Fluoro-Jade C (F-Jade) positive cells as described previously (Schmuck & Kahl 2009, Noraberg et al. 1999). For F-Jade measurements the cell culture were fixed in cold ethanol (4°C) for 10 min. Cells were then treated with detergent (0.3% Triton X-100 in PBS, Sigma, Deisenhofen, Germany) for 10 min and washed twice with distilled water. F-Jade is stable in a stock solution in distilled water (0.01%), which can be stored at 4°C. The final concentration for staining was 0.001% in distilled water. The cells were covered with the dye and gently shaken for 30 min in the dark. The dye was then removed from the cell cultures, the cells were washed, and fluorescence was determined with an upright microscope equipped with epifluorescence (Eclipse E800, Nikon).
Data analysis and statistics
For each data group, results were expressed as mean ± S.E., and n refers to the number of independent experiments. For statistical analysis, each treatment was compared with its respective control, and significance was determined using a one-way ANOVA followed by a Tukey post hoc test. For multiple group treatments, significance was determined using a two-way ANOVA followed by a Bonferroni post hoc test.
Results
Aβ-treated microglia potentiate the changes in connexin-based channels induced by hypoxia/reoxygenation in cortical astrocytes
Recently, it was reported that 3 h of hypoxia in high glucose induces a transient increase in hemichannel activity and decrease in gap junctional communication in astrocytes during reoxygenation (Orellana et al. 2010). In order to evaluate if microglia and other pro-inflammatory agents could potentiate these effects on Cx based channels, astrocytes alone or in co-culture with microglia were treated with a fragment of Aβ peptide (Aβ25-35) previously shown to be toxic for neurons and astrocytes (Pike et al. 1995, Assis-Nascimento et al. 2007) and retains most of neurotoxic and pro-inflammatory effects of Aβ [1–42] peptide found in vivo (Yankner et al. 1990; Meda et al. 1995). To examine hemichannel and gap junction channel activities Etd uptake and scrape loading/LY transfer technique were employed, respectively. As reported previously (Orellana et al. 2010), control astrocytes exhibited a low Etd uptake (Supplementary Fig. 1A) and high LY intercellular diffusion (Supplementary Fig. 2A). However, astrocytes subjected to 3 h hypoxia in high glucose (27 mM) showed at 1 h reoxygenation a transient increase in Etd uptake (743.5 ± 57.5% normalized to control; n=4) (Fig. 1A and Supplementary Fig. 1B) and decrease in LY diffusion (53.7 ± 9.4% normalized to control; n=4) (Fig. 2A and Supplementary Fig. 2B). In contrast, normal glucose (5 mM) during hypoxia was unable to induce the effects mentioned above (Fig. 1A and 2A). The changes in Etd uptake and LY diffusion induced by hypoxia in high glucose were abolished in astrocytes co-cultured for 24 h with resting microglia (Fig. 1B and 2B and Supplementary Figs. 1E and 2E). However, astrocytes co-cultured with microglia for 24 h in the presence of 10 μM Aβ25-35 showed a prominent increase in Etd uptake (Fig. 1C and Supplementary Fig. 1H) and decrease in LY diffusion (Fig. 2C and Supplementary Fig. 2H) approaching a plateau at ~1 h reoxygenation that persisted for at least 3 h (Supplementary Figs. 1I and 2I). These responses were substantially greater with higher than normal glucose for Etd uptake (at 1 h of reoxygenation 882.8 ± 142.1% and 579.8 ± 95.1%, respectively normalized to control; n=4) and LY diffusion (55.3 ± 7.4% and 21.8 ± 6.4%, respectively, normalized to control; n=4). It is noteworthy that the abovementioned responses in Etd uptake and LY diffusion were not produced by the inverted sequence of toxic Aβ (Aβ35-25, not shown).
Figure 1. Aβ25-35-treated microglia potentiate astroglial Etd uptake induced by hypoxia in high glucose.

(A–F) Averaged data normalized to control (dashed line) of Etd uptake by astrocytes alone (Ast) or co-cultured for 24 h with microglia (MG) in presence or absence of 10 μM Aβ25-35 (Aβ) and then exposed to 3 h hypoxia in 5 mM (○) or 27 mM (●) glucose followed by several periods of reoxygenation. It is shown the Etd uptake of astrocyte pre-treated for 24 h with 10 μM Aβ25-35 or with conditioned media from microglia exposed for 24 h to 10 μM Aβ25-35 (CM-Aβ) and then subjected to hypoxia/reoxygenation. It is alsoshown the Etd uptake of astrocytes pre-treated for 24 h with TNF-α and IL-1β (10 pg/ml of each). ** p < 0.005, *** p < 0.001, (●) vs (○) at each time point. Each value corresponds to mean ± S.E. of four independent experiments.
Figure 2. Aβ25-35-treated microglia potentiate the reduction of astroglial coupling induced by hypoxia in high glucose.

(A–F) Averaged data normalized to control (dashed line) of area of LY diffusion in astrocytes alone (Ast) or astrocytes co-cultured for 24 h with microglia (MG) in presence or absence of 10 μM Aβ25-35 (Aβ) and then exposed to 3 h hypoxia in 5 mM (○) or 27 mM (●) glucose followed by several periods of reoxygenation. It is shown the LY diffusion between astrocyte pre-treated for 24 h with 10 μM Aβ25-35 or with conditioned media from microglia exposed for 24 h to 10 μM Aβ25-35 (CM-Aβ) and then subjected to hypoxia/reoxygenation. It is also shown the LY diffusion between astrocytes pre-treated for 24 h with TNF-α and IL-1β (10 pg/ml of each).** p < 0.005, *** p < 0.001, (●) vs (○) at each time point. Each value corresponds to mean ± S.E. of four independent experiments.
