

Abstract
One of the most fundamental problems in cell biology concerns how cells communicate with their surroundings through surface receptors. The last few decades have seen major advances in understanding the mechanisms of receptor-ligand recognition and the biochemical consequences of such encounters. This review describes the emergence of solution nuclear magnetic resonance (NMR) spectroscopy as a powerful tool for the structural characterization of membrane-associated protein domains involved in transmembrane signaling. We highlight particularly instructive examples from the fields of immunoreceptor biology, growth hormone signaling, and cell adhesion. These signaling complexes comprise multiple subunits each spanning the membrane with a single helical segment that links extracellular ligand-binding domains to the cell interior. The apparent simplicity of this domain organization belies the complexity involved in cooperative assembly of functional structures that translate information across the cellular boundary.
Introduction
Cell surface receptors that transmit signals across the membrane are dynamic molecules consisting of multiple functional regions. In many cases a complex capable of signaling must be assembled from multiple single-pass membrane proteins that consist of a ligand-binding extracellular (EC) domain, a membrane-spanning transmembrane (TM) domain, and a cytoplasmic region capable of initiating a biochemical signal. Dynamic interaction of the EC domain with signaling partners results in chemical modification of the cytoplasmic region, and in many cases this can be attributed to global conformational changes that must propagate through the membrane. Crystallization of an intact receptor that includes all of the functional domains has proven to be extremely challenging, and this is likely due to the complexity and dynamic nature of the receptors. Therefore, past decades of crystallographic studies on this type of multi-domain receptor complex have taken a “divide and conquer” approach, focusing primarily on obtaining the structures of soluble domain fragments. Crystallization of fragments containing the TM domains, on the other hand, has been largely unsuccessful. Extensive biochemical analyses in many different receptor systems have shown that the TM helices are not merely inert anchors but in fact play essential roles in the oligomerization of receptor subunits within the membrane. Thus, the modes of association between the TM helices directly link the conformational state of EC domains to that of the juxtamembrane (JM) regions and the cytoplasmic-signaling motifs.
In 1997 a seminal study by Engelman and coworkers (MacKenzie et al., 1997) demonstrated the utility of solution nuclear magnetic resonance (NMR) spectroscopy in determining the high-resolution structure of the TM domain of glycophorin A (GpA), the primary sialoglycoprotein of human erythrocyte membranes that forms non-covalent dimers through the association of the TM helices. This study not only revealed unique physical and chemical features governing the assembly of two hydrophobic helices in a membrane-like environment but also represented a proof-of-principle that it is possible to reconstitute oligomeric TM domains in detergent micelles for structure determination by NMR. The membrane-associated regions of cellular receptors have historically occupied a major “blind spot” in structural biology, and the effectiveness of solution NMR in measuring structural and dynamic parameters for small protein domains in the presence of detergent and lipid now puts it in a unique position to fill crucial gaps in our structural knowledge of TM receptor systems. The general application of NMR in structural characterization of membrane proteins has grown rapidly in the past decade, and this progress has been comprehensively reviewed in a recent article from Sanders and colleagues (Kim et al., 2009). Likewise, many excellent reviews are available addressing the proposed mechanisms of signal transduction in individual receptor systems. Rather than attempt to provide a comprehensive overview of the role of membrane-associated protein domains in signaling, we have chosen to focus this review on a selection of recent studies in which the application of solution NMR techniques has provided significant new information on the structure and function of trans-bilayer signaling complexes in important biological systems. To this end, in the following sections we highlight several instructive examples from the fields of immunoreceptor biology, growth hormone signaling, and cell adhesion.
Immunoreceptor Assembly, Structure, and Signaling
Immunoreceptors are molecular sensors that alert the cells of the immune system to the presence of potential pathogens and injured or transformed host cells. These sensors are multi-subunit membrane protein complexes comprising ligand-binding modules that are non-covalently coupled to signal-transducing modules bearing cytosolic kinase-recruiting domains (Figure 1) (Call and Wucherpfennig, 2007). Biophysical methods, including both X-ray crystallography and solution NMR, have made important contributions to our current understanding of immunoreceptor-ligand interactions and the formation of intra-cellular-signaling complexes (Kuhns et al., 2006; Love and Hayes, 2010). However, the mechanisms that physically link these events across the plasma membrane remain unknown, and no structures of intact immunoreceptor complexes have yet been reported. The most complex of these structures, the heterodimeric αβ T cell antigen receptor (Figure 1A), is expressed at the lymphocyte surface as part of an obligate complex with four different signal-transducing proteins: CD3γ, CD3δ, CD3ε, and the ζ chain (CD247); collectively referred to as the TCR-CD3 complex (Samelson et al., 1985; Sussman et al., 1988; van Agthoven et al., 1981). Together, these six subunits form four different functional modules: the ligand-binding TCRαβ heterodimer; and the CD3δε, CD3γε, and ζζ-signaling dimers. Reports from several groups in the late 1980s and early 1990s identified basic and acidic residues in the TM domains of TCR and CD3 proteins as critical determinants of assembly and cell-surface expression (reviewed in Ashwell and Klausner, 1990; Call and Wucherpfennig, 2005). In fact the TM domains themselves were found to be sufficient for assembly of receptor subunits with signaling modules (Call et al., 2002; Cosson et al., 1991; Manolios et al., 1990). The interactions among potentially ionized TM residues governing assembly involved a complex electrostatic network in which two acidic residues in the TM domains of dimeric-signaling modules were required for each contact with a single basic residue in the receptor TM domains (Figure 1A) (Call et al., 2002, 2004). These studies suggested a model of assembly in which a triad of one basic and two acidic TM residues was co-localized at a three-helix interface in the membrane for each TCR-CD3 interaction. But how is the close apposition of two acidic groups in a signaling dimer stabilized within the hydrophobic lipid bilayer interior? And what exactly is the three-dimensional arrangement of the intact triad assembly motif?
Figure 1. Intramembrane Assembly of Activating Immunoreceptor Complexes.

