

Summary
Autophagy is a key cytoplasmic biomass and organellar quality and quantity control pathway of the eukaryotic cell. It is particularly suited to capture and degrade large, multi-macromolecular cytosplasmic targets earmarked for degradation or turnover. Typical autophagic cargos represent large swaths of cytosol as a source of energy and anabolic precursors at times of growth restrictions imposed by the absence of growth factors, nutrient limitation or hypoxia. Autophagy is the only effective mechanism for removal of whole organelles such as leaky or surplus mitochondria, disposal of potentially toxic protein aggregates too large for proteasomal removal, and elimination of intracellular microbes including bacteria, protozoa and viruses. Recent studies have shown that human immunodeficiency virus (HIV) is targeted for eliminated by autophagy but that this is countered by the viral protein Nef. Here we review these relationships and underscore the untapped potential of autophagy as a druggable antiviral process.
Introduction to autophagy
The sensu stricto autophagy refers to the process of macroautophagy whereby cytoplasmic targets are captured within organelles of endomembranous origin, termed autophagosomes, which subsequently mature into autolysosomes where the captured cargo is degraded or otherwise eliminated [1]. The physiological functions of autophagy include providing a cell-autonomous source (by auto-digestion of cytosol) of energy and amino acids at times of cellular metabolic crisis or nutritional deprivation, prevention of cell death or senescence due to accumulation of faulty organelles and large macromolecular aggregates [1] and the still debated potential cell death modality [2]. These classical roles of autophagy have recently been amended to include a wide range of innate and adaptive immunity functions [3]. All cells rely on constitutive autophagy to carry out the basal housekeeping role of eliminating sporadically damaged organelles due to normal wear and tear, for example occasional depolarized mitochondria that cannot rejoin the mitochondrial network [4]. The baseline, housekeeping autophagy can be augmented by elicited autophagic responses to nutritional, differentiation, and danger signals [5]. Autophagy in principle involves three morphological stages (Fig. 1): (i) initiation (formation of crescent membranes termed phagophores), (ii) elongation and closure (increase of the phagophore and its closure into a completed autophagosome containing the sequestered cargo), (iii) and maturation (conversion of autophagosomes into degradative organelles termed autolysosomes by fusion with late endosomal and lysosomal organelles or trafficking carriers).
Fig. 1. Macroautophagy.
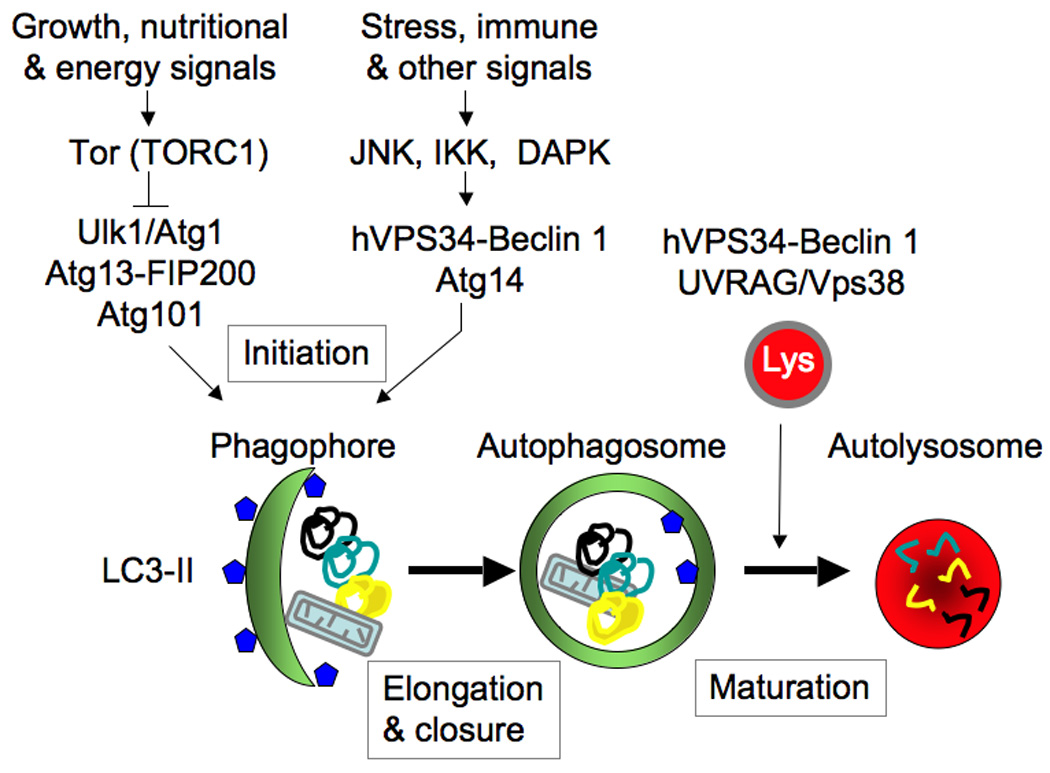
Shown are morphological stages of autophagy, different autophagic organelle intermediates and only key signaling and regulatory systems. The Atg conjugation cascade is deliberately omitted as it has been covered in numerous reviews. Lys, lysosome. LC3-II, lipidated (by phosphatidylethanolamine at the C terminus) human Atg8.