These actions of Aβ25-35 required the presence of microglia because they were not observed in only Aβ25-35-treated astrocytes (Figs. 1D and 2D). Supporting this view, astrocytes incubated for 24 h with conditioned medium from Aβ25-35-treated microglia (CM-Aβ) and then exposed to hypoxia in high glucose exhibited elevated Etd uptake (at 1 h of reoxygenation 722.4 ± 94.1%, normalized to control; n=4) (Fig. 1E) and decreased LY diffusion (at 1 h of reoxygenation 31.2 ± 8.5% normalized to control; n=4) (Fig. 2E). Interestingly, similar results were observed at 1 h reoxygenation on Etd uptake (666.1 ± 63.8% normalized to control; n=4) (Fig 1F) and LY diffusion (34.1 ± 6.8% normalized to control; n=4) (Fig. 2F) in astrocytes pre-treated for 24 h with TNF-α and IL-1β (10 pg/ml). These data suggested to TNF-α and IL-1β are putative candidates to mediate the effect of Aβ25-35-treated microglia on astroglial hemichannels and gap junction channels during reoxygenation. To address this hypothesis, we evaluated whether sTNF-aR1 (soluble form of the TNF-α receptor that binds TNF-α) and IL-1ra (recombinant receptor antagonist for IL-1β) affect the abovementioned responses. Treatment with sTNF-aR1 and IL-1ra for 24 h completely prevented the increase in Etd uptake (from 641.7 ± 44.7% to 115.9 ± 21.9% normalized to control; n=3) and the reduction in LY diffusion (from 31.7 ± 5.8% to 95.6 ± 16.8% normalized to control, n=3) induced by CM-Aβ during reoxygenation (Fig. 3). When these agents were applied alone (each at 100 ng/ml), partial prevention was observed (Fig. 3).
Figure 3. TNF-α and IL-1β account entirely for the changes in astroglial hemichannels and gap junctions induced by CM-Aβ.

Astrocyte cultures were treated for 24 h with CM-Aβ followed by 3 h hypoxia in 27 mM glucose and 3 h reoxygenation. In some experiments the CM-Aβ treatment was in the presence of 100 ng/ml of IL-1ra, a recombinant antagonist of the IL-1β receptor, or of 100 ng/ml of sTNF-aR1, a soluble form of the TNF-α receptor that binds TNF-α. Graphs of Etd uptake (left, white bars) and the area of LY diffusion after scrape loading (right, black bars) normalized to their respective controls (dashed lines). * p < 0.05, ** p < 0.005, effect of the respective antagonist compared to treatment effect (CM-Aβ + hypoxia). Each value corresponds to mean ± S.E. of four independent experiments.
Notably, protocols that produced persistent (CM-Aβ or TNF-α/IL-1β) increase in Etd uptake (Fig. 4A) and reduction in intercellular LY diffusion (not shown) during reoxygenation were unable to generate similar responses when applied alone (24 h before hypoxia). Thus, treatments for 24 h with CM-Aβ or lower TNF-α/IL-1β concentrations were unable to change hemichannel activity by themselves, indicating that hypoxia/reoxygenation is necessary to observe an increase in hemichannel activity. Finally, the decrease in intercellular LY diffusion was not due to LY leakage through astroglial hemichannels, since similar results were observed in experiments performed in the absence and presence of La3+ (200 μM), a connexin hemichannel blocker (not shown).
Figure 4. Increase in astroglial uptake induced by Aβ25-35-treated microglia is mediated through Cx43 hemichannels.

(A) Averaged data normalized to control (dashed line) of Etd uptake by astrocytes (Ast) co-cultured for 24 h with microglia (MG) without (white bar) or with 10 μM Aβ25-35 (black bar). Also is shown the Etd uptake by astrocytes treated for 24 h with 10 μM Aβ25-35 (gray bar), with CM-Aβ (lighter cross hatched bar) or 10 pg/ml each of TNF-α + IL-1β (darker cross hatched bar). (B) Time-lapse measurements of Etd uptake in astrocytes under control conditions (gray line) or exposed for 24 h to CM-Aβ (black line) and then subjected to 3 h of hypoxia in 27 mM glucose followed by 3 h of reoxygenation.. (C) Graphs representing Etd uptake normalized to control (dashed line) by astrocytes pretreated or not (left) for 24 h with CM-Aβ (middle) or 10 pg/ml of TNF-α + IL-1β (right) and then subjected to 3 h of hypoxia in 27 mM glucose followed by 3 h reoxygenation. For each group is shown the effect on dye uptake of the connexin hemichannel blockers La3+ (200 μM), Gap26 (200 μM), Gap27 (200 μM), or Cx43E2 (1:500 dilution); or pannexin hemichannel blockers 10panx1 (200 μM), Eb1 (200 μM) or probenecid (1 mM). All blockers were used co-incubated with Etd. Moreover, Cx43−/− astrocytes (open bars) subjected to the abovementioned protocols exhibited low uptake like that of wild type cells treated with connexin blockers. *** p < 0.001, compared to the respective treatment. Each value corresponds to mean ± S.E. of four independent experiments.
To investigate the identity of the astroglial hemichannels behind the CM-Aβ-induced Etd uptake during reoxygenation, we examined the effect of several connexin hemichannel blockers. In “time-lapse” experiments, the CM-Aβ-induced Etd uptake during reoxygenation was rapidly blocked by 200 μM La3+(from 0.62 ± 0.02 AU/min to 0.05 ± 0.008 AU/min, n=4) (Fig. 4B) and by 200 μM Gap26 or 200 μM Gap27 in “snapshot” experiments (from 727.7 ± 104.1% to 48.9 ± 15.6% or 44.5 ± 38.8%, respectively; n=5) (Fig. 4C); Gap26 and Gap27 are mimetic peptides of the first and second extracellular loop of Cx43 hemichannels, respectively (Evans & Leybaert 2007). Moreover, to elucidate more specifically the contribution of Cx43 hemichannels in this response, we employed an antibody directed to the second extracellular loop of Cx43 (Cx43E2), which block specifically Cx43 hemichannels, but not Cx43 gap junction channels (Siller-Jackson et al. 2008). We found that this antibody completely inhibited the CM-Aβ-induced Etd uptake (from 727.7 ± 104.1% to 78.9 ± 12.1%; n=3) (Fig. 4C). As expected, the Etd uptake induced by low concentrations of TNF-α and IL-1β during reoxygenation was blocked as well by the abovementioned blockers (Fig. 4C). Since it has been shown that astrocytes express functional Panx1 hemichannels in vitro (Iglesias et al. 2009), we studied their possible contribution on the CM-Aβ-induced Etd uptake observed during reoxygenation. For this purpose, we used two mimetic peptides of the second extracellular loop of Panx1 (10panx1 and E1b) and probenecid, which blocks Panx1 hemichannels (Pelegrin & Surprenant 2006). 10panx1 (200 μM), E1b (200 μM), or probenecid (1 mM), failed to reduce the Etd uptake elicited during reoxygenation in astrocytes pre-treated with CM-Aβ (from 727.7 ± 104.1% to 731.4 ± 62.3%, 718.1 ± 71.3% and 723.2 ± 117.9%, respectively; n=5) or TNF-α and IL-1β (from 718.2 ± 103.9% to 712.1 ± 90.3%, 703.8 ± 101.4% and 732.1 ± 60.1%, respectively; n=5) (Fig. 4C). Furthermore, protocols that induced Etd uptake in wild type astrocytes were unable to induce these responses in astrocytes cultured from Cx43−/− mice (44.2 ± 12.9% normalized to control, n=3) (Fig. 4C), indicating that astroglial Etd uptake occurred largely if not exclusively through Cx43 hemichannels.
