

Abstract
Much of the world’s genomic data are available to the community through networked databases that are accessed via Web interfaces. Although this paradigm provides browse-level access and has greatly facilitated linking between databases, it does not provide any convenient mechanism for programmatically fetching and integrating data from diverse databases. We have created a library and an application programming interface (API) named AcePerl that provides simple, direct access to ACEDB databases from the Perl programming language. With this library, programmers and computer-savvy biologists can write software to pose complex queries on local and remote ACEDB databases, retrieve the data, integrate the results, and move data objects from one database to another. In addition, a set of Web scripts running on top of AcePerl provides Web-based browsing of any local or remote ACEDB database. AcePerl and the AceBrowser Web browser run on Unix systems and are available under a license that allows for unrestricted use and redistribution. Both packages can be downloaded from URL http://stein.cshl.org/AcePerl. A Microsoft Windows port of AcePerl is in the planning stages.
The ACEDB database engine is an object-oriented system capable of storing and retrieving complex biological information (Durbin and Thierry-Mieg 1991). Originally designed for the storage of data from the Caenorhabditis elegans sequencing project, it has been adapted by multiple groups for use with various model organisms. In addition to its continuing role as the primary repository of all C. elegans sequencing and mapping data (Waterston and Sulston 1995), ACEDB is currently being used as the engine behind the Saccharomyces cerevisiae genome database (SGD; Cherry et al. 1998), the human sequencing projects at the Sanger Centre and the Washington University Genome Sequencing Center (Waterston and Sulston 1998; C. elegans sequencing consortium 1998), and a variety of crop plant and livestock genome analysis projects (Bigwood 1997).
ACEDB provides an intuitive object-oriented view of biological data, rapid response times, and the ability to store very large objects, such as megabases of contiguous DNA sequence. It also provides a graphical user interface complete with many specialized data visualization tools, such as a genetic map viewer and a sequence annotation display. However, the original ACEDB architecture was hampered by the lack of a client/server architecture and the difficulty in adapting the hard-coded graphical displays to new purposes; therefore, some groups have abandoned ACEDB in favor of commercial relational database systems.
These deficiencies have been corrected over the past 2 years by the development of a client/server version of ACEDB. Client/server ACEDB allows both graphical and text-only clients to connect to remote ACEDB databases from across the Internet, send database queries to the server, and retrieve whole or partial data objects in response. In addition, clients with sufficient privileges can update a remote database by adding new objects or modifying existing ones.
This paper announces the availability of AcePerl, a Perl programming language interface to ACEDB. Perl (Wall et al. 1996) is used widely in the bioinformatics community for everything from local data management, to data format conversion, to access to remote Web-based databases and is easily learned even by those without formal training in programming. This interface allows direct access to both local and remote ACEDB databases via an elegant programmer’s interface. Biologists and novice programmers can write simple scripts to fetch and display ACEDB data objects, whereas the more experienced can formulate batch queries to perform complex data mining operations. The interface allows multiple ACEDB databases to be opened simultaneously and data objects compared, integrated, and moved from one to the other. In addition to the basic interface, a set of Perl CGI scripts known collectively as AceBrowser allows any networked or local ACEDB database to be browsed via World Wide Web (WWW) pages, and serves as a template for the design of application-specific Web pages.
RESULTS
API Design
In ACEDB, all data are organized into a series of “models” that represent familiar biological objects. For example, there is a Genome_Sequence model that contains all of the structural and annotation information about a large stretch of genomic sequence, a Protein model that stores functional information about proteins, and a Genetic_Map model, containing information about a particular genetic map. In addition, most ACEDB databases hold nonbiological information as well, such as Author and Citation models. The data types stored by an ACEDB database are completely under the control of the database designer.
A typical Genome_Sequence object is shown in Figure 1, which is a screen shot taken from the AceBrowser application described below. An object consists of a unique identifier, in this case “C01C4,” and a series of tags, in this case “DNA,” “Structure,” “DB_info,” and so forth. Tags may have subtags, as seen, for instance, to the right of the Structure tag, which has the subtags “From” and “Subsequence.” Subtags may be nested to any degree, forming hierarchical structures similar to the outline of a paper.
Figure 1.