(A) Schematic diagram of the T cell receptor (TCR)-CD3 complex in the plasma membrane. The ligand-binding TCRαβ heterodimer assembles with three different dimeric-signaling modules (CD3δε, CD3γε, and ζζ) through polar interactions deep within the hydrophobic lipid bilayer. In each assembly event, two acidic TM residues in the signaling module form a specific binding site for a single basic TM residue in the partner receptor. Ribbon diagrams were prepared using crystal structures of a human TCR structure (Kjer-Nielsen et al., 2002) (PDB ID 1KGC) and the CD3 crystal structures 1XIW (human δε) (Arnett et al., 2004) and 1SY6 (human γε) (Kjer-Nielsen et al., 2004).
(B) Solution NMR structure of the human ζζTM dimer fragment embedded in DPC-SDS mixed detergent micelles (PDB ID 2HAC) (Call et al., 2006). The structure is shown as a bundle of the 20 lowest-energy structures (left) and a ribbon diagram of a representative structure (right). Non-interface side chains are omitted for clarity. Key features discussed in the text are highlighted and labeled using three-letter amino acid code.
(C) Schematic diagram of the natural killer (NK) cell-activating receptor complex NKG2C-CD94-DAP12. Many activating NK cell receptors assemble using a structural arrangement within the membrane that is very similar to that observed for TCR-CD3. The EC domain structure of NKG2C-CD94 is represented by the NKG2A-CD94 crystal structure (Sullivan et al., 2007) (PDB ID 3BDW). NKG2A and NKG2C are almost identical in this region. CD94 is not required for assembly of NKG2C with DAP12, but it is required for ligand binding and receptor function.
(D) Solution NMR structure of the human DAP12TM dimer fragment in TDPC-SDS mixed detergent micelles (PDB ID 2L34) (Call et al., 2010). Key features are highlighted as in (B) for the bundle of 15 lowest-energy structures (left) and one representative structure (right).
Solution NMR Structure of the ζζTM Homodimer
The first atomic-scale structural information to shed light on this problem came with the 2006 report of the solution NMR structure of the TCR-associated, membrane-embedded ζζTM homodimer (Figure 1B) (Call et al., 2006). This structure, determined in mixed detergent micelles composed of dodecylphosphocholine (DPC) and dodecylsulfate (SDS) in a 5:1 molar ratio, revealed extensive helix-helix contacts along the TM interface responsible for formation of the left-handed coiled-coil dimer. The two critical aspartic acids in ζζ were closely juxtaposed within the dimer interface and stabilized in a particular conformation by several key features, including an inter-helical disulfide bond near the EC lipid head group region and a pair of tyrosine-threonine hydrogen bonds that locked the helix-packing mode in the carboxyterminal half of the interface. In this conformation one oxygen from each aspartic acid side chain pointed out of the helix dimer interface where it would presumably be available for contact with a basic receptor TM residue. This structure provided strong support for the basic-acidic-acidic triad model of TM assembly by demonstrating that the two aspartic acids adopted a conformation in which both could theoretically contact a lysine or arginine. However, the geometry of the receptor-binding site offered no obvious solution for how a basic residue could be positioned to contact both aspartic acids simultaneously.
Solution NMR Structure of the NKG2CTM-DAP12TM Complex
A more recent study provided the structure of another, functionally homologous TM-signaling dimer, DAP12, both alone and with a receptor TM domain in an assembled trimeric complex (Call et al., 2010). DAP12 was originally identified based on the presence of cytosolic immunoreceptor tyrosine-based activation motifs (ITAMs) similar to those that are phosphorylated in the CD3 and ζζ signal-transducing components upon TCR triggering (Lanier et al., 1998a; Tomasello et al., 1998). Although the two proteins exhibit no further sequence identity outside of these cytosolic motifs, DAP12 shares two key features with the ζ chain (Figures 1C and 1D): a disulfide linkage (here, within a 15-aa EC stalk region); and a pair of aspartic acids in the TM domains that direct its assembly with more than a dozen different activating receptors carrying basic TM residues (Lanier, 2009). The structure of the DAP12 dimer in tetradecylphosphocholine (TDPC)-SDS micelles (Figure 1D) revealed that the critical aspartic acids were juxtaposed in a manner that was strikingly similar to the arrangement observed in the ζζTM structure. No additional inter-helical hydrogen bonds were identified, and this helix-helix interface appeared to be stabilized primarily through van der Waals packing and the disulfide bond in the flexible EC stalk region. The major new feature observed in the DAP12TM dimer structure was the presence of a threonine residue that formed an intra-helical hydrogen bond to the aspartic acid and turned out to be critical for assembly with receptor TM domains (Call et al., 2010). Therefore, the electrostatic network organizing this TM assembly was more extensive than previously recognized.
In order to provide the three-dimensional structure of an assembled immunoreceptor complex, the TM domain of the type II, C-type lectin-like receptor NKG2C (Lanier et al., 1998b) was fused to the C-terminal end of one strand of the disulfide-linked DAP12TM dimer such that it could pack in its natural anti-parallel orientation with respect to the signaling dimer (Figures 1C and 2) (Call et al., 2010). In this study, engineering of a covalent trimer was critical for NMR experiments. This structure, also determined in TDPC-SDS micelles, provided the first look at a heterotrimeric complex of TM domains from single-spanning membrane proteins. It revealed a loosely defined groove along the DAP12TM dimer interface where the receptor TM helix was accommodated such that the lysine residue was ideally situated to interact favorably with only one of the aspartic acid residues in DAP12 (Figure 2B). So why were both aspartic acids in the motif absolutely required for assembly, as previously demonstrated in biochemical studies (Feng et al., 2005)? The answer appears to lie not in a direct tripartite interaction, but in the formation of an extensive membrane-embedded electrostatic network in which the distribution of electronegativity among multiple polar residues is the key determinant. Notably, the variability in the low-energy structure bundle suggested that the lysine amino group may contact the aspartic acid and the threonine simultaneously, offering a structural explanation for the threonine’s important role in assembly. The occurrence of this DxxxT/S sequence in signaling module TM domains turned out to be a critical feature for at least two other important receptor complexes, TCR-CD3 and NKG2D-DAP10 (Call et al., 2010), and therefore, it may represent a common structural element in this extended family of activating immunoreceptors.