We have previously demonstrated that 3 h hypoxia in high glucose followed by reoxygenation causes a transient increase in surface astroglial Cx43 hemichannels that could explain the transient increase in Etd uptake observed under these conditions (Orellana et al. 2010). Both changes were transient and simultaneous and showed maximal values at 1 h of reoxygenation, but were back to normal at 3 h of reoxygenation and were not modified by reoxygenation in cells exposed to 3 h hypoxia in normal glucose (Orellana et al. 2010). To study if a similar association might occur in the present study, we determined the effect of CM-Aβ on total and surface levels of Cx43 during reoxygenation. Comparable levels of total Cx43 were detected in astrocytes under control conditions or after treatment for 24 h with CM-Aβ and then exposed to 3 h of hypoxia in normal or high glucose followed by 3 h reoxygenation (113.2 ± 14.8% or 95.4 ± 13.2%, respectively, normalized to control; n=3) (Fig. 5A and C). Moreover, surface levels of Cx43 were comparable to control levels at 3 h reoxygenation after the hypoxia period in high or normal glucose at (117.4 ± 8.4% and 105.4 ± 10.8%, respectively, normalized to control; n=3) (Fig. 5B and C). However, pre-treatment with CM-Aβ for 24 h followed by hypoxia in high glucose or even in normal glucose induced a prominent increase in surface levels of Cx43 at 3 h reoxygenation (319.2 ± 33.9% and 306.5 ± 36.7%, respectively, normalized to control; n=3) (Fig. 5B and C), suggesting summation of stimuli (hypoxia in low glucose and CM-Aβ).
Figure 5. Aβ25-35-treated microglia induces increase in surface levels of Cx43 after hypoxia.
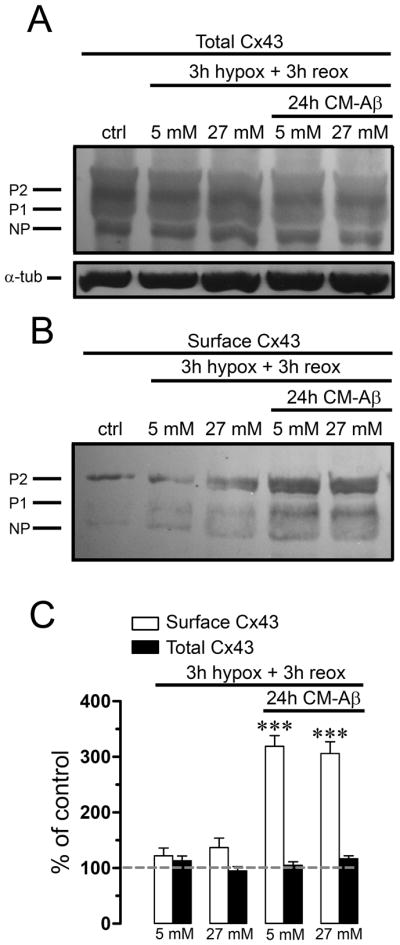
(A and B) Astrocytes cultures were controls or were subjected to 3 h hypoxia in 5 or 27 mM glucose followed by 3 h reoxygenation. Other cultures were pre-incubated for 24 h in CM-Aβ and then subjected to 3 h hypoxia in 5 or 27 mM glucose followed by 3 h reoxygenation. Levels of total Cx43 and surface Cx43 isolated by biotinylation were measured by Western blot analysis. (A) Western blot of total Cx43 present in homogenates. None of the treatments affected quantities of the phosphorylated (P1–P2) and nonphosphorylated (NP) forms of Cx43 (markers on the left). (B) Western blot of surface Cx43 from astrocytes under the same conditions. (C) Quantification of surface and total Cx43 normalized to the control in the treatments mentioned above. *** p < 0.001, compared to control. Each value corresponds to mean ± S.E. of at least 3 independent experiments.
Astroglial Cx43 hemichannel activity induced by Aβ-treated microglia promotes neuronal death by opening neuronal Panx1 hemichannels
To explore whether astrocytes can potentiate neuronal vulnerability via hemichannels, astrocytes alone or co-cultured with neurons were treated with CM-Aβ and then subjected to hypoxia in normal glucose. Firstly, we evaluated if 3 h hypoxia induce cell death in neuronal cultures. Under control conditions a small amount of neuronal death was observed by measure of F-Jade staining (5.9 ± 2.9% normalized to total cells, n=4) (Fig. 6A), whereas after hypoxia in normal glucose neuronal death was prominently increased only after 24 h reoxygenation (49.9 ± 10.6% normalized to total cells, n=4) (Fig. 6A). Importantly, this death was associated with beading of neuronal process, but not with Etd uptake (Supplementary Fig. 3C), suggesting that this was a process independent of hemichannel activity. In contrast, the abovementioned protocol was unable to decrease neuronal viability and change morphology at 24 h reoxygenation when neurons were co-cultured with astrocytes (6.9 ± 1.9% normalized to total cells, n=5) (Fig. 6A and Supplementary Fig. 3I), indicating that under these conditions astrocytes were neuroprotective. Interestingly, when neurons were co-cultured with astrocytes in the presence of CM-Aβ for 24 h a significant increase in neuronal death (66.8 ± 6.2% normalized to total cells, n=4) (Fig. 6A) and neuronal Etd uptake (Supplementary Fig. 3K) was observed at 1 h reoxygenation. Nevertheless, neurons treated for 24 h with CM-Aβ presented similar Etd uptake (Supplementary Fig. 3F) and neuronal death (51.1 ± 12.7% normalized to total cells, n=4) (Fig. 6A and Supplementary Fig. 3F) than neurons not treated with CM-Aβ (Fig. 6A and Supplementary Fig. 3C). Importantly, Gap26 decreased the CM-Aβ-induced neuronal death (11.9 ± 8.2% normalized to total cells, n=4) (Fig. 6A) and Etd uptake in neurons and astrocytes at 1 h reoxygenation (Supplementary Fig. 3N–O). The neuroprotective effect of Gap26 is likely due to its blocking effect on astroglial hemichannels since it did not affect the reduction in astroglial dye coupling induced by hypoxia/reoxygenation in high glucose (62 ± 4.2% and 61.2 ± 5.1%, respectively, normalized to control, n=3) (Fig. 6B). Further indication of the role of astroglial Cx43 hemichannels in neuronal death was obtained by blocking these channels with a specify antibody. In co-cultures treated with the Cx43E2 antibody during reoxygenation neuronal death was drastically reduced (Fig. 6A). Moreover, a similar reduction in neuronal death was observed in co-cultures of Cx43−/− astrocytes and wild type neurons. In the latter cultures, treatment for 24 h with CM-Aβ followed by hypoxia and 1 h reoxygenation did not significantly affect neuronal survival compared to control (4.5 ± 2.2% normalized to control, n=3) (Fig. 6A).