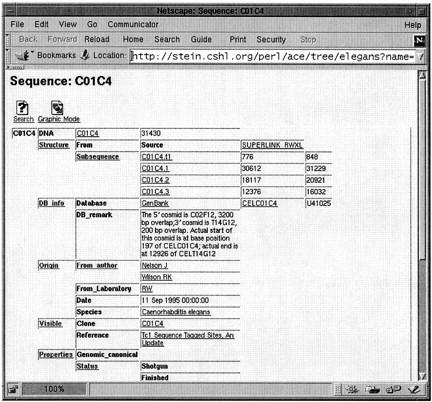
An ACEDB database object is a hierarchical set of tags and data values.
The tags act like field names to identify the position of the actual data. For example, to the right of Subsequence are entries for four predicted genes (or other transcriptional units), along with their starting and ending positions. The data can be a simple value, as for instance, the text to the right of the tag “DB_remark,” or it can be a link to another database object. Examples of links are the four predicted genes, which are database Sequence objects.
Our goal in designing the AcePerl application programming interface (API) was to closely mirror the structure of the ACEDB database. Biological data are fetched, modified, and written as whole objects, and the objects provide specialized functions for retrieving their tags and tag data. Box B1 shows a short Perl script that fetches the C01C4 sequence from a remote database, retrieves and prints the value of the DB_remark field, and prints out the names and brief identifications of each of the genes in the sequence. The intent is not to teach Perl programming but to demonstrate the simplicity of the API.
Box 1.

This program fetches and displays information about a C. elegans genomic sequence and its predicted genes.
#!/usr/bin/perl
The first line of the program identifies it as a Perl script, and is required by the Unix operating system.
use Ace;
This loads the ACDB interface, making its functionality available to the script.
$db=Ace->connect( -host=>′wormsrv1.sanger.ac.uk′,-port=>210201);
This line attempts to connect to a public access C. elegans database located at Internet address wormsrv1.sanger.ac.uk and at Internet port 210201. There may be several different databases running simultaneously on a given host, and the port allows them to be distinguished from one another. If successful, a database object is created and stored in the Perl variable $db ($ is the Perl language indication that this is a single-valued variable). Scripts may connect to multiple databases simultaneously if they wish.
$sequence = $db->fetch(Genome_Sequence=>′C01C4′);
Using the database object stored in $db, this line attempts to fetch the Genome_Sequence named C01C4. If successful, a new sequence object is created and stored in a Perl variable named $sequence. This is the simplest way to fetch objects, by referring to them specifically by model name and identifier. Users of the API may also fetch objects by using a key word search, a partial name match, or an ACEDB query language statement. The latter allows the database to be searched for objects that satisfy one or more conditions or that satisfy more complex relationships between objects.
print $sequence->DB_remark, ″\n″;
This statement fetches the contents of the DB_remark tag and prints it out. Note that this works even though DB_remark is a subtag of DB_info. The ″\n″is a Perl notation that prints a newline character at the end of the line.
@genes=$sequence->Subsequence; foreach $gene (@genes) { print $gene, ″″,$gene=>Brief_identification, ″\n″; }
These four lines display information about each of the predicted genes in the sequence. The first line fetches the contents of the Subsequence tag and stores it into the Perl variable @genes. In Perl, the @ symbol indicates that multiple values are expected. In fact, this operation retrieves a list of four Sequence objects.
The next line begins a foreach loop, in which the program iterates over each member of the @genes list in turn, storing the object into a variable named $gene with each iteration. (This is a feature of Perl and is not directly related to the AcePerl API.) Within the body of the loop, the program fetches the contents of the Brief_identification tag, which indicates the database curator’s best guess as to the identity of the gene. The value returned by Brief_identification is printed, along with the gene’s database identification, separated by a space. Note that this operation has transparently performed several database accesses to fetch each of the sequence objects in turn.
The output of this script is shown below. From this we learn that C01C4.t1 is a transcription unit for tRNAMet, C01C4.1 is weakly similar to the Aplysia californica buccalin protein, C01C4.3 is a serine/threonine protein kinase, and C01C4.2 is as yet unidentified:
The 5′ cosmid is C02F12, 3200 bp overlap; 3′ cosmid is T14G12, 200 bpoverlap. Actual start of this cosmid is at base position 197 of CELC01C4; actual end is at 12926 at CELT14G12
C01C4.t1 tRNA-Met
C01C4.1 A. californica buccalin precursor (weak)
C01C4.2
C01C4.3 serine/threonine protein kinase
Moving Objects between Databases
If the author of the script wished to store copies of the four retrieved genes into a local database, he/she could add the following two lines to the script:
$localdb=Ace−>connect (−path=>‘/home/acedb/mydata‘);
$localdb−>put (@genes);
The first line opens up a connection to a local non-networked database (the -path argument points to the location of the database directory in the file system), and the second copies the list of genes into the newly opened database. The sequence objects are now incorporated into the user’s private database, where he/she is free to examine and modify them or view them using ACeDB’s graphical tools. It is equally straightforward to create new objects, modify existing ones, or to delete objects entirely.