Figure 2. Trimeric Assembly of the NKG2C-DAP12 TM Peptide Complex.

The bundle of the 15 lowest-energy structures (A) and one representative ribbon structure (B) of a covalently linked NKG2C-DAP12 trimeric TM peptide complex in TDPC-SDS mixed micelles are shown (PDB ID 2L35) (Call et al., 2010). The disulfide-linked dimer of type I DAP12TM peptides (green ribbons) was engineered with the type II NKG2CTM peptide attached as an in-frame fusion to the C terminus of one DAP12TM strand through a flexible linker. Key features discussed in the text are highlighted in (B) and labeled using three-letter amino acid code.
An interesting footnote to this story is that DAP12 contains two tandem GpA-like GG4 motifs (GVLAGIVMG) that do not participate in helix-helix packing within the assembled trimeric complex (glycine positions highlighted in orange in Figures 1D and 2B). These sequences are conserved in higher vertebrates, and whether they bear any functional significance remains to be seen. But perhaps a cautionary lesson can be extracted here with regard to the modeling of helix-helix interactions based on predicted packing motifs.
Collectively, these studies set the stage for functional investigations into what type of conformational changes accompany transmission of a signal through the lipid bilayer, e.g., reorientation of TM helices around a fixed point formed by the focused polar contact (Engelman, 2003). Several other modular receptor systems, such as the B cell receptor (BCR)-Igαβ complex and particular immunoglobulin Fc receptor complexes, are known to require TM interactions for assembly or function but do not have the familiar basic and acidic TM motifs. Further biochemical and NMR-based studies in the vein of those described above could shed light on what structural elements direct these important interactions and how they affect receptor function.
Solution NMR Structure of a Membrane-Associated Intracellular-Signaling Domain
Studies of TM interactions within immunoreceptor complexes have provided a structural framework for understanding assembly and imposed important new restraints on how we envision the molecular architecture in intact structures. But how EC-ligand binding regulates phosphorylation of intracellular-signaling domains remains an open question. An important new clue to this problem has come from a series of studies examining the behavior of cytoplasmic-signaling domains in the presence of lipid membranes. Aivazian and Stern (2000) first demonstrated in a 2000 study that the cytoplasmic tail of the ζ chain bound to negatively charged lipid vesicles in vitro and exhibited an increased helical content as measured by circular dichroism spectroscopy. Later studies reported similar observations for CD3ε and Fc receptor γ chain cytoplasmic domains (Deford-Watts et al., 2009; Sigalov et al., 2006; Xu et al., 2008). These sequences were previously assumed to be unstructured in solution, and the new results prompted the hypothesis that intracellular-signaling domains in non-triggered TCR-CD3 complexes may be bound to the negatively charged inner leaflet of the plasma membrane and at least partially structured. Phosphorylation of the critical tyrosine residues in ITAM-containing peptides was greatly reduced in the presence of negatively charged lipid vesicles in vitro (Aivazian and Stern, 2000; Xu et al., 2008), suggesting that lipid binding could regulate early signaling events in T cells (Kuhns and Davis, 2008). Importantly, these experiments were all performed using purified peptides and negatively charged lipids in vitro, and so the specificity and physiological relevance of lipid binding was not immediately apparent.
A recently published structure-function study of the ITAM-containing CD3ε cytoplasmic tail sequence (Xu et al., 2008) provided two key pieces of information that led to a new model for regulation of ITAM signaling through lipid binding. First, a FRET-based assay confirmed that the CD3ε cytoplasmic tail was bound to the plasma membrane in live T cells in a charge-dependent manner. Second, a solution NMR structure confirmed that specific regions were partially structured in the lipid-bound form of the 57-amino acid CD3ε cytoplasmic peptide (CD3εcyt) (Figure 3). The consensus ITAM consists of two repeats of a YxxI/L sequence spaced six to 12 residues apart in which the tyrosine residues and conserved hydrophobic residues would be spaced one turn apart in a helix. The NMR structure of CD3εcyt bound to POPG-containing phospholipid-detergent bicelles (Figure 3A) revealed that the two hemi-ITAM sequences did, in fact, adopt locally helical conformations that resulted in localization of these side chains to the lipid-facing surface. Furthermore, observation of specific NOEs between peptide and lipid protons provided solid evidence that the conserved tyrosine, leucine, and isoleucine side chains were deeply embedded within the hydrophobic interior of the phospholipid bilayer (Figure 3B), explaining the inaccessibility of the tyrosines to kinases that was observed in vitro. Lipid binding has yet to be demonstrated for CD3γ, CD3δ, or DAP12 cytoplasmic ITAM sequences; however, the data for CD3ε, ζ chain, and FcR γ chain suggest a model in which receptor triggering must cause a shift in the equilibrium from bound to unbound ITAMs (Kuhns and Davis, 2008; Xu et al., 2008). Whether this event is as simple as an applied mechanical force or involves a more complex mechanism such as exclusion of negatively charged lipids from engaged receptors remains to be determined.
Figure 3. Regulation of Immunoreceptor Signaling through Dynamic Membrane Binding of Cytoplasmic Signaling Sequences.

(A) Solution NMR structure of the membrane-bound mouse CD3ε cytoplasmic tail sequence (PDB ID 2K4F) (Xu et al., 2008). A view from the perspective of the cell interior is shown with the inner leaflet of the plasma membrane bilayer represented by the beige disc. Membrane binding is energetically dominated by the polybasic region (blue) directly C-terminal to the TM domain. The proline-rich region (yellow) and the ITAM (green) follow.
(B) In the membrane-bound structure, the ITAM region adopts a partially helical conformation and inserts the canonical tyrosine and aliphatic residues into the lipid bilayer interior where they are protected from phosphorylation by Src-family kinases.