The keys to initiation of autophagy are the regulation of (i) Atg1 (in yeast) or its equivalent Ulk1 (in mammals) complexes [6] and (ii) the phosphatidylinositol (PI) 3-kinase hVPS34-Atg6 (Beclin 1) complex with Atg14 (complex I) and additional interacting components [7]. To control autophagy in response to growth factor and nutritional signals (Fig. 1), the Atg1/Ulk1 complex is coupled to Tor complex 1 (TORC1). In mammals, mTORC1 during growth factor and nutrient-replete conditions associates with the Ulk1-Atg13-FIP200-Atg101 complex (with mammalian FIP200 being a functional equivalent of yeast Atg17), thus inhibiting autophagy. Upon receiving starvation signals, mTORC1 dissociates from the Ulk1-Atg13-FIP200-Atg101 complex, which appears to translocate [6] to (still elusive in mammalian cells) preautophagosomal membranes that may possibly involve rough endoplasmic reticulum (rER) [8] areas that can be visualized by a marker, DFCP-1 [9]. There, the Ulk1 complex, in cooperation with the PI 3-kinase hVPS34 complex I and its lipid product PI 3-phosphate (PI3P) along with the PI3P-binding effector proteins WIPI-1 and WIPI-2 (equivalents of yeast Atg18) lead to the formation of nascent autophagosomes [6]. The phagophore elongation and autophagosomal closure phase requires Atg9 (the sole integral membrane Atg protein whose cyclical trafficking between peripheral membrane pools and the growing phagophore is controlled by Atg1/Ulk1) and Atg8 (in mammalian cells represented by a whole family of Atg8 proteins: LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2/GATE16). LC3s (Atg8) are C-terminally conjugated, in a process assisted by the Atg12–Atg5-Atg16L complex. Atg8 proteins are required for phagophore membrane growth and eventual closure to complete the double-membrane autophagosome [10]. Maturation is the final, degradative stage of the pathway, whereby closed autophagosomes fuse with late endosomal/lysosomal organelles or carrier intermediates, generating autolysosomes, delimited by a single membrane. This is where the captured cargo (cytosol, ribosomes, protein aggregates, mitochondria, microbes) is degraded by hydrolytic enzymes [1], or alternatively (with only a few examples known to date) expelled by a process akin to exocytosis [11].
Autophagy in innate and adaptive immunity
Autophagy has many roles in innate and adaptive immunity [3] and infection [12]. First, autophagosomes can directly destroy microbial pathogens (bacteria, protozoa and viruses). For example, autophagosomes can directly capture microbes that are free in the cytosol coated with poly-ubiquitin complexes via a number of adapter proteins (e.g. p62, NDP52) that in turn bind to LC3 (Fig. 2). In this process, the pathogen is being reeled into a phagophore, like a fish on a hook-line [13–16]. In a mirror image of this process, autophagy can deliver antimicrobial or neo-antimicrobial peptides (the latter being derived from cytosolic components, such as ribosomal proteins or ubiquitin, by limited digestion in autolyosomes) to microbes still safely ensconced inside their preferred membranous niches, such as the Mycobacterium tuberculosis phagosome (Fig. 2) [17]. Second, autophagy is an effector and a regulator of innate immunity responses via pattern recognition receptors (such as Toll-like receptors, Nod-like receptors, and RIG-I-like receptors) [18–21] and systems sensing damage-associated molecular patterns such as ATP and self-DNA-containing complexes [22,23]. Third, atophagy is a co-effector of cell-autonomous defenses dependent on immunity related GTPases [24–26] including human IRGM [27], which is a risk factor in Crohn’s disease [28] and tuberculosis [29]. Fourth, autophagy is controlled by T cell polarization, with IFN-γ (a Th1 cytokine, protecting against intracellular pathogens) inducing autophagy, whereas IL-4 and IL-13 (Th2 cytokines, permissive for intracellular microbes) inhibit autophagy [30]. Fifth, autophagy enhances endogenous cytoplasmic antigen (including viral) presentation via MHC II [31] and MHC I molecules [32], with implications for selection of T cell repertoires [33] and vaccine development [34].
Fig. 2. Two principal ways of how autophagy directly kills microbes.
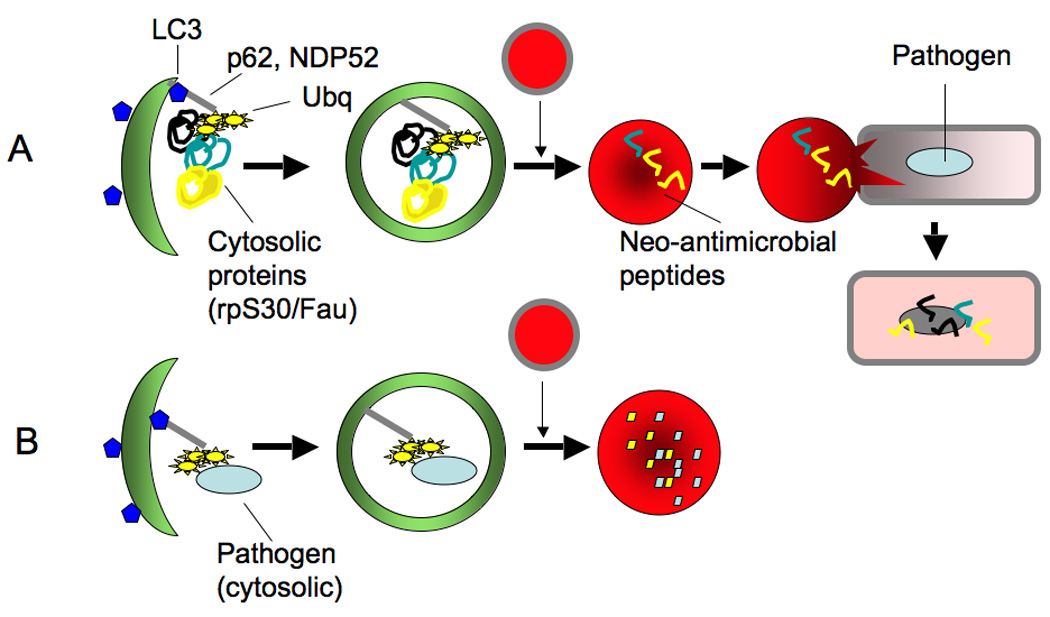
(A) It brings cytoplasmic components, which serve as the source of antimicrobial or neo-antimicrobial peptides (the latter derived by proteolytic processing of e.g. ribosomal protein S30 precursor Fau and ubiquitin (Ubq) processing into peptides in autolysosomes) and delivers them to membranous compartments harboring intracellular bacteria. (B) It captures microbes in the cytosol and brings them into the autophagosome for killing in autolysosomes. In both cases autophagic adapter proteins such as p62 and NDP52 (they interact with LC3 via a WXXL motif) are used to capture polyubiquitinated cargo for delivery to autophagosomes.