Retrieving Meta-Information about Objects
Unlike some other database APIs, the AcePerl interface does not require the script author to understand the design of the database before accessing it. Information about which models are defined by the database, how the objects are structured, and other schema information is available through a few function calls.
For example, to obtain the list of models contained within a database, the programmer could write the following piece of code:
@models=$db−>models
This will return a list of available data models such as “Allele,” “Author,” “Clone,” and so forth. These models can be explored further to discover their internal structure. It is similarly easy to discover the structure of a data object. For example, the tags function will return a list of all tags in an object:
@tags=$sequence−>tags
These functions make it possible for a programmer to work productively with a database that he/she is encountering for the first time.
Format Conversion
Because ACeDB databases must live in a heterogeneous environment of many database engines and file formats, the AcePerl API has a rich set of function for interconverting ACeDB objects with other formats. A sampling follows:
$sequence−>asDNA
This converts Sequences and objects that contain sequence data into FASTA format for analysis by use of BLAST, FASTN, and other nucleotide analysis tools. It also facilitates moving objects into other nucleotide databases. An asPeptide function provides the equivalent conversion for protein sequences.
$sequence−>asTable
This converts the object into tab-delimited text, for importation into spreadsheets, relational databases, and proprietary databases such as Microsoft Access.
$sequence−>asString
This converts the object into a pretty-printed string representation of the object, preserving its hierarchical structure.
$sequence−>asHTML
This converts the object into HTML for incorporation into web pages. Hypertext links are automatically created when appropriate. This is how Figure 1 was generated.
$sequence−asGIF
This converts the object into a graphical representation of the data and returns a GIF image for display in a web page or other graphical application. An example of this format is shown in Figure 2.
Figure 2.
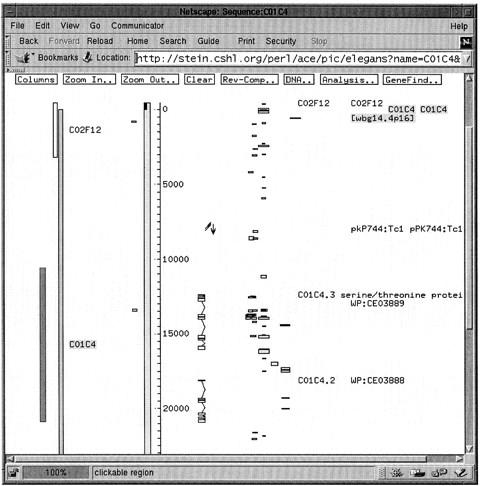
Graphical representation of the C01C4 Genome_Sequence object returned by the asGIF function.
$sequence−>asAce
This converts the object into the flat-file representation of ACEDB data known as “.ace” format. Another function is available to reverse the operation and import .ace format files.
AceBrowser Web Interface
We have built a set of WWW Common Gateway Interface (CGI) scripts on top of the AcePerl API, both as a demonstration of how to use the interface and as a useful service in itself. When these CGI scripts are installed on a Web server, remote and local ACEDB databases can be browsed via a series of dynamically generated Web pages. Figures 1 and 2 show sample output from the CGI scripts in text and graphical modes, respectively. The pages are fully active and linked. In text mode, selecting a data field that corresponds to another object in the database will link to a new page that displays the contents of the object. Selecting a tag will cause a portion of the hierarchy to collapse or expand like an outline in a word processing program. In graphic mode, all buttons and objects are links, allowing the user to change the view and to display related database objects in much the way that he/she could with the original graphical ACEDB. Graphical objects that are stored in the database, such as JPEG pictures, are displayed in the pages as inline images.
The AceBrowser scripts provide four database search interfaces. An object-browsing mode allows the user to retrieve objects by class, optionally restricting the search to names that match a pattern. A text-browsing mode allows the user to search the entire database for matching text or key words (see Fig. 3) for an example of a search for the word “pathogen.” A sequence-similarity search allows the user to search the database for matching Sequence entries using BLASTN or BLASTP. Finally, we provide direct access to the Ace query language for more sophisticated searches.
Figure 3.