Geometry and Dynamics in Activation of Growth Factor Receptors
The epidermal growth factor receptors (EGFRs) belong to the receptor tyrosine kinase family of cell-surface receptors that are essential components of signaling pathways governing cell growth and differentiation (Schlessinger, 2002). Proteins in the EGFR family are single-pass membrane proteins consisting of an EC domain, a TM domain, an intracellular JM region, and a cytoplasmic kinase domain. Receptor activation results in auto-phosphorylation of specific tyrosine residues in the kinase activation loop, the JM region, and the flexible region C-terminal to the kinase domain. EGFR forms dimers in the cell membrane to function, but unlike the immunoreceptors, whose assembly is required in order to acquire the capacity for ligand-triggered signaling, EGFR dimer assembly is induced by ligand binding to the EC domain and is an active component of signal transduction. Crystal structures of the EGFR EC domain in the absence and presence of ligand revealed that binding of EGF to the receptor caused a major conformational change resulting in symmetric dimerization of the EC domain (Burgess et al., 2003). The cytoplasmic kinase domain also dimerizes, but in an asymmetric arrangement with the C-terminal lobe of the activator kinase abutting the N-terminal active site of the receiver kinase (Figure 4A) (Zhang et al., 2006), and this particular conformation was required for auto-phosphorylation. These studies provided a clear view of the trigger and its consequence: the trigger is the EGF-induced formation of a symmetric dimer in the EC domain, and the consequence is the formation of an asymmetric dimer in the kinase domain that leads to auto-phosphorylation. But the region connecting the EC domain to the kinase domain, which includes as many as 60 residues of TM and JM sequence, has been missing from the crystallographic studies. Therefore, the relationship between symmetric dimer formation outside the cell and asymmetric dimer formation inside the cell remained unclear.
Figure 4. The TM Domain of EGFR Shows Close Helix-Helix Packing via the GG4-like Sequence Motif.

(A) The overall model of the activating state of EGFR made after that shown in Figure 5C of Jura et al. (2009).
(B) The solution NMR structure of the TM domain of ErbB2 receptor tyrosine kinase (PDB ID 2JWA) presumably corresponding to the receptor active state (Bocharov et al., 2008).
(C) The solution NMR structure of the TM domain of the GpA protein (PDB ID 1AFO).
Solution NMR Studies of TM Domain Interactions in EGFR Signaling
This is where solution NMR provided critical structural information for the membrane-embedded region of the receptor (Figure 4B). Arseniev and coworkers (Bocharov et al., 2008) demonstrated that a peptide corresponding to the membrane-spanning region of ErbB2 spontaneously forms homodimers in detergent/lipid mixed micelles, and they have determined the high-resolution structure of the TM dimer by solution NMR. The 1H-15N correlation spectrum of peptide reconstituted in dimyristoylphosphocholine/dihexanoyl phosphocholine (DMPC/DHPC, molar ratio 1:4) displayed a homogeneous set of resonances, indicating that the peptide concentration used for NMR (0.5–1 mM) supported essentially quantitative dimer formation. The structure shows a right-handed helical-packing mode very similar to that of the glycophorin A TM (GpATM) dimer (Figure 4C). Both structures exhibited tight packing in the N-terminal region through the GG4-like motif comprising two small amino acids (S) such as glycine, serine, or threonine separated by three typically hydrophobic amino acids (x). The SxxxS sequence allows the apposition of the backbone of two α helices to within van der Waals distances, so close that Cα-H···O hydrogen bonds across the dimeric interface are in principle possible (Engelman, 2003; Mackenzie, 2006). In the GpATM dimer the nine residue GG4-like sequence that dominates the free energy of assembly is G79xxxG83xxxT87. The corresponding GG4-like sequence motif in ErbB2TM is T652xxxS656xxxG660. In both cases the helices cross at the middle of the GG4-like sequence at an angle of approximately −40°, resulting in a divergence of the two monomers near the C-terminal end (MacKenzie et al., 1997). However, residues near the C-terminal end of the TM helices in ErbB2 also make important contacts. Hydrophobic residues such as leucines, valines, and isoleucines form a hydrophobic cluster at the dimer interface that may contribute significantly to stabilization of the structure (Bocharov et al., 2008). Moreover, the aromatic residues flanking the hydrophobic cluster are probably involved in partitioning of the TM helices at the apolar-polar interface of the lipid bilayer. The NMR data and the structure of ErbB2TM strongly suggest that the TM helix alone in the cell membrane would spontaneously assemble into a stable homodimer. This result then raises a number of puzzling questions that call for further investigations. Is dimerization of the TM helix in the absence of the EC domain sufficient to activate the receptor because it should facilitate the formation of the asymmetric cytoplasmic kinase domain dimer? If dimerization of the TM domain is all that is required for kinase activation, then what is the inactive state of the TM domain? If the inactive state of the TM domain is not dimeric, then what is preventing its dimer formation? There have also been reports of preformed, inactive dimers of EGFR (Yu et al., 2002)—Does this mean there could be a different, inactive dimeric conformation of the TM domain?
NMR-Derived Insights into the Role of the EGFR JM Domain
In addition to the TM helices, the cytoplasmic JM sequence (residues 645–682) connecting the TM domain and the kinase domain has recently been shown to play an important role in guiding the formation of the active asymmetric dimer, although the structural details are still not crystal clear. Two independent crystal structures of the asymmetric kinase dimer revealed that JM residues 664–682 of the receiver kinase adopted an extended structure, wrapping around the C-terminal lobe of the activator kinase and forming hydrogen bonds with several polar residues in this domain (Jura et al., 2009; Red Brewer et al., 2009). Nonconservative substitution of key interacting residues on both sides of the interface severely affected formation of the active, asymmetric dimer, providing compelling evidence that the JM segment is involved in stabilizing the relevant structure. Another segment of the JM region (residues 645–663) that is immediately C-terminal to the TM helix and proximal to the membrane has different structural properties. This JM segment, denoted JM-A, has an amphipathic sequence pattern that suggests the formation of a helical structure that could interact with the phosphate groups in the membrane bilayer. Indeed, solution NMR measurements recorded on an isolated 15-residue peptide spanning the JM-A segment indicated transient formation of an α helix (Jura et al., 2009). Mutagenesis and NOE analyses suggested that the JM-A helix likely dimerizes in an antiparallel manner on the surface of the membrane, which would be consistent with the activated conformation of the flanking JM region in the asymmetric dimer. The crystal structure of the cytoplasmic region including both the kinase domain and the full JM region also showed that JM-A was helical, but its position in the activator and receiver kinase chain did not seem to support JM-A dimerization. The JM-A segment is clearly important for signaling because its deletion results in ~10-fold reduction in the catalytic activity of the kinase domain (Jura et al., 2009).