HIV-1 infection and pathogenesis
Recent studies have implicated autophagy in HIV biology [35–38] and pathogenesis [39,40] (Fig. 3). Before we review these reports, here we briefly summarize the understanding of HIV-1 infection and progression to AIDS based on the “pre-autophagy era” studies. The complex infectious cycle, which includes acute and chronic infection, of human immunodeficiency virus HIV-1 leading to AIDS starts with penetration of the incoming virions through mucosal surfaces during sexual intercourse [41,42], where they become subject to uptake by, or surface association with, dendritic cells (DC) [42]. DCs transfer the virus to lymph nodes where infection spreads to both antigen presenting cells (such as macrophages and DCs) and CD4+ T cells [43,44], through a highly efficient cell-to-cell spread process (including transfer between T cells) dependent on virological synapse or polysinapses [45–47]. In these early events, the incoming HIV-1 overpowers an amazing array of cell-autonomous anti-retroviral defense mechanisms such as the polynucleotide cytidine deaminase RNA editing APOBEC3 proteins (countered by the viral product Vif, which assembles the elongin-cullin-Rbx ubiquitin ligase and commits APOBEC3s to degradation) [48] and an (unfortunately) inefficient human version of the retroviral resistance factor TRIM5α [49], which is far less potent in defense against HIV-1 than its monkey orthologs due to its unusually fast molecular evolution as a retroviral restriction factor [50]. The infection of activated CD4+ T cells results in a profound loss of HIV-infected T cells, nearly depleting gut lymph nodes, where 80% of all lymphoid tissues and cells in the human body are located [51,52]. Despite CD8+ T cell and natural killer (NK) cell surveillance, with MHC-I alleles HLA-B, HLA-C and NK cell receptor KIR polymorphisms with disease progression [53–56] and elimination of infected cells, the virus escapes (by many mechanisms such as downregulation of MHC-I molecules on the cell surface as well as escape mutations in viral proteins targeted by CD8+ cells) these rather efficient but imperfect effectors of immunological control. Eventually, the virus establishes a chronic infection marked by postacute set point level of plasma viremia, a parameter termed set point viremia, which represents the initial “watermark” for the outcome of the HIV-host defense battles during the acute infection. Progression to AIDS is a complex process that correlates with the set point viremia and its increase in early years post-sero-conversion. If unchecked by antiretroviral therapy, HIV-1 leads to continuous CD4+ T cell decline, partly due to apoptosis of infected cells, partly due to elimination of infected CD4+ T cells by CD8+ cytotoxic T cells, and partly due to cell death of bystander, uninfected T cells [57–60]. The loss of CD4+ T cells is further exacerbated by the co-receptor usage switch from R5, or M-tropic, to X4, or T-tropic, which is more pathogenic to T cells but less fit for transmission, since X4 can lead to cell death even prior to completion of reverse transcription [61]. Thus, HIV-1 variants emerging in the patient during late-stage disease are associated with more aggressive T cell depletion, resulting in AIDS.
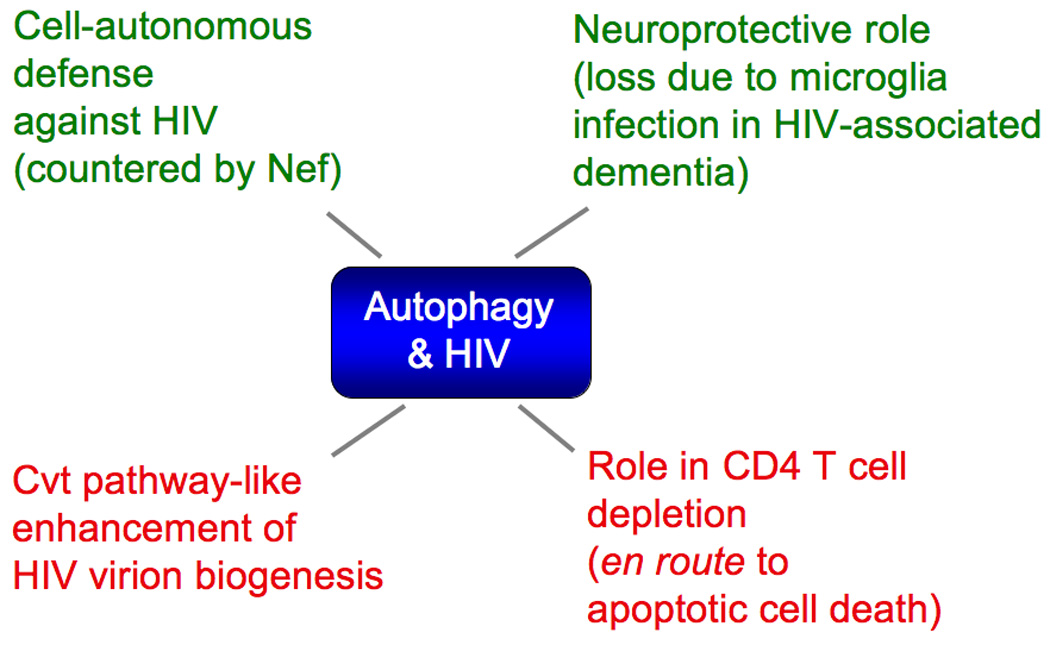
Fig. 3. Known intersections between autophagy and HIV.