Figure 6. Astroglial Cx43 hemichannel activity induced by Aβ25-35-treated microglia followed by hypoxia/reoxygenation accelerates neuronal death caused by opening of neuronal Panx1 hemichannels.

(A) Cell death was monitored as percent of neurons positive to F-Jade staining. Neurons alone (N), or co-cultured with astrocytes (N+A) were treated or untreated with CM-Aβ for 24 h and then subjected to 3 h hypoxia in 5 mM glucose followed by several periods of reoxygenation (0, 1, 12 or 24 h). In some experiments 200 μM Gap26 or Cx43E2 (1:500 dilution) was applied during the reoxygenation period. Also it is shown data from neurons co-cultured with astrocytes from Cx43−/− mice. (B) LY diffusion (normalized to control) by astrocytes incubated for 1 h with 200 μM Gap26 (white bar) or by astrocytes exposed to CM-Aβ for 24 h and then subjected to 3 h hypoxia in 5 mM glucose (black bars) followed by 1 h reoxygenation without (middle bar) or with 200 μM Gap26 (right bar). (C) Etd uptake in neurons normalized to control conditions (dashed line) or subjected to 3 h hypoxia in 5 mM glucose followed by 1 h reoxygenation in CM-Ast or not. Also it is shown the effect of La3+, Gap26, probenecid (Prob) or 10panx1 (200 μM each) on Etd uptake of control or treated neuronal cultures. Gap26 (*); effect on neuronal Etd uptake of CM-Ast made in presence of Gap26 during reoxygenation. In some experiments, the effect on neuronal Etd uptake of CM-Ast made in the presence of Cx43E2 (1:500 dilution) during reoxygenation was studied. (D) Cell death in cultures of neurons subjected to 3 h hypoxia in 5 mM glucose followed by 1 h reoxygenation with the same treatments as in C. *** p < 0.001, ** p < 0.005, compared to control; ### p < 0.001, compared to hypoxia plus Ast-CM effect. Each value corresponds to mean ± S.E. of at least 4 independent experiments.
Since the above data suggested that astrocytes release neurotoxins via Cx43 hemichannels, conditioned medium was prepared from astrocytes treated for 24 h with CM-Aβ and then exposed to 3 h hypoxia followed by 3 h reoxygenation. Thus, neuronal cultures were subjected to hypoxia in normal glucose and then reoxygenated in the abovementioned conditioned media (CM-Ast). After 3 h hypoxia followed by 1 h of reoxygenation in CM-Ast, prominent neuronal death was observed (44.1 ± 12.1% normalized to total cells, n=4) and neurons that remained alive exhibited a greater Etd uptake (1521 ± 218.6% normalized to control, n=5) (Fig. 6C and D). Interestingly, CM-Ast made from astrocytes exposed to Gap26 or Cx43E2 antibody during reoxygenation was unable to increase neuronal Etd uptake either in normal or high glucose (102.8 ± 34.8% or 98.2 ± 24.2%, respectively, normalized to control, n=5). Moreover, Gap26 also reduced neuronal death at 1 h reoxygenation under the same protocol (8.8 ± 12.3% normalized to total cells, n=4) (Fig. 6C and D). These observations suggested that soluble factors released from astroglial Cx43 hemichannels affect hemichannel activity and viability of neurons. In agreement with the previous interpretation, 10panx1 or probenecid, but not 200 μM La3+ or Gap26, prevented the neuronal Etd uptake (114.9 ± 29.9%, 119.8 ± 34.7%, 1565.6 ± 278.8% and 1531.8 ± 255.2%, respectively, normalized to control, n=4) and death in neurons (3.2 ± 1.7%, 6.9 ± 2.8%, 46.9 ± 8.1% and 43.1 ± 6.1%, respectively, normalized to total cells, n=4) induced during reoxygenation in CM-Ast (Fig. 6C and D).
Death of cortical neurons is mediated by opening of Panx1 hemichannels activated via P2 and NMDA receptors
Since it has been proposed that glutamate (Thompson et al. 2008) and ATP (Pelegrin & Surprenant 2006), acting through NMDA and P2 receptors, respectively, activate Panx1 hemichannels, we investigated if CM-Ast alone could induce neuronal dye uptake and death and if so, whether this effect could be prevented by glutamate and/or P2 receptor blockers.
In neuronal cultures treatment with CM-Ast alone for 1 h, we observed increased Etd uptake (1127.1 ± 74.1% normalized to control, n=4) and cell death (50.3 ± 9.8% normalized total cells, n=4) evaluated with F-Jade (Fig. 7 and Supplementary Fig. 4B). The increase in Etd uptake and neuronal death induced by CM-Ast was reduced partially by degradation of extracellular ATP with apyrase (376.7 ± 69.9% and 39.2 ± 8.2%, respectively, n=4) or treatment with blockers of P2X receptors: oATP (445.9 ± 64.3% and 40.1 ± 5.2%, respectively, n=4), suramin (389.8 ± 74.8% and 41.8 ± 7.2%, respectively, n=4), and BBG (421.3 ± 73.2% and 42.7 ± 6.3%, respectively, n=4) or a blocker of the NMDA receptor: CPP (523.8 ± 96.3% and 30.8 ± 10.7%, respectively, n=4) and was almost completely blocked when activation of both glutamate and P2 receptors was inhibited (Fig. 7 and Supplementary Fig. 4C). Similarly, both the Etd uptake and neuronal death were abolished by probenecid (115.1 ± 24.6% and 8.1 ± 2.6%, respectively, n=4) or 10panx1 (114.8 ± 16.3% and 5.6 ± 2.1%, respectively, n=4) (Fig. 7).