The available crystal and solution structures all converge on a central theme of EGFR activation that is based on dimerization of domains at different levels: the ligand-induced symmetric dimerization of the EC domain, the right-handed packing of the TM helices, the specific interaction of the JM region of the receiver kinase chain with the C-lobe of the activator kinase domain, and the asymmetric interaction between the kinase domains themselves. Dimerization of the entire receptor is clearly cooperative because dimerization of each domain reduces the entropic cost of dimerization of other domains. The remaining uncertainty is in the dimerization of the JM-A membrane-proximal segment. Because the arrangement of the JM-A segment relative to the TM helix has a direct consequence on the positioning of the two kinase domains, it is important to determine the exact conformation of the JM-A segment while attached to the TM helix in a model membrane medium such as detergent/lipid bicelles. Single-pass membrane proteins with a TM helix flanked by an amphipathic helix are common in nature, and recent solution NMR studies have shown that it is entirely possible to determine the high-resolution structures of such systems in their oligomeric state (Pielak and Chou, 2010; Schnell and Chou, 2008). A more complete view of the membrane-associated region of EGFR would allow the construction of a more definitive model of the entire receptor in the active dimeric form while also providing insights into the inactive state of the JM segments. Furthermore, other growth hormone and cytokine receptor systems may transmit signals using related structural solutions in which conformational alterations in TM domains are required. One example is the erythropoietin (Epo) receptor, which is known to undergo specific changes in dimeric conformation concomitant with activation (Lemmon and Schlessinger, 2010). TM sequences do participate in this conformational transition (Cammett et al., 2010; Kubatzky et al., 2005), but the structural basis of this role is unclear, and the system is likely to benefit from more detailed, NMR-based studies of the TM structure and dynamics.
Conformational Change and Bi-Directional Signaling through Integrins
Integrins are cell-surface adhesion receptors that mediate intercellular contacts and dynamic interactions between cells and the EC matrix. These functions are critical for maintaining tissue integrity and directing cell migration, and they play a major role in diverse biological processes including development, tumor metastasis, and immune function, to name but a few (Hynes, 1992). Members of the integrin family are heterodimers of α and β subunits, each comprising a multi-domain EC segment of up to 150 kDa, a single α-helical TM domain and a cytoplasmic tail of variable length (Luo et al., 2007). Binding of the integrin “head” domains to EC ligands is accompanied by clustering of integrin heterodimers and alterations in the TM and cytoplasmic domains that transmit information to cytoskeletal and signaling molecules inside the cell. At the same time, the interactions of proteins with the cytoplasmic tails can induce the activated conformation of the integrin EC domains in response to several different signaling pathways and thereby transmit information in the “inside-out” direction. Therefore, integrins are known as “bi-directional” signaling complexes, and the mechanisms of regulation and interplay between these two signaling modes are of great interest to biologists across a wide range of disciplines. Integrin activation is associated with a dramatic transition between two conformations that correspond to low-affinity and high-affinity phases of cell adhesion (Figure 5). This transition has been observed by cryoelectron microscopy and FRET techniques and is reminiscent of a “jack-knife” motion (Chigaev et al., 2003; Kim et al., 2003; Takagi et al., 2002). A number of biochemical and biophysical studies have demonstrated that the dynamic association and dissociation of integrin-α and -β TM domains and the binding of cytosolic proteins at the inner membrane surface are key events in activation of integrin-mediated cellular adhesion (reviewed in Shattil et al., 2010). Studying these events that take place at the membrane has proven exceedingly difficult, and solution NMR has made several major contributions to the field over the last decade.
Figure 5. Regulation of Integrin Adhesion through Reversible Association of TM and Cytoplasmic Sequences.
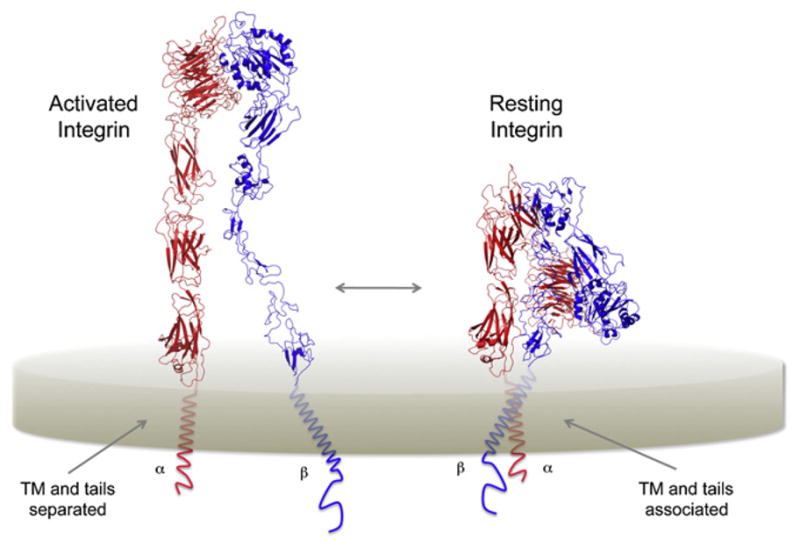
Schematic representation of the activated (left) and resting (right) forms of αβ-integrin heterodimers. In the resting conformation the TM α helices and cytoplasmic JM regions are closely associated. Separation of these domains is accompanied by a dramatic conformational change in the EC domains resembling a “jack-knife” motion. Ribbon diagrams were prepared using the crystal structure of the closed conformation of αxβ2-integrin and a model of the open form (Xie et al., 2010) kindly provided by Professor Can Xie (Peking University). The inserted I-domain in αx has been omitted for clarity.