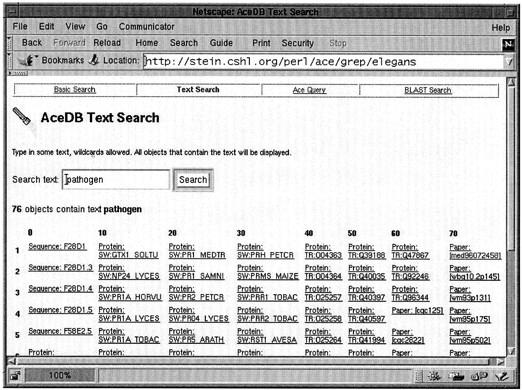
The results of a text search for the word “pathogen” with the AceBrowser interface.
AceBrowser scripts are similar in many respects to WebAce, a set of Perl scripts originally written in Douglas Bigwood’s laboratory at the U.S. Department of Agriculture and now supported by the Sanger Centre (WebAce 1998). AceBrowser can be used side-by-side with WebAce or as a template on which to build application-specific Web interfaces to ACEDB data sources. AceBrowser and WebAce both lack some of the user interface features of the stand-alone X-windows Ace client (such as DNA analysis) but provide all of the basic functionality for searching and navigating ACEDB databases.
Performance
The performance of the AcePerl API depends on the speed of the network connecting the client program to the server database. To reduce the impact of network speed, a great deal of effort has been made to minimize the amount of information that must be transferred between server and client. For example, when database objects are first retrieved from the remote database, only their identifiers are transferred. The operation of fetching the body of the object is deferred until the data are requested, and then only the portion of the object that is needed is transferred.
Operations that do not require the transmission of large objects are fast. For example, it takes just 5 sec to retrieve the database identifiers of all 3782 Genomic_Sequence objects from a server located at the Sanger Centre in the United Kingdom and transmit them to a client located at Cold Spring Harbor in New York. However, fetching the DNA sequences associated with those objects (average size, 35 kb) takes significantly longer, ∼3 sec per object.
DISCUSSION
Before the advent of the WWW, biologists who wished to mine the resources of biological databases faced formidable challenges. If the amount of data was small, one option was to copy the database onto the researcher’s own machine, an operation that usually required the installation of specialized software and a certain degree of technical expertise. Another option, suitable for larger databases, was to use Telnet to log into the database host machine from across the Internet and to access the database via a specialized command-line interface. Unfortunately, most biologists found these barriers to be inconvenient at best, and insurmountable at worst.
The web altered this situation radically by introducing a paradigm in which biological data could be retrieved in a universal format, HTML, displayed by a widely available software viewer, the web browser, and fetched by use of a standardized object naming nomenclature, the Uniform Resource Locator (URL). It became possible for biologists to access large comprehensive biological databases such as GenBank (Benson 1998) and Genome Data base (GDB) (Letovsky 1998), as well as smaller organism-specific databases such as FlyBase (FlyBase Consortium 1998) and SGD (Cherry 1998), all without installing new software or learning an unfamiliar command-line interface. Almost as significantly, this paradigm made it possible for diverse biological databases to achieve a first-order level of integration by providing cross-reference style links to connect objects in the different databases.
Although web-based front ends to biological databases do provide interactive, browse-level access to biological data, they are deficient in at least one aspect: There is no programmable, batch-level interface. This is important when the questions the biologist seeks to answer move beyond object-at-a-time retrieval to more sophisticated problems.
To give a concrete example, the National Center for Biotechnology Information’s (NCBI’s) excellent Web Entrez system (Benson et al. 1998) makes it easy to retrieve the GenBank entry for a genomic sequence given its accession number. Once the GenBank entry is retrieved, one can follow links to the proteins encoded by the sequence and to the position of the sequence on any of several genome maps. However, as soon as the researcher needs to interact with the data in a more complex way, things get difficult. For example, what if the researcher wishes to find all sequences located on chromosome 3, retrieve their predicted ORFs, and return the protein sequences as a list sorted by position? This could not be done easily with Web Entrez, because like other web-based systems, the types of questions that the researcher is allowed to ask of the system are limited to a set of prepackaged queries that are hard-coded into the scripts that drive the web pages. To move beyond this, researchers must be able to go around the web interface and “talk” directly to the underlying database. Furthermore, they must engage in some programming, either directly by writing a Perl script themselves or indirectly by hiring someone to do it for them.