One of the important factors leading to AIDS in HIV-1 infected humans (as opposed to SIV-infected sooty mangabey monkeys, who do not develop AIDS in part due to preserved mucosal integrity and muted innate immunity responses among other factors) is the permeability of the gastrointestinal tract during HIV infection that allows microbial product translocation (leading to, for example, increased plasma LPS levels) and chronic immune activation [62] with a component that may involve programmed death 1 receptor increase on monocytes and associated CD4+ T cell dysfunction [63]. This sustains the inexorable loss off infected and uninfected CD4+ T cells and emergence of acquired immunodeficiency syndrome, characterized by opportunistic infections with other microbes and malignancies that can otherwise be controlled by a competent immune system. The virus also seeds latent reservoirs and establishes persistent HIV infection primarily in long-lived cells such as resting T cells (e.g., memory CD4+ T cells) and macrophages (both being spared from virus-induced cell death and depletion), microglia, and other cell types. The reservoirs of the persistent virus, with possible re-seeding by viral exchange between T cells and antigen presenting cells, become evident as patient’s viremia returns upon interruption of, otherwise very effective, so-called highly active antiretroviral therapy (HAART) [64,65].
Autophagy and HIV
Several lines of investigations in recent years have made inroads in exploring the multi-pronged role of autophagy in HIV infection (Fig. 3) and pathogenesis as: (i) a previously unappreciated function of autophagy as a cell-autonomous defense against HIV-1 [37,38], (ii) a pathway that during HIV-1 biogenesis enhances virion assembly, production, or release from cells [35,37,38]; (iii) a process participating in phenomena leading up to cell death of CD4+ T lymphocytes, including demise of bystander (uninfected) T cells [38,39,60,66–70]; (iv) a neuroprotective process in neurons, compromised due to HIV-1 infection of microglia/macrophages of the central nervous system, as a contributing factor in HIV-associated dementia [40,71,72]; and (v) an innate immunity process countered by HIV-1 [36,37] through Nef, acting as a specific HIV-1 factor targeting Atg6 (Beclin 1) complexes [37].
HIV is a target for elimination by autophagy if not counteracted by the viral accessory protein Nef
As a cell-autonomous innate defense against HIV-1, autophagy eliminates HIV in macrophages [37]. However, this is detectable only after the virus is disarmed by elimination of the nef gene. Nef, with the name originating from a misnomer (negative replication factor), is a 27-kDa, myristoylated HIV protein, lumped into the category of accessory viral proteins along with Vif, Vpu, Vpr (or Vpx in HIV-2), since these HIV proteins are not essential for in vitro viral growth. Nevertheless, Nef is required for maintaining high viral loads during infection in humans and contributes to the progression to fully blown AIDS. Incidentally, people infected with HIV-1 strains with deletions of the nef gene do not progress to AIDS symptoms, or progress with considerable delay compared to infections with standard HIV-1 strains. Nef is required for efficient viral replication and AIDS pathogenicity of humans infected with HIV-1, as cohorts of patients infected with strains deleted for nef have been found with normal CD4+ T cell counts 14 years after infection [73,74]. Deletions in the nef gene in SIV have similar effects in the attenuation of an otherwise highly pathogenic model in Rhesus monkeys [75].
The mechanisms by which Nef acts as a pathogenic factor in vivo are not fully understood, and those that have been identified until very recently, while important, are mostly indirect. The indirect effects include downregulation of two key surface molecules, MHC class I (thus evading recognition of infected cells by CD8+ cytotoxic lymphocytes) and CD4 (possibly promoting dissemination of infectious virus progeny away from the already infected cell). The latter role of Nef may overlap or be redundant with the effects of Vpu, which antagonizes the host restriction factor named tetherin. Another proposed indirect effect of Nef is the ability of Nef from non-pathogenic SIV (but not from HIV-1 and SIVcpz) to downregulate TCR-CD3; hence, the failure of HIV-1 and SIVcpz Nefs to downregulate T cell responses (normally seen with Nefs from non-pathogenic viruses) may contribute to excessive T cell activation, CD4+ T cell loss, and progression to AIDS [76–79]. The most recent work on Nef and autophagy [37] has now uncovered a direct function of Nef in protection against innate immunity and cell-autonomous defenses (Fig. 4), since it acts as an inhibitior of autophagic maturation thus protecting HIV from destruction in autolysosomes. The published data now indicate that Nef binds to Beclin 1 protein complexes, and thus blocks autophagy maturation (but not autophagy induction) in infected macrophages, as an avoidance of autophagic elimination and degradation [37]. The inhibition by Nef of autophagic maturation may help explain several puzzling but unexplained effects of Nef action on cells, such as the less understood Nef-induced accumulation of multivesicular body endosome-like organelles [80,81] and emergence of large vacuoles [82], all of which may have some role in the autophagic flux, and potentially representing pathway intermediates enriched due to the Nef-imposed block in autophagic maturation.
Fig. 4. Autophagy can capture and destroy Nef-less HIV-1.