Dynamics in TM, JM, and Cytoplasmic Domain Associations
Several lines of evidence had suggested that the JM and cytoplasmic domains of integrin-α and -β subunits were closely associated in the resting state and that integrin activation was accompanied by a separation of tail domains (reviewed in Shattil et al., 2010). A 2003 study from the Springer laboratory (Kim et al., 2003) provided direct support for this model in an experimental system where a robust FRET signal could be observed between donor and acceptor fluorophores attached to the cytoplasmic tails of surface-expressed αLβ2-integrin (LFA-1) in live cells. This signal was specifically lost upon integrin activation, indicating that the EC conformational change was indeed accompanied by a separation of membrane-proximal cytosolic domains. That separation of the cytosolic domains was necessary for integrin activation was evidenced by another study demonstrating that replacement of TM and cytosolic regions with constitutively associating domains such as basic-acidic coiled-coil sequences resulted in blockage of activation (Lu et al., 2001).
Several solution NMR studies in the early 2000s sought to define the structural basis of this interaction. The Qin (Vinogradova et al., 2002) and Vogel (Weljie et al., 2002) groups measured heterotypic associations between integrin-αIIb and β3-tail peptides and published NOE-derived solution structures of the αIIbβ3-tail complex. In the Qin structure the αIIb tail adopted a partially α-helical conformation that extended for approximately two turns and terminated at a proline residue (P998). This was followed by a non-helical turn that allowed the C-terminal segment to fold back onto the N-terminal JM region through electrostatic interactions. The β3 tail adopted an extended helical conformation in its N-terminal 23 amino acid region but was then unstructured for the remaining 25 residues extending to the native C terminus. The primary αIIbβ3 interface was located in the N-terminal (membrane-proximal) helical regions and comprised a cluster of methyl and aromatic van der Waals contacts followed by a trio of electrostatic contacts between the αIIb R995 side chain and several β3 interfacial residues. The Vogel study (Weljie et al., 2002) used shorter, C-terminally truncated peptides and identified two conformations of the complex that differed primarily in the disposition of the membrane-distal portion of the β3 peptide. Both studies provided plausible rationalizations for a subset of the known disruptive mutations in this region (such as alterations in the conserved αIIb GFFKR motif) based on the contacts observed in their structures. However, the differences among the structural models and the absence of TM domains and membrane lipids left their physiological relevance in question.
Nonetheless, the spectroscopic, biochemical and live-cell imaging data in support of a specific interaction between the cytosolic sequences represented strong evidence that the α and β TM domains must be closely associated in the nonactivated conformation. This was also consistent with the crystal structure of the 12-domain αVβ3 EC fragment in which the α and β C termini abutting the predicted TM domains were separated by only a few angstroms (Xiong et al., 2001). Accordingly, a disulfide-scanning study of TM sequences revealed a specific heterodimerization interface within the N-terminal halves of the αIIb and β3 TM α helices in nonactivated cells (Luo et al., 2004). This interface was lost upon activation, and locking the association through a membrane-embedded disulfide bond blocked activation, in agreement with previous results using engineered cytosolic constraints (Luo et al., 2004; Zhu et al., 2007). Two recent solution NMR structures of αIIbβ3 TM complexes in phospholipid bicelles (Lau et al., 2009) or organic solvent (deuterated acetonitrile/water) (Yang et al., 2009) provided confirmation that the primary contacts were located in the N-terminal portion of the TM helices (Figure 6). These studies defined a helix-helix interface based largely on methyl- and glycine-dependent packing. However, as with previous studies reviewed above, these reports differ significantly regarding the disposition of the membrane-proximal cytosolic regions, particularly the intracellular TM to JM transition in αIIb. The structure solved in organic solvent (Yang et al., 2009) used constructs extending through the entire tail regions of both subunits. In this structure the GFFKR motif was helical and did not participate in extensive contacts with the β subunit (Figure 6A). The TM portions in this structure exhibited significant departures from ideal helical conformations, a feature that is not unusual in membrane proteins, but considering the extent of the distortions observed here, may reflect the lack of lipid-imposed geometric constraints in organic solvent. In the bicelle-embedded structure (Figure 6B), a sharp turn in the αIIb sequence exiting the lipid bilayer resulted in localization of the αIIb GFFKR diphenylalanine sequence inside the membrane where it formed specific contacts with the cytosolic end of the β3 TM domain through aromatic interactions with W715. The observed difference in the extent of lipid-embedded sequence in this structure results in a significant tilt in the β3 TM helix with respect to both the membrane normal and αIIb, which is consistent with previous NMR studies of bicelle-embedded monomeric α and β TM peptides (Lau et al., 2008a, 2008b). Both studies of the heterodimeric TM complex proposed a salt bridge between αIIb R995 and β3 D723, but it should be noted that a non-spectroscopic restraint enforcing this contact was included in the calculation of the bicelle-embedded structure (Lau et al., 2009) based on the known disruptive effects of a R995A mutation. The biochemical and functional data on proposed interactions in this region are mixed (Shattil et al., 2010), and care should be taken when utilizing biochemical restraints in combination with NMR-derived measurements.
Figure 6. Solution NMR Structures of the αIIbβ3 TM Peptide Complexes.

Bundle (left) and ribbon (right) representations of the αIIbβ3 TM complex solved in (A) deuterated acetonitrile (CD3CN):H2O (PDB ID 2KNC) (Yang et al., 2009) or (B) dihexanoylphosphocholine: palmitoyl-oleoylphosphocholine:palmitoyl-oleoyl-phosphoserine (DHPC:POPC:POPS) bicelles (PDB ID 2K9J) (Lau et al., 2009). Glycines involved in helix-helix packing (yellow), putative salt-bridge interactions, and GFFKR aromatic interactions discussed in the text are highlighted and labeled in three-letter amino acid code. Additional C-terminal sequences in the structure in (A) were omitted for clarity in comparing the two structures.
A study published at the same time as the NMR structures used disulfide crosslinking-derived distance restraints to map the αIIbβ3 TM complex interface using the full-length proteins in the cell membrane (Zhu et al., 2009). Their modeling arrived at a packing arrangement and helix-helix orientation that were globally similar to both NMR structures. This model also predicted the non-helical reverse-turn orientation in αIIb and resulted in an aromatic packing arrangement similar to that observed in the bicelle structure (Lau et al., 2009). These studies illustrate the great promise of solution NMR in examining the dynamic interactions underpinning integrin signaling but also emphasize the great care that should be taken when considering the design of protein constructs and the experimental conditions under which they are studied. Whether the discrepancies among the structures described here derive strictly from differences in experimental conditions or reflect a degree of biologically relevant conformational complexity remains to be determined.