Figure 7. Neuronal death induced by astrocytes is prevented by Panx1 hemichannel blockers but not by connexin hemichannel blockers; protection by P2X and NMDA receptor blockers.

Averaged Etd uptake (white bars) and death (black bars) normalized to control of neurons treated for 1 h with CM-Ast alone or after 20 min pretreatment and continued application of 20 μM CPP (NMDA receptor blocker), 300 μM oATP (P2X receptor blocker), 200 μM suramin (P2 receptor blocker), 10 μM BBG (P2X7 receptor blocker) or 10 U/ml apyrase (ATPase). It is shown the effect of probenecid (Prob) and 10panx1 in the abovementioned responses. Prob and 10panx1 were applied at 200 μM during reoxygenation. *p < 0.001, compared to Ast-CM effect. Each value corresponds to mean ± S.E. of at least 4 independent experiments.
To elucidate the possible role of glutamate and ATP in this response, we measured the concentration of these molecules in CM-Ast generated by wild type astrocytes under control condition or treated with inhibitors of Cx or Panx1 hemichannel blockers during reoxygenation as well as in the CM-Ast generated by Cx43−/− astrocytes. In the extracellular medium of astrocytes maintained under control conditions, levels of glutamate and ATP were 23.4 ± 5.3 nmol/106 cells (n=3) and 11.3 ± 1 nmol/106 cells (n=3), respectively (Fig. 8). Conditioned media obtained during reoxygenation from astrocytes not treated with CM-Aβ presented similar levels of glutamate and ATP than control astrocytes (not shown). However, levels of glutamate and ATP were much higher in CM-Ast compared to control (82.2 ± 7.8 nmol/106 cells and 62.5 ± 5.6 nmol/106, respectively, n=4) (Fig. 8). Interestingly, in CM-Ast made from astrocytes exposed to Gap26 during reoxygenation the levels of glutamate and ATP were similar to control conditions (18.1 ± 2.3 nmol/106 cells and 10.2 ± 1.4 nmol/106, respectively, n=4) (Fig. 8). But, neither 10panx1 nor probenecid applied during reoxygenation decreased the levels of glutamate (77.1 ± 7.9 nmol/106 cells and 73.5 ± 4.9 nmol/106, respectively, n=3) or ATP (60.7 ± 9.2 nmol/106 cells and 58.1 ± 7.2 nmol/106, respectively, n=3) (Fig. 8).
Figure 8. Aβ25-35-treated microglia increase the release glutamate and ATP from astroglial Cx43 hemichannels during reoxygenation.

Release of glutamate (black bars) and ATP (gray bars) by astrocytes under control conditions or treated with CM-Aβ for 24 h and then subjected to 3 h hypoxia in 5 mM glucose followed by 1 h reoxygenation. In some experiments 200 μM Gap26, 500 μM probenecid (Prob), 200 μM 10panx1 and 10 μM brefeldin A (Bref A) were applied during the reoxygenation period. Also is shown data from astrocytes from Cx43−/− mice subjected to the same abovementioned protocols.
The above data suggest that glutamate and ATP released by astrocytes occurred via Cx43 hemichannels. In support of this notion, low levels of extracellular glutamate and ATP were detected in the extracellular mediun of Cx43−/− astrocytes (16.9 ± 1.6 nmol/106 cells and 9.3 ± 2.5 nmol/106, respectively, n=3) (Fig. 8). To study the possible role of vesicular ATP release, the effect of 10 μM brefeldin A, an inhibitor of vesicular transport, was studies and it was found that this compound did not affect the levels of glutamate and ATP in CM-Ast (70.1 ± 2.1 nmol/106 cells and 54.2 ± 7.2 nmol/106 cells, respectively, n=3) (Fig. 8), suggesting that under these conditions the main pathway of ATP and glutamate release occurs via hemichannels.
ATP and glutamate open pannexin hemichannels in neurons
Because ATP and glutamate probably mediated the CM-Ast-induced Etd uptake and death in neurons, we also investigated whether both molecules could affect the activity of neuronal hemichannels. ATP or glutamate alone increased neuronal hemichannel activity after 1 h incubation but with different concentration/response relationship (Fig. 9A). Glutamate increased the Etd uptake in a concentration dependent manner with two ascending steps, whereas ATP increased Etd uptake with the maximal effect at 100 μM (373.1 ± 87.9% normalized to control, n=4) and its effect was progressively weaker at higher concentrations (Fig. 9A). To exclude the possibility that the effects could be mediated by breakdown products of ATP (e.g., ADP and adenosine), we treated neurons for 1 h with several concentrations of ADP or adenosine. Under these conditions no changes in Etd uptake were observed (Supplementary Fig. 5A). Additionally, the ATP-induced Etd uptake was inhibited after inhibiting P2X7 receptors with oATP and BBG (Orellana et al. 2010; Iglesias et al. 2009) but not with DPCPX, an A1 adenosine receptor blocker (Cechova et al. 2010) (Supplementary Fig. 5B) When glutamate and ATP were co-applied to neurons, Etd uptake was increased synergistically up to 100 μM, but for higher concentrations, the effect of both molecules became progressively weaker (Fig. 9A). The increase in neuronal Panx1 hemichannel activity induced by 100 μM ATP/glutamate (1012.7 ± 143.7% normalized to control, n=3) was prevented by 1 mM probenecid (123.1 ± 9.1% normalized to control, n=3), but was not affected by 200 μM La3+ (886.9 ± 83.1% normalized to control, n=3) (Fig. 9B). Similarly, the neuronal death induced by ATP or glutamate alone or in combination was prevented by Panx1 but not Cx43 hemichannel blockers (Fig. 9C).