A number of technologies have the potential to provide direct access to biological databases. Relational databases such as SyBase (Anderson 1997), Oracle (Hipsley 1996), and mySQL (T.c.X 1998) provide client/server APIs that allow software programs to send structured query language (SQL) requests to remote databases and to retrieve the results. Although these APIs provide some very compelling advantages, including operating system independence and the ability to interact with databases from different vendors, they are rarely used to provide public access to biological databases. The primary reason for this is that in a typical relational database, a biological data type, such as a DNA sequence and its associated information, must be split across multiple tables in the interests of efficiency. The result of “fracturing” the object in this way is that the database design becomes large and difficult for outsiders to understand, making casual public access impractical.
The Common Object Request Broker Architecture (CORBA) (Orfali and Harkey 1998) technology solves the problem of fractured data types by providing a standard for sharing whole data objects across the Internet. CORBA includes a data definition language, a data access protocol, and common services for naming and locating objects. CORBA has the potential to overcome the limitations of relational database APIs by keeping biological data types intact and providing a uniform cross-platform interface to heterogeneous databases and other services (Rodriguez-Tome et al. 1997; Barillot 1998). Although CORBA will likely be the ultimate solution to the problem of biological database access, in the short term, CORBA-based interfaces have been slow to appear because of the complexity of CORBA application programming and the need to agree on standardized biological data types (OiB 1998).
At least as far as ACEDB databases are concerned, AcePerl provides direct access to biological databases in a way that preserves the original biological object. It allows simple queries, such as name searches and full text searches, as well as complex ones with the full ACEDB query language. For example, the task proposed earlier of fetching a genome’s worth of predicted peptides and presenting them in mapped order can be performed with a Perl script ∼20 lines long. Multiple databases can be opened simultaneously, and objects can be moved between them, facilitating the integration and consolidation of diverse sources of biological information. Finally, a rich set of conversion routines allow ACEDB objects to be interconverted with other common data formats, making it possible to move data between ACEDB and other databases and data analysis programs.
AcePerl was designed with the intent of making the rich information sources stored in various ACEDB databases more available to the public, as well as making it easier to set up and maintain private ACEDB databases in researchers’ own laboratories. To that end, the programmer’s interface is simple, consistent, and well documented. The process of building and installing the AcePerl library itself is done in an automated fashion by use of tools that are familiar to Perl programmers. The AcePerl interface can be readily learned by novice Perl programmers, and, it is hoped, by computer-savvy biologists.
AcePerl is complementary to JADE, an ACEDB interface for the Java programming language (Stein et al 1998). In a production setting, JADE can be used to develop fast graphical displays for ACEDB objects, whereas AcePerl can be used to provide data entry, batch processing, scripted queries, and other nongraphical tasks.
There are a number of issues that still need to be addressed in the AcePerl API. Currently it is only possible to move a data object from one ACEDB database to another if both databases share the same model for the object. A future version of AcePerl will relax this requirement so that objects with substantially similar models (e.g., a model that is a subset of another) can be transferred as well. A related issue is object naming. The AcePerl library currently makes no attempt to resolve naming conflicts when objects are transferred from one database to another. A future version of the library will resolve naming conflicts by optionally qualifying copied objects with the name of its original database.
We also intend to augment the format conversion routines to increase the flexibility of the interface. One current project is to provide import and export routines for the NCBI ASN.1 and GenBank flat file formats (Ostell and Kans 1998). Another is to provide conversion routines for the BoulderIO format used at the Whitehead Institute/MIT Center for Genome Research (Stein et al 1994).
A final important task is to port AcePerl to the Microsoft Windows architecture, so that it can be used from Windows 95/98 and Windows NT systems. Currently, it is only practical to use AcePerl from Unix machines, and although the increasingly popular freeware Linux operating system is supported, this will present an obstacle for many laboratories. We are currently collaborating with Richard Bruskiewich (University of British Columbia, Vancouver) the author of the Windows port of ACEDB, to achieve this goal.
AcePerl and AceBrowser are freely available under terms that allow unrestricted use and redistribution. Source code, documentation, demos and example scripts, as well as lists of publicly accessible ACEDB databases, are available at http://stein.cshl.org/AcePerl.
METHODS
AcePerl operates on top of the ACEDB 4.6 distribution, available at ftp://ncbi.nlm.nih.gov/repository/acedb/ace.4_6/. It will also interoperate with ACEDB version 4.5e after applying source code patches that are included in the AcePerl package. AcePerl requires a machine running the Unix or Linux operating systems and Perl version 5.004 or higher. You will also need a working C compiler and an Internet connection. These are standard features of most Unix distributions.