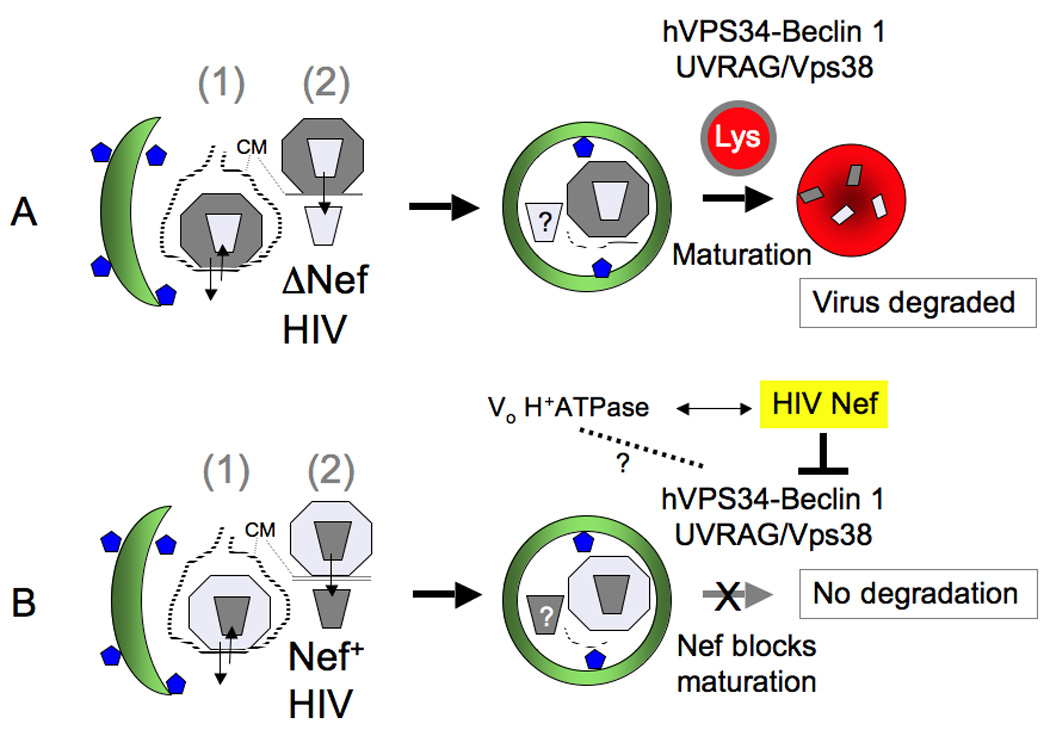
(A) Virus budding into endomembranous compartments or being taken up into endosomes can be captured by autophagosomes, and if Nef is absent the virus can get digested in autolysosomes. (B) Nef blocks maturation by interacting with Beclin 1 complexes (a motif that has been shown to be important for Nef-Vo H+-ATPase interaction is also required for Nef-Beclin 1 complex interactions; dotted line and question mark indicate that these complexes could be related or cooperating). Depicted are: (1) HIV-1 virions in the process of uptake into endosomes with subsequent fusion or en route to in-trans infection through virological synapse, or newly budded virions into endomembranous compartments connected to the plasma membrane via a narrow channel (this occurs in macrophages and dendritic cells). These options are indicated by arrows going in or out of the cell. (2) Upon virus fusion at the plasma membrane with the target cell (e.g., CD4+ T cells) and release of the capsid into the cytoplasm, it may become a target for autophagic removal (question mark indicates that this has not been established yet).
HIV Nef-Beclin 1 interactions depend on the diacidic 174DD175 motif in Nef
The previously reported Nef mutant with changes in the di-acidic motif (174DD175 → 174AA175) responsible for interactions with the V1 domain of vacuolar H+-ATPase and required for CD4 downregulation [77], is essential for the ability of Nef to co-immunoprecipitate with Beclin 1 (mammalian Atg6) [37]. In contrast, the mutation 154EE155 → 154QQ155, in the region of Nef with the di-acidic motif required for β-COP interactions [77,83], does not abrogate Nef co-immunoprecipitation with Beclin 1. Surprisingly, the 2G → 2A mutation, preventing Nef N-terminal myristoylation and assumed to be a sine qua non for Nef association with membranes and many of its functions [77], does not prohibit Nef binding to Beclin 1 complexes. It should be noted that the NefG2A mutant does not completely lose membrane association and instead shifts from plasma membrane to undefined endomembranes [84], which is compatible with Nef’s action on autophagosomal organelles. Importantly, the Nef D174A, D175A mutant that lost the ability to bind to Beclin 1 has no ability to block the autophagic flux [37]. This is of key significance since HIV infection induces autophagy via TLR8 signaling [18], and thus the Nef-dependent action of prohibiting progression of autophagosomes into autolysosomes may spare the virus from early degradation. The dependence of the inhibition of the autophagic flux on the di-acidic 174DD175 motif in Nef is of particular interest, since 174DD175 is also responsible for interactions with the V1 domain of the vacuolar H+-ATPase [77]. Inhibitors of vacuolar H+ ATPase, such as bafilomycin A1 are routinely used in the laboratory to inhibit autophagic flux when it is necessary to differentiate autophagy induction from autophagy maturation [5]. Hence, one possible model emerging from these studies is that Beclin 1, when in the complex with hVpS34 – UVRAG/Vps38 responsible for autophagic maturation, might affect the vacuolar H+-ATPase, and that this is targeted by Nef via its 174DD175 motif (Fig. 4).
Nef action resembles the function of other viral anti-autophagic factors
The inhibitory role of Nef on autophagic flux, strongly suggests that this function of Nef may be of significance and needs to be explored. This is further underscored by the reports that the same cellular defense machinery affected by Nef is targeted by other viral pathogens employing specific viral proteins for this purpose [85]. For example, just like HIV Nef [37], the influenza A virus M2 protein binds Beclin 1 and inhibits autophagic maturation [86]. The herpes simplex virus 1 protein ICP34.5 binds to Beclin 1 but it appears to shut down autophagy altogether [87]. Beclin 1 is also targeted by γ herpesvirus Bcl-2-like proteins (vBcl-2) via binding to the BH3–like domain of Beclin 1 [88–90]. In addition to Beclin 1, other autophagic proteins are targeted by viral factors, as in the case of Kaposi's sarcoma-associated herpesvirus, which encodes viral FLIP (vFLIP) that acts in a fashion similar to cellular FLIP (cFLIP) by preventing Atg3 (an upstream conjugation factor in the LC3-II lipidation cascade) from interacting with LC3 [91]. Thus, Nef is not an isolated case of a viral factor targeting autophagy, and this action of Nef should be of interest for developing drugs that would inhibit progression to AIDS, and in the context of HAART might help in elimination of residual sources of persistent virus.