Figure 9. ATP and glutamate act together to permeabilize and kill neurons in neuronal cultures.

(A) Etd uptake ratio normalized to control (dashed line) in neuronal cultures exposed for 1 h to various concentrations of ATP (white circles), glutamate (black circles) or ATP plus glutamate (gray circles). (B) Etd uptake normalized to control (dashed line) in neuronal cultures exposed for 1 h to 100 μM ATP (white bars), 100 μM glutamate (black bars) or 100 μM ATP plus 100 μM glutamate (gray bars). Etd uptake was unaffected by La3+ (200 μM, middle bars) but blocked by probenecid (200 μM, right bars). (C) Cell death measured as percent of F-Jade positive cells in neuronal cultures treated with glutamate and/or ATP as in B. Probenecid and 10panx1 provided complete protection, but not La3+ and Gap26. # p < 0.001; compared to control; * p < 0.001, compared to the respective treatment. Each value corresponds to mean ± S.E. of 4 independent experiments.
Discussion
In this study we have demonstrated that Aβ-treated microglia potentiate the increase in astroglial hemichannel activity and reduction in gap junctional communication induced by hypoxia in high glucose. In addition, the extracellular media of inflamed astrocytes was neurotoxic due to its glutamate and ATP content, molecules which activated neuronal Panx1 hemichannels via NMDA/P2X receptors leading to neuronal death. Therefore, neurons could be efficiently protected from ischemia and neurotoxicity by blocking NMDA and P2X receptors as already proposed, but also by targeting either glial or neuronal hemichannels composed by Cx43 and Panx1, respectively.
When microglia are activated with Aβ25-35 they release pro-inflammatory cytokines (Block et al. 2007). Accordingly, medium conditioned by microglia treated with Aβ25-35 (CM-Aβ) induced a persistent change in Cx based channels of astrocytes subjected to a 3 h hypoxia in high glucose, suggesting the action of soluble factors present in the CM-Aβ. In agreement with this interpretation, simultaneous neutralization of TNF-α and IL-1β with IL-1ra and sTNF-aR1 completely prevented the changes induced by CM-Aβ. Persistent and opposite responses of astroglial Cx43 hemichannels and gap junction channels was recently demonstrated during reoxygenation after 6 h of hypoxia (Orellana et al. 2010) and after LPS treatment of co-cultured microglia (Froger et al. 2009). Here, a similar persistent response was observed in astrocytes pre-treated with CM-Aβ or low concentrations of TNF-α and IL-1β followed by only 3 h of hypoxia. None of these conditions alone caused detectable or persistent changes, but CM-Aβ-treated astrocytes presented similar responses during reoxygenation to that induced by 6 h of hypoxia (Orellana et al. 2010), indicating convergence and addition of their effects at the level of astroglial Cx based channels. Supporting this interpretation, the additive effect was also evident in the increase of Cx43 levels at the cell membrane, which has been shown to account for the increase in hemichannel activity evoked by pro-inflammatory conditions (Orellana et al. 2010). In addition, astroglial Etd uptake induced by CM-Aβ or TNF-α/IL-1β after 3 h hypoxia was exclusively dependent on Cx43 hemichannels, because: 1) Etd uptake was not observed in Cx43−/− astrocytes, 2) pharmacological treatments known to block Cx43, but not Panx1 hemichannels (e.g., La3+ and Gap26), inhibited this response and 3) two Panx1 mimetic peptides, 10panx1 and E1b, as well probenecid, a Panx1 hemichannel blocker, failed to block the increase in Etd uptake.
Microglia not stimulated with Aβ25-35 prevented the increase in astroglial hemichannel activity observed at 3 h reoxygenation after 3 h hypoxia in high glucose. The latter is in agreement with the known neuroprotective effect of resting microglia (Block et al. 2007). In fact, microglia suppress both the effect of H2O2 on astroglial gap junctional communication and its toxicity (Rouach et al. 2004). In contrast, activated microglia and pro-inflammatory cytokines induce astroglial uncoupling (Faustmann et al. 2003), astroglial hemichannel opening and neuronal death (Retamal et al. 2007, Block et al. 2007).
It is well established that astrocytes under resting conditions can prevent neuronal damage induced by hypoxia during reoxygenation (Trendelenburg & Dirnagl 2005). In contrast, astrocytes treated with pro-inflammatory conditions promote neuronal damage (Thornton et al. 2006). We here show that this process, in addition to involving increased opening of astroglial Cx43 hemichannels, is also associated with the activation of neuronal Panx1 hemichannels. Indeed, Gap26 directly prevented astroglial hemichannel activity but also had a secondary preventative action on Panx1 hemichannel activity in neurons. The functional role of gap junctional communication in animal models of stroke remains controversial (Orellana et al. 2009). By selectively blocking astroglial hemichannels without interfering with their gap junctions, we have shown that inhibition of hemichannels is neuroprotective. However, as gap junctional communication is reduced in our conditions it remains to be demonstrated if recovery of intercellular communication, independently of changes in hemichannel activity, could increases neuronal resistance to pro-inflammatory conditions.
The concentration of both glutamate and ATP was much higher in CM-Ast generated by astrocytes with functional Cx43 hemichannels. Due to the enhanced activity of astroglial Cx43 hemichannels, more glutamate and ATP could have been released through this signaling pathway (Ye et al. 2003, Kang et al. 2008). Accordingly, the presence of Cx43 hemichannel blockers during conditioning of the culture media by astrocytes treated with CM-Aβ or astrocytes without Cx43 expression (astrocytes from Cx43−/− mice) prevented the increase in glutamate and ATP concentration observed in the extracellular media harvested from untreated wild type astrocytes. These findings and the inability of brefeldin A to inhibit the glutamate/ATP release rule out the possible participation of vesicular release of these molecules (Rossi & Volterra 2009).