Interactions of Cytoplasmic Proteins with Integrin Tails
Activation of integrin-driven cell adhesion can result from signaling pathways that do not involve binding of ligands to integrin EC domains. This is achieved through the action of cytoplasmic regulatory proteins that interact directly with the integrin tail domains to induce conformational changes (Shattil et al., 2010). Specifically, the cytoskeletal protein talin binds to integrin tails at the inner face of the plasma membrane to regulate cell adhesion through the “inside-out” signaling pathway. In addition to the structural studies cited above, NMR-based experiments have helped to define this interaction and the mechanism by which talin binding induces integrin activation. In the 2002 study from the Qin laboratory (Vinogradova et al., 2002), addition of talin head domain (talin-H) to the αIIbβ3-tail complex resulted in significant chemical shift perturbations and line broadening in β3, but not αIIb, suggesting the formation of a large complex between talin H and the β3 cytoplasmic domain. Furthermore, combination of 15N-labeled αIIb tail with a mixture of unlabeled β3 and talin-H resulted in a 15N-1H correlation spectrum that was indistinguishable from that of the free αIIb tail, indicating that formation of the tail complex had been blocked. The crystal structure of a talin-β3 chimeric protein showed that the primary talin-binding site was distant from the most membrane-proximal β3 sequence (Garcia-Alvarez et al., 2003). However, the β3-tail sequence used in the chimeric protein was N-terminally truncated more than 20 residues away from the TM domain, and NMR data from the same study showed talin-induced chemical shift perturbations in residues that overlap the major αIIb-β3 tail contacts at the TM to cytoplasmic transition. A recently published NMR and MD simulation study of cooperative talin-integrin binding at the inner plasma membrane surface (Kalli et al., 2010) suggested that a combination of talin-induced alterations in the β3 TM helix tilt angle and competition for the αIIb R995-β3 D723 salt bridge may be responsible for the disruption of TM and cytoplasmic interactions and stabilization of the activated state. The combination of all of these techniques exemplifies the increasingly sophisticated approaches that are required to address such difficult problems in trans-bilayer signaling.
The Way Forward: Major Challenges and New Techniques
The above examples collectively illustrate that, given the successful reconstitution of the native oligomeric state of receptor TM domains in model membrane media, solution NMR can be used to study almost any type of small TM oligomers. We anticipate that many more such structures from other receptor systems will be determined using approaches similar to those described above. Although the quality of NMR spectra is an important criterion in choosing a membrane mimic for reconstituting TM oligomers, investigators need to make proper judgments on the relevance of the artificial reconstitution systems, which is often protein dependent. The above examples have demonstrated the use of a variety of model membrane media including micelles, bicelles, and organic solvent. Among them, the fast-tumbling bicelles can in principle best represent the cell membrane because they contain a significant patch of lipid bilayer for accommodating membrane proteins when q (molar ratio of lipid to detergent) is greater than 0.5 (Sanders and Prosser, 1998; Vold et al., 1997). An important technical issue with bicelles is that the detergent component is not very tunable for adjusting solubility of the protein-bicelle complex. Detergent micelles with phosphate head groups have been the most commonly used membrane mimic for solution NMR studies of membrane proteins owing to their smaller size (relative to bicelles) and overall tunability, e.g., they support doping with detergents of different structures and charge properties. The most obvious physical difference between micelles and bicelles is the strong curvature of detergent micelles that deviates from the ideal membrane-like surface that may be required for interactions with cytoplasmic sequences (Chou et al., 2002). Therefore, the conformations of membrane surface domains determined in micellar environments must be viewed with care. For the TM domains, where helix-helix packing forces dominate assembly, the differences in the effects of micelles and bicelles are much less pronounced. At present, there have been no reports of major structural differences observed at atomic resolution between TM assemblies reconstituted in micelles versus those reconstituted in bicelles. Organic solvents have also been used in studying hydrophobic peptides. Although apolar solvents appear to support formation of native-like oligomers in some instances (Yang et al., 2009), they are a poor mimic of the polar/apolar interface in lipid bilayers that is often important in defining the hydrophilic ends of TM domains.
What is the next step forward? Separating the domains for X-ray or NMR studies has provided structural details that have fundamentally advanced our understanding of receptor signaling. However, this deconstructionist approach misses the spatial and angular relationships among domains that will define the mechanics of receptor activation, and these features remain to be described in the context of intact receptors. Solubilizing an intact receptor complex for liquid-phase NMR studies is no trivial task because the detergents commonly used for reconstituting TM domains can partially denature the water-soluble folded domains. An alternative is to use the more stably assembled nanodisc, developed originally by Bayburt and Sligar (2003). Membrane nanodiscs are self-assembled patches of lipid bilayer surrounded by a ring of amphipathic membrane scaffold protein (MSP). The MSP is typically apolipoprotein, which can be overexpressed in E. coli and purified using standard biochemistry protocols. Probably the most significant advantage of the nanodisc system is that there is no detergent involved. The large size of nanodiscs (150–200 kDa) has, in the past, discouraged solution NMR spectroscopists, but a number of recent studies have revived the promise of these lipid discs for structural investigation. A membrane-associated region of human CD4 (a co-receptor for TCR) consisting of both TM and a membrane-associated amphipathic helix was reconstituted in nanodiscs and resulted in a high-quality 2D 1H-13C correlation spectrum exhibiting dispersion and resolution that were comparable to that observed in DPC micelles (Gluck et al., 2009). Another study incorporated a 30 kDa human voltage-dependent anion channel (VDAC) into nanodiscs and showed that these large protein-disc complexes (as confirmed by EM-negative stain images) yielded promising 2D spectra (Raschle et al., 2009). In both studies, relaxation optimization using protein deuteration (Gardner and Kay, 1998) and TROSY spectroscopy (Pervushin et al., 1997) was necessary for achieving the reported NMR spectra. These studies suggest that with further improvements, such as reduction in disc size (Raschle et al., 2010), it may be possible to generate intact, soluble receptor-lipid complexes for NMR measurements.