AcePerl was developed on an Intel Pentium 300 running Slackware Linux version 2.0.33. Memory requirements are generally small, but 32 Mb is recommended. More memory will be required to run a local ACEDB database. At least 96 Mb is recommended for the current C. elegans release.
Acknowledgments
We gratefully acknowledge the contributions and insights of the following individuals during the design of AcePerl and the preparation of this manuscript: LaDeana Hillier, Richard Durbin, Richard Bruskiewich, James Gilbert, Mark Yandell, Tim Hubbard, Michael Hoffman, and Sam Cartinhour. This work was supported by National Institutes of Health grant 5P50 HG01458-03.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL stein@cshl.org; FAX (516) 751-8461.
REFERENCES
- Anderson GW. Client/server database design with sybase: A high-performance and fine-tuning guide. New York, NY: McGraw-Hill; 1997. [Google Scholar]
- Barillot E, Guyon F, Cussat-Blanc C, Viara E, Vaysseix G. HuGeMap: A distributed and integrated Human Genome Map database. Nucleic Acids Res. 1998;26:106–1077. doi: 10.1093/nar/26.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF. GenBank. Nucleic Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigwood D. Databases at the National Agricultural Library. 1997. http://probe.nalusda.gov:8000/alldbs.html http://probe.nalusda.gov:8000/alldbs.html. . [Google Scholar]
- C. elegans sequencing consortium. 1998. Genome sequence of the nematode Caenorhabditis elegans: a platform for investigating biology. Science (in press). [DOI] [PubMed]
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, Weng S, Botstein D. SGD: Saccharomyces genome database. Nucleic Acids Res. 1998;26:73–80. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Thierry-Mieg J. A C. elegans database. 1991. Documentation, code and data available from anonymous FTP servers at lirmm.lirmm.fr, cele.mrc-lmb.cam.ac.uk and ncbi.nlm.nih.gov. [Google Scholar]
- FlyBase Consortium. FlyBase: A drosophila database. Nucleic Acids Res. 1998;26:85–88. doi: 10.1093/nar/26.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipsley P. Developing client/server applications with Oracle Developer/2000. Indianapolis, IN: SAMS Publishing; 1996. [Google Scholar]
- Letovsky SI, Cottingham RW, Porter CJ, Li PWD. GDB: The human genome database. Nucleic Acids Res. 1998;26:94–100. doi: 10.1093/nar/26.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OiB. Objects in bioinformatics 1998. 1998. http://www.ebi.ac.uk/oib98/ http://www.ebi.ac.uk/oib98/. . [Google Scholar]
- Orfali R, Harkey D. Client/server programming with JAVA and CORBA. New York, NY: John Wiley & Sons; 1998. [Google Scholar]
- Ostell JM, Kans JA. The NCBI data model. In: Baxevanis A, Ouellette Y, editors. Bioinformatics: A practical guide to the analysis of genes and proteins. New York, NY: John Wiley & Sons; 1998. pp. 121–144. [Google Scholar]
- Rodriguez-Tome P, Helgesen C, Lijnzaad P, Jungfer K. A CORBA server for the Radiation Hybrid DataBase. Intell Systems Mol Biol. 1997;5:250–253. [PubMed] [Google Scholar]
- Stein LD, Marquis A, Dredge E, Reeve MP, Daly MJ, Rozen S, Goodman N. Splicing UNIX into a genome mapping laboratory. USENIX Summer Technical Conference. 1994;2:221–229. http://www.usenix.org/publications (available at http://www.usenix.org/publications). ). [Google Scholar]
- Stein LD, Cartinhour S, Thierry-Mieg D, Thierry-Mieg J. JADE: An approach for interconnecting bioinformatics databases. Gene. 1998;209:GC39–GC43. [PubMed] [Google Scholar]
- T.c.X. (Data Konsult AB) MySQL home page. 1998. http://www.tcx.se/ http://www.tcx.se/. . [Google Scholar]
- Wall L, Christiansen T, Schwartz R. Programming Perl. 2nd ed. Sabastopol, CA: O’Reilly and Associates; 1996. [Google Scholar]
- Waterston R, Sulston J. The genome of Caenorhabditis elegans. Proc Natl Acad Sci. 1995;92:10836–10840. doi: 10.1073/pnas.92.24.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The human genome project: Reaching the finish line. Science. 1998;282:53–54. doi: 10.1126/science.282.5386.53. [DOI] [PubMed] [Google Scholar]
- WebAce. WebAce home page. 1998. http://webace.sanger.ac.uk/ http://webace.sanger.ac.uk/ [Google Scholar]