Positive role of the early stages of autophagy in HIV-1 biogenesis
The interactions between HIV-1 and autophagy are a bit more complicated than just a straightforward autophagic role as a cell-autonomous viral restriction factor. It turns out that the early stages of viral assembly and budding, and possibly virion egress may be assisted by the autophagic proteins and complexes participating in the early, nondegradative stages of autophagy (Fig. 5) Although the currently accepted model is that HIV in phagocytic cells buds, as in T cells, at the plasma membrane, in macropahges at least a subset of HIV-1 virions may bud into intracellular endomembranous compartments connected via narrow channels to the plasma membrane [92]. Intracellular compartments, potentially corresponding to the above endomembranes, intersect with the autophagy pathway [37]. Gag p55 protein is found in protein complexes with LC3-II by co-immunoprecipitation and can be seen in membranes carrying autophagic markers by subcellular organelle fractionation on sucrose gradients. These Gag p55- and LC3-II-positive membranes represent intermediates most likely en route to virion assembly sites. Finally, intracellular viral proteins and virions in macrophages colocalize with LC3 by imunofluorescence and immunoelectron microscopy [37]. Inhibition of autophagy using Atg protein knockdowns or pharmacological inhibitors results in lower yields of the virus released from the macrophage [37]. Thus, it appears that autophagy, albeit primarily considered to act as a degradative process, assists in virion trafficking, assembly, maturation or release. This statement comes with one caveat, and that is that the process of autophagy must be prevented to progress through the maturation stages, which is precisely what HIV-1 does in phagocytic cells using Nef as described above. Although this “biosynthetic” role of autophagy in assisting HIV biogenesis appears paradoxical, given the perils of progressing into degradative autophagic vacuoles, it has informative counterparts in some other well studied roles of autophagy. For example, autophagic machinery is important for the biosynthetic cytoplasm-to-vacuole targeting (Cvt) pathway which delivers two important enzymes, aminopeptidase I and α-mannosidase, to the yeast vacuole [93,94]. The observation that Gag associates with LC3 should thus be of considerable interest not only as a specific cellular mechanism enhancing HIV-1 biogenesis [35,37] but also as the first example of a biosynthetic role of the autophagic pathway in mammalian cells, with the Cvt pathway in yeast being the only known precedent thus far for the utilization of the core autophagic machinery in biogenesis [94].
Fig. 5. Autophagic pathway plays a mild positive role in early HIV-1 biogenesis.
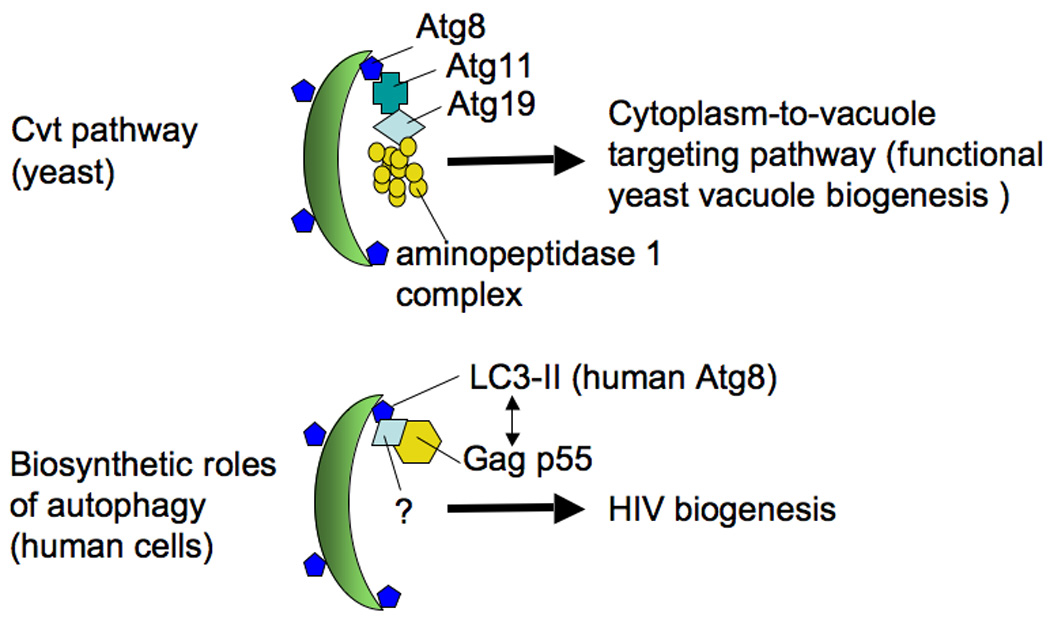
Published evidence indicates that when a virus encodes Nef, induction of autophagy promotes viral yields or egress. An interaction of HIV-1 Gag (the p55 precursor protein, giving rise through proteolytic processing to the capsid, matrix, nucleocapsid and budding regulator p6) and LC3-II has been reported, as well as colocalization by fluorescence microscopy and electron microscopy. An analogy has been made to the Cvt (cytoplasm-to-vacuole targeting) pathway in yeast.
Conclusions
Autophagy plays a role in HIV-1 biology and pathogenesis at many stages: (i) HIV-1 biogenesis; (ii) cell-autonomous defense against Nef-less HIV variants; (iii) depletion of CD4+ T cells; and (iv) HIV-associated dementia. Of particular interest is the multifaceted nature of overlaps between HIV and host cellular homeostasis of many cells. All observations regarding the roles of autophagy in HIV-1 replication or elimination, and AIDS pathogenesis reported so far are consistent with autophagy’s role as a cell-autonomous defense and a major decision crossroads of cell survival or death. A surprise came from the realization that autophagy can also play a biogenesis role in mammalian cells (in the case of HIV) as evidenced by complexes between LC3-II and Gag (precursor protein Pr55Gag), which is a 55-kDa precursor to several key mature virion proteins (matrix, capsid, nucleocapsid and p6) that play roles in virus assembly and budding and as major constituents of the mature HIV virion’s inner structure. Whether Gag and LC3-II require an adapter protein to bridge them and how exactly autophagy or LC3-II-positive membranes assist Gag in its trafficking or processing is not known. Nevertheless, a conceptual parallel may exist with the well-known Cvt pathway, which co-opts autophagic machinery for biogenesis purposes using the core Atg factors for delivery of cytosolic precursors to the lumen of the yeast vacuole where they carry out their biological function. Lastly, it should be obvious that the observed interactions of autophagy and HIV may present potential targets for new interventions, but that more work is needed before appropriate strategies may be contemplated.