A striking finding of the present work was that in the presence of Aβ25-35, astroglial hemichannels are implicated in neuronal death induced by pro-inflammatory conditions. Accordingly, the CM-Ast was deleterious only when it was harvested from astrocytes not treated with Cx43 hemichannel blockers applied at time zero of reoxygenation. These findings suggested that during reoxygenation soluble effectors were released via astroglial Cx43 hemichannels. This interpretation is supported by the following findings: 1) the increase in neuronal Etd uptake and death were partially prevented with inhibition of NMDA or P2X receptors, but was completely prevented by inhibition of both receptor types, 2) exogenous ATP and glutamate mimicked the Etd uptake and neuronal death induced by CM-Ast and 3) higher ATP and glutamate concentrations were detected in CM-Ast generated by astroglial cells expressing functional Cx43 hemichannels. These findings provide an explanation of a recent report in which we observed that Etd uptake occurred in neurons cocultured with astrocytes treated with proinflammatory cytokines (TNF-α and IL-1β) and we showed that hemichannels of inflamed astrocytes worsen the NMDA excitotoxicity in neurons (Froger 2010).
The in vitro neurotoxic effect of glutamate has been well documented (Lau & Tymianski). In addition, the extracellular ATP concentration is known to increase in ischemic brain (Melani et al. 2005) and ATP could be neurotoxic acting directly on neurons (Amadio et al. 2005) or indirectly by inducing astroglial release of glutamate (Fellin et al. 2006). In the neurodegeneration model used in the present work excessive extracellular ATP and glutamate enhance neuronal mortality via Panx1 hemichannels. This notion implies that persistent activation of Panx1 hemichannels is more deleterious to neurons than prolonged activation of P2 and/or NMDA receptors. In support of this possibility, studies in knock out animals for P2X7 receptors show similar neuronal sensitivity in ischemic and excitotoxic brain (Le Feuvre et al. 2003) and knock out animals for the NMDA subunits NR1 and NR2 show only reduction in the affected brain area (Morikawa et al. 1998). Previous evidence indicates that Panx1 hemichannels can be activated by extracellular ATP through P2X7 receptors (Pelegrin & Surprenant 2006, Locovei et al. 2006) as well as through glutamate receptors (Thompson et al. 2008). However, in hippocampal pyramidal cells inhibitors of glutamate receptors, but not inhibitors of Panx1 hemichannels, prevent the anoxic depolarization (Madry et al 2010.), suggesting that opening of Panx1 hemichannel might depend on experimental conditions.
We propose that microglia activated by Aβ25-35 release pro-inflammatory cytokines for which an increase in astroglial hemichannel activity has been demonstrated (Retamal et al. 2007) (Fig. 10). The activation of such hemichannels allows the release of neurotoxic molecules including glutamate and ATP, which can act on microglia inducing further cytokine release (Block et al. 2007) (Fig. 10). Then, opening of neuronal Panx1 hemichannels could be triggered due to the rise in [Ca2+]i via activation of NMDA and P2X receptors by glutamate and ATP, respectively (Fig. 10). Panx1 hemichannels are likely to contribute to the intracellular Ca2+ overload that activates neurotoxic intracellular cascades during brain ischemia and excitotoxicity (Szydlowska & Tymianski 2010). The complete neuronal death inhibition elicited by Panx1 hemichannel blockade suggests that other mechanisms known to contribute to the Ca2+ overload, including ionotropic receptors and channels (e.g., NMDA, AMPA and kainate receptors, TRPM, and P2X receptors and CaV1.2 channels) and membrane transporters (Szydlowska & Tymianski 2010), may also act as activators of Panx1 hemichannels or participate downstream of Panx1 hemichannels.
Figure 10. Signaling that leads to neuronal death through the contribution of inflamed glial cells.

Initially, microglia upon activation with Aβ (1) release pro-inflammatory cytokines (TNF-α/IL-1β) (2), which increase astroglial hemichannel activity when it is followed by hypoxia in high glucose (3). Then, astrocytes release glutamate and ATP via Cx43 hemichannels, which activate opening of Panx1 hemichannels in neurons (4). ATP released as a result of Panx1 hemichannel opening could contribute in the progression of neuronal death by a vicious cycle since it will activate more P2X receptors leading to more Ca2+ entry and activation of intracellular neurotoxic cascades (5).
Neurodegenerative processes, as in DM and AD, which are accompanied by neuro-inflammation, including micro strokes (Pasquier et al. 2006, Snowdon et al. 1997), might cause enhanced astroglial and neuronal hemichannel activity, leading to cell death and impairment of CNS function. AD and DM are two of the most common and devastating health problems in the elderly. Several studies have shown associations between DM and moderate cognitive impairment of both memory and executive functions (Pasquier et al. 2006, Takeda et al. 2010). Moreover, the risk of both vascular dementia and AD is greater in patients with type 2 DM (Pasquier et al. 2006). Interestingly, increase in Cx43 expression has been observed in reactive astrocytes from AD patients (Nagy et al. 1996) and a double transgenic mouse developing Aβ plaques (Mei et al. 2010). Thus, dysregulation of Cx43 and Panx1 based channels may contribute to the development of CNS pathologies and Cx as well as Panx1 hemichannels might represent potential and alternative targets for therapeutic intervention in neuro-inflammatory diseases.
Supplementary Material
Acknowledgments
This work was partially supported by the CRPCEN and INSERM (France; to CG); CONICYT 24080055 (to JAO); FONDECYT 1070591 (to JCS); FONDEF DO7I1086 (to JCS); ANILLO ACT-71 (to JCS), NIH (NS55363 to MVLB), INSERM (France; Département des Relations Internationales to JAO) and the Heart & Stroke Foundation of BC & Yukon (CCN). Conception and design were performed by JAO, CG and JCS and most acquisition of data was performed by JAO. Moreover part of acquisition of data and analysis of data were performed by NF and PE.
Footnotes
All authors contributed in equal form on interpretation of data, drafting the article and revising critically in it intellectual content.
All authors declare no conflict of interest.
The data of this work was presented by Dr. Juan A. Orellana as partial fulfillment of the requirements to obtain the degree of PhD in Physiological Sciences at the Pontificia Universidad Católica de Chile.