Supplementary Material
Aknowledgments
This work was supported by grant AI069345 from National Institutes of Health, 107160-44-RGRL from amfAR, a Bill and Melinda Gates Grand Challenge Explorations grant. C. Dinkins was supported by National Institutes of Health Biology of Infectious Diseases and Inflammation training grant T32AI007538.
References
- 1.He C, Klionsky DJ. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu Rev Genet. 2009 doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. Embo J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010 doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 7.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010 doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009 doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 9.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–4659. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deretic V. Autophagy in infection. Curr Opin Cell Biol. 2010 doi: 10.1016/j.ceb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 14.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 15.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 16.Orvedahl A, Macpherson S, Sumpter R, Jr, Tallóczy Z, Zou Z, Levine B. Autophagy protects against sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HW, IV, Kyei GB, Johansen T, Vergne I, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010 doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. Embo J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2009 doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 21.Tormo D, Checinska A, Alonso-Curbelo D, Perez-Guijarro E, Canon E, Riveiro-Falkenbach E, Calvo TG, Larribere L, Megias D, Mulero F, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell. 2009;16:103–114. doi: 10.1016/j.ccr.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA. ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol. 2008;9:35. doi: 10.1186/1471-2172-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 25.Feng CG, Weksberg DC, Taylor GA, Sher A, Goodell MA. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell. 2008;2:83–89. doi: 10.1016/j.stem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 28.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, et al. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 2009;5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–517. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 31.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 32.English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippe R, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009 doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- 34.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–276. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 35.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 36.Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. Aids. 2008;22:695–699. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Espert L, Varbanov M, Robert-Hebmann V, Sagnier S, Robbins I, Sanchez F, Lafont V, Biard-Piechaczyk M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One. 2009;4:e5787. doi: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One. 2008;3:e2906. doi: 10.1371/journal.pone.0002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai SK, Hida K, Shukair S, Wang YY, Figueiredo A, Cone R, Hope TJ, Hanes J. Human immunodeficiency virus type 1 is trapped by acidic but not by neutralized human cervicovaginal mucus. J Virol. 2009;83:11196–11200. doi: 10.1128/JVI.01899-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 464:217–223. doi: 10.1038/nature08757. [DOI] [PubMed] [Google Scholar]
- 43.Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu HJ, Reuter MA, McDonald D. HIV traffics through a specialized, surface-accessible intracellular compartment during trans-infection of T cells by mature dendritic cells. PLoS Pathog. 2008;4:e1000134. doi: 10.1371/journal.ppat.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piguet V, Sattentau Q. Dangerous liaisons at the virological synapse. J Clin Invest. 2004;114:605–610. doi: 10.1172/JCI22812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol. 2007;9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prevost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J Virol. 2009;83:6234–6246. doi: 10.1128/JVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364:675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 50.Johnson WE, Sawyer SL. Molecular evolution of the antiretroviral TRIM5 gene. Immunogenetics. 2009;61:163–176. doi: 10.1007/s00251-009-0358-y. [DOI] [PubMed] [Google Scholar]
- 51.Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lackner AA, Mohan M, Veazey RS. The gastrointestinal tract and AIDS pathogenesis. Gastroenterology. 2009;136:1965–1978. doi: 10.1053/j.gastro.2008.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344:1668–1675. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 54.Thomas R, Apps R, Qi Y, Gao X, Male V, O'HUigin C, O'Connor G, Ge D, Fellay J, Martin JN, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alter G, Altfeld M. NK cells in HIV-1 infection: evidence for their role in the control of HIV-1 infection. J Intern Med. 2009;265:29–42. doi: 10.1111/j.1365-2796.2008.02045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, et al. HLA alleles associated with delayed progression to AIDS contribute strongly to the Initial CD8+ T cell response against HIV-1. PLoS Med. 2006;3:e403. doi: 10.1371/journal.pmed.0030403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perfettini JL, Castedo M, Roumier T, Andreau K, Nardacci R, Piacentini M, Kroemer G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005;12 Suppl 1:916–923. doi: 10.1038/sj.cdd.4401584. [DOI] [PubMed] [Google Scholar]
- 58.Nishimura Y, Brown CR, Mattapallil JJ, Igarashi T, Buckler-White A, Lafont BA, Hirsch VM, Roederer M, Martin MA. Resting naive CD4+ T cells are massively infected and eliminated by X4-tropic simian-human immunodeficiency viruses in macaques. Proc Natl Acad Sci U S A. 2005;102:8000–8005. doi: 10.1073/pnas.0503233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muthumani K, Choo AY, Hwang DS, Premkumar A, Dayes NS, Harris C, Green DR, Wadsworth SA, Siekierka JJ, Weiner DB. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood. 2005;106:2059–2068. doi: 10.1182/blood-2005-03-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varbanov M, Espert L, Biard-Piechaczyk M. Mechanisms of CD4 T-cell depletion triggered by HIV-1 viral proteins. AIDS Rev. 2006;8:221–236. [PubMed] [Google Scholar]