References
- Amadio S, D’Ambrosi N, Trincavelli ML, Tuscano D, Sancesario G, Bernadi G, Martini C, Volonte C. Differences in the neurotoxicity profile induced by ATP and ATPgammaS in cultured cerebellar granule neurons. Neurochem Int. 2005;47:334–342. doi: 10.1016/j.neuint.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Assis-Nascimento P, Jarvis KM, Montague JR, Mudd LM. Beta-amyloid toxicity in embryonic rat astrocytes. Neurochem Res. 2007;32:1476–1482. doi: 10.1007/s11064-007-9335-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Cechova S, Elsobky AM, Venton BJ. A1 receptors self-regulate adenosine release in the striatum: evidence of autoreceptor characteristics. Neuroscience. 2010;171:1006–1015. doi: 10.1016/j.neuroscience.2010.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Evans WH, Leybaert L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun Adhes. 2007;14:265–273. doi: 10.1080/15419060801891034. [DOI] [PubMed] [Google Scholar]
- Faustmann PM, Haase CG, Romberg S, Hinkerohe D, Szlachta D, Smikalla D, Krause D, Dermietzel R. Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia. 2003;42:101–108. doi: 10.1002/glia.10141. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pozzan T, Carmignoto G. Purinergic receptors mediate two distinct glutamate release pathways in hippocampal astrocytes. J Biol Chem. 2006;281:4274–4284. doi: 10.1074/jbc.M510679200. [DOI] [PubMed] [Google Scholar]
- Froger N, Orellana JA, Cohen-Salmon M, Ezan P, Amigou E, Sáez JC, Giaume C. Cannabinoids prevent the opposite regulation of astroglial connexin43 hemichannels and gap junction channels induced by pro-inflammatory treatments. J Neurochem. 2009;111:1383–1397. doi: 10.1111/j.1471-4159.2009.06407.x. [DOI] [PubMed] [Google Scholar]
- Froger N, Orellana JA, Calvo CF, Amigou E, Kozoriz MG, Naus CC, Sáez JC, Giaume C. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Molecular and Cellular Neuroscience. 2010;45:37–46. doi: 10.1016/j.mcn.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Genever PG, Skerry TM. Regulation of spontaneous glutamate release activity in osteoblastic cells and its role in differentiation and survival: evidence for intrinsic glutamatergic signaling in bone. FASEB J. 2001;15:1586–1588. doi: 10.1096/fj.00-0594fje. [DOI] [PubMed] [Google Scholar]
- Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J Neurosci. 2009;29:7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagansky N, Levy S, Knobler H. The role of hyperglycemia in acute stroke. Archives of neurology. 2001;58:1209–1212. doi: 10.1001/archneur.58.8.1209. [DOI] [PubMed] [Google Scholar]
- Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M. Connexin 43 hemichannels are permeable to ATP. J Neurosci. 2008;28:4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Le Feuvre RA, Brough D, Touzani O, Rothwell NJ. Role of P2X7 receptors in ischemic and excitotoxic brain injury in vivo. J Cereb Blood Flow Metab. 2003;23:381–384. doi: 10.1097/01.WCB.0000048519.34839.97. [DOI] [PubMed] [Google Scholar]
- Locovei S, Wang J, Dahl G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006;580:239–244. doi: 10.1016/j.febslet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Madry C, Haglerod C, Attwell D. The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain. doi: 10.1093/brain/awq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–50. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Mei X, Ezan P, Giaume C, Koulakoff A. Astroglial connexin immunoreactivity is specifically altered at beta-amyloid plaques in beta-amyloid precursor protein/presenilin1 mice. Neuroscience. 2010;171:92–105. doi: 10.1016/j.neuroscience.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Melani A, Turchi D, Vannucchi MG, Cipriani S, Gianfriddo M, Pedata F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int. 2005;47:442–448. doi: 10.1016/j.neuint.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Morikawa E, Mori H, Kiyama Y, Mishina M, Asano T, Kirino T. Attenuation of focal ischemic brain injury in mice deficient in the epsilon1 (NR2A) subunit of NMDA receptor. J Neurosci. 1998;18:9727–9732. doi: 10.1523/JNEUROSCI.18-23-09727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Li W, Hertzberg EL, Marotta CA. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996;717:173–178. doi: 10.1016/0006-8993(95)01526-4. [DOI] [PubMed] [Google Scholar]
- Naus CC, Bechberger JF, Zhang Y, Venance L, Yamasaki H, Juneja SC, Kidder GM, Giaume C. Altered gap junctional communication, intercellular signaling, and growth in cultured astrocytes deficient in connexin43. J Neurosci Res. 1997;49:528–540. doi: 10.1002/(SICI)1097-4547(19970901)49:5<528::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Bruggencate GT, Steinberg R, Stockle H. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc Natl Acad Sci U S A. 1977;74:1287–1290. doi: 10.1073/pnas.74.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noraberg J, Kristensen BW, Zimmer J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc. 1999;3:278–290. doi: 10.1016/s1385-299x(98)00050-6. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Hernandez DE, Ezan P, Velarde V, Bennett MV, Giaume C, Sáez JC. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia. 2010;58:329–343. doi: 10.1002/glia.20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Sáez PJ, Shoji KF, Schalper KA, Palacios-Prado N, Velarde V, Giaume C, Bennett MV, Sáez JC. Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid Redox Signal. 2009;11:369–399. doi: 10.1089/ars.2008.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes mellitus and dementia. Diabetes Metab. 2006;32:403–414. doi: 10.1016/s1262-3636(07)70298-7. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- Retamal MA, Froger N, Palacios-Prado N, Ezan P, Sáez PJ, Sáez JC, Giaume C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007;27:13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Volterra A. Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull. 2009;80:224–232. doi: 10.1016/j.brainresbull.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Rouach N, Calvo CF, Duquennoy H, Glowinski J, Giaume C. Hydrogen peroxide increases gap junctional communication and induces astrocyte toxicity: regulation by brain macrophages. Glia. 2004;45:28–38. doi: 10.1002/glia.10300. [DOI] [PubMed] [Google Scholar]
- Sáez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Schmuck G, Kahl R. The use of Fluoro-Jade in primary neuronal cell cultures. Arch Toxicol. 2009;83:397–403. doi: 10.1007/s00204-008-0360-4. [DOI] [PubMed] [Google Scholar]
- Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, Sprague E, Jiang JX. Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J Biol Chem. 2008;283:26374–26382. doi: 10.1074/jbc.M803136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–1559. doi: 10.1126/science.1165209. [DOI] [PubMed] [Google Scholar]
- Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem. 2006;98:258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;50:307–320. doi: 10.1002/glia.20204. [DOI] [PubMed] [Google Scholar]
- Whitehead SN, Cheng G, Hachinski VC, Cechetto DF. Progressive increase in infarct size, neuroinflammation, and cognitive deficits in the presence of high levels of amyloid. Stroke. 2007;38:3245–3250. doi: 10.1161/STROKEAHA.107.492660. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.