- 61.Zhou Y, Shen L, Yang HC, Siliciano RF. Preferential cytolysis of peripheral memory CD4+ T cells by in vitro X4-tropic human immunodeficiency virus type 1 infection before the completion of reverse transcription. J Virol. 2008;82:9154–9163. doi: 10.1128/JVI.00773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 63.Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, Hill BJ, Noto A, Ancuta P, Peretz Y, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4(+) T cell activation during HIV infection. Nat Med. 2010 doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maldarelli F, Palmer S, King MS, Wiegand A, Polis MA, Mican J, Kovacs JA, Davey RT, Rock-Kress D, Dewar R, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007;3:e46. doi: 10.1371/journal.ppat.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Lunzen J, Hoffmann C. Virological rebound and its consequences during treatment interruption. Curr Opin HIV AIDS. 2007;2:1–5. doi: 10.1097/COH.0b013e328011aab1. [DOI] [PubMed] [Google Scholar]
- 66.Denizot M, Varbanov M, Espert L, Robert-Hebmann V, Sagnier S, Garcia E, Curriu M, Mamoun R, Blanco J, Biard-Piechaczyk M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy. 2008;4:998–1008. doi: 10.4161/auto.6880. [DOI] [PubMed] [Google Scholar]
- 67.Molina L, Grimaldi M, Robert-Hebmann V, Espert L, Varbanov M, Devaux C, Granier C, Biard-Piechaczyk M. Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics. 2007;7:3116–3130. doi: 10.1002/pmic.200700306. [DOI] [PubMed] [Google Scholar]
- 68.Espert L, Biard-Piechaczyk M. Autophagy in HIV-induced T cell death. Curr Top Microbiol Immunol. 2009;335:307–321. doi: 10.1007/978-3-642-00302-8_15. [DOI] [PubMed] [Google Scholar]
- 69.Espert L, Codogno P, Biard-Piechaczyk M. What is the role of autophagy in HIV-1 infection? Autophagy. 2008;4:273–275. doi: 10.4161/auto.5211. [DOI] [PubMed] [Google Scholar]
- 70.Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy and CD4+ T lymphocyte destruction by HIV-1. Autophagy. 2007;3:32–34. doi: 10.4161/auto.3275. [DOI] [PubMed] [Google Scholar]
- 71.Alirezaei M, Kiosses WB, Fox HS. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy. 2008;4:963–966. doi: 10.4161/auto.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gougeon ML, Piacentini M. New insights on the role of apoptosis and autophagy in HIV pathogenesis. Apoptosis. 2009;14:501–508. doi: 10.1007/s10495-009-0314-1. [DOI] [PubMed] [Google Scholar]
- 73.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995;332:228–232. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- 74.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 75.Daniel MD, Kirchhoff F, Czajak SC, Sehgal PK, Desrosiers RC. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science. 1992;258:1938–1941. doi: 10.1126/science.1470917. [DOI] [PubMed] [Google Scholar]
- 76.Peterlin BM. Nef: out and in? Nat Immunol. 2006;7:229–230. doi: 10.1038/ni0306-229. [DOI] [PubMed] [Google Scholar]
- 77.Roeth JF, Collins KL. Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol Mol Biol Rev. 2006;70:548–563. doi: 10.1128/MMBR.00042-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schindler M, Munch J, Kutsch O, Li H, Santiago ML, Bibollet-Ruche F, Muller-Trutwin MC, Novembre FJ, Peeters M, Courgnaud V, et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell. 2006;125:1055–1067. doi: 10.1016/j.cell.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 79.Schindler M, Schmokel J, Specht A, Li H, Munch J, Khalid M, Sodora DL, Hahn BH, Silvestri G, Kirchhoff F. Inefficient Nef-mediated downmodulation of CD3 and MHC-I correlates with loss of CD4+T cells in natural SIV infection. PLoS Pathog. 2008;4:e1000107. doi: 10.1371/journal.ppat.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stumptner-Cuvelette P, Jouve M, Helft J, Dugast M, Glouzman AS, Jooss K, Raposo G, Benaroch P. Human immunodeficiency virus-1 Nef expression induces intracellular accumulation of multivesicular bodies and major histocompatibility complex class II complexes: potential role of phosphatidylinositol 3-kinase. Mol Biol Cell. 2003;14:4857–4870. doi: 10.1091/mbc.E03-04-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sandrin V, Cosset FL. Intracellular versus cell surface assembly of retroviral pseudotypes is determined by the cellular localization of the viral glycoprotein, its capacity to interact with Gag, and the expression of the Nef protein. J Biol Chem. 2006;281:528–542. doi: 10.1074/jbc.M506070200. [DOI] [PubMed] [Google Scholar]
- 82.Sanfridson A, Hester S, Doyle C. Nef proteins encoded by human and simian immunodeficiency viruses induce the accumulation of endosomes and lysosomes in human T cells. Proc Natl Acad Sci U S A. 1997;94:873–878. doi: 10.1073/pnas.94.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piguet V, Gu F, Foti M, Demaurex N, Gruenberg J, Carpentier JL, Trono D. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell. 1999;97:63–73. doi: 10.1016/s0092-8674(00)80715-1. [DOI] [PubMed] [Google Scholar]
- 84.Bentham M, Mazaleyrat S, Harris M. Role of myristoylation and N-terminal basic residues in membrane association of the human immunodeficiency virus type 1 Nef protein. J Gen Virol. 2006;87:563–571. doi: 10.1099/vir.0.81200-0. [DOI] [PubMed] [Google Scholar]
- 85.Deretic V. Strange bedfellows expose ancient secrets of autophagy in immunity. Immunity. 2009;30:479–481. doi: 10.1016/j.immuni.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gannage M, Dorman D, Albrecht R, Dengjel J, Torrosi T, Ramer P, Lee M, Strowig T, Arrey F, Conenello G, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host and Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib D, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host and Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 88.Ku B, Woo JS, Liang C, Lee KH, Hong HS, E X, Kim KS, Jung JU, Oh BH. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine γ-herpesvirus 68. PLoS Pathog. 2008;4:e25. doi: 10.1371/journal.ppat.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the γ-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4:989–997. doi: 10.4161/auto.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiaofei E, Hwang S, Oh S, Lee JS, Jeong JH, Gwack Y, Kowalik TF, Sun R, Jung JU, Liang C. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of γ herpesvirus 68. PLoS Pathog. 2009;5:e1000609. doi: 10.1371/journal.ppat.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol. 2007;177:329–341. doi: 10.1083/jcb.200609050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shintani T, Huang W-P, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell. 2002;3:825–837. doi: 10.1016/s1534-5807(02)00373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010 doi: 10.1016/j.febslet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.