These promising advances notwithstanding, successful reconstitution of the receptor-nanodisc complex is not sufficient because it is unrealistic to resolve and assign resonances of such large proteins when uniformly isotope labeled. One general strategy is to segmentally isotope label regions of interest. If, for example, it were possible to isotope label only the JM-A segment of EGFR in the context of the full-length receptor, it would allow NMR measurement of the conformation and dynamic properties of this segment in the ligand-free and ligand-bound states. Along these lines, isotope labeling of an internal segment of the maltose-binding protein for NMR spectroscopy has been demonstrated (Otomo et al., 1999). However, the intein ligation method used in this study requires refolding of the polypeptide chain, which is unlikely to work for the much more complex membrane-embedded receptors. Therefore, new peptide ligation protocols that do not require denaturation of the protein need to be developed.
Now that atomic resolution structural studies for many TM receptors are rapidly advancing, it has become clear that the new challenge is to develop improved experimental systems that can provide structural information on the arrangement of functional domains relative to one another and insights into how this may be altered during signaling. Increasingly sophisticated biochemical and biophysical approaches will be required to rise to this challenge, and the application of solution NMR techniques as described in this review will continue to have an important impact in this arena. Together with the development of novel methods for preparation of membrane-embedded protein samples, these techniques are capable of providing a clearer view into what has long constituted a major blind spot in structural biology: the dynamic molecular interactions that control signaling events occurring at the cellular membrane.
Acknowledgments
The authors thank Melissa J. Call for critical review of the manuscript and for help with figures.
References
- Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7:1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- Arnett KL, Harrison SC, Wiley DC. Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc Natl Acad Sci USA. 2004;101:16268–16273. doi: 10.1073/pnas.0407359101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwell JD, Klausner RD. Genetic and mutational analysis of the T-cell antigen receptor. Annu Rev Immunol. 1990;8:139–167. doi: 10.1146/annurev.iy.08.040190.001035. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 2003;12:2476–2481. doi: 10.1110/ps.03267503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG, Arseniev AS. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem. 2008;283:6950–6956. doi: 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]
- Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- Call ME, Wucherpfennig KW. The T cell receptor: critical role of the membrane environment in receptor assembly and function. Annu Rev Immunol. 2005;23:101–125. doi: 10.1146/annurev.immunol.23.021704.115625. [DOI] [PubMed] [Google Scholar]
- Call ME, Wucherpfennig KW. Common themes in the assembly and architecture of activating immune receptors. Nat Rev Immunol. 2007;7:841–850. doi: 10.1038/nri2186. [DOI] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wucherpfennig KW. Stoichiometry of the T-cell receptor-CD3 complex and key intermediates assembled in the endoplasmic reticulum. Embo J. 2004;23:2348–2357. doi: 10.1038/sj.emboj.7600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Wucherpfennig KW, Chou JJ. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol. 2010;11:1023–1029. doi: 10.1038/ni.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Schnell JR, Xu C, Lutz RA, Chou JJ, Wucherpfennig KW. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell. 2006;127:355–368. doi: 10.1016/j.cell.2006.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammett TJ, Jun SJ, Cohen EB, Barrera FN, Engelman DM, Dimaio D. Construction and genetic selection of small transmembrane proteins that activate the human erythropoietin receptor. Proc Natl Acad Sci USA. 2010;107:3447–3452. doi: 10.1073/pnas.0915057107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigaev A, Buranda T, Dwyer DC, Prossnitz ER, Sklar LA. FRET detection of cellular alpha4-integrin conformational activation. Biophys J. 2003;85:3951–3962. doi: 10.1016/S0006-3495(03)74809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JJ, Kaufman JD, Stahl SJ, Wingfield PT, Bax A. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J Am Chem Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- Cosson P, Lankford SP, Bonifacino JS, Klausner RD. Membrane protein association by potential intramembrane charge pairs. Nature. 1991;351:414–416. doi: 10.1038/351414a0. [DOI] [PubMed] [Google Scholar]
- Deford-Watts LM, Tassin TC, Becker AM, Medeiros JJ, Albanesi JP, Love PE, Wulfing C, van Oers NS. The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J Immunol. 2009;183:1055–1064. doi: 10.4049/jimmunol.0900404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman DM. Electrostatic fasteners hold the T cell receptor-CD3 complex together. Mol Cell. 2003;11:5–6. doi: 10.1016/s1097-2765(03)00016-9. [DOI] [PubMed] [Google Scholar]
- Feng J, Garrity D, Call ME, Moffett H, Wucherpfennig KW. Convergence on a distinctive assembly mechanism by unrelated families of activating immune receptors. Immunity. 2005;22:427–438. doi: 10.1016/j.immuni.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- Gardner KH, Kay LE. The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu Rev Biophys Biomol Struct. 1998;27:357–406. doi: 10.1146/annurev.biophys.27.1.357. [DOI] [PubMed] [Google Scholar]
- Gluck JM, Wittlich M, Feuerstein S, Hoffmann S, Willbold D, Koenig BW. Integral membrane proteins in nanodiscs can be studied by solution NMR spectroscopy. J Am Chem Soc. 2009;131:12060–12061. doi: 10.1021/ja904897p. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli AC, Wegener KL, Goult BT, Anthis NJ, Campbell ID, Sansom MS. The structure of the talin/integrin complex at a lipid bilayer: an NMR and MD simulation study. Structure. 2010;18:1280–1288. doi: 10.1016/j.str.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Magn Reson Spectrosc. 2009;55:335–360. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- Kjer-Nielsen L, Clements CS, Brooks AG, Purcell AW, McCluskey J, Rossjohn J. The 1.5 A crystal structure of a highly selected antiviral T cell receptor provides evidence for a structural basis of immunodominance. Structure. 2002;10:1521–1532. doi: 10.1016/s0969-2126(02)00878-x. [DOI] [PubMed] [Google Scholar]
- Kjer-Nielsen L, Dunstone MA, Kostenko L, Ely LK, Beddoe T, Mifsud NA, Purcell AW, Brooks AG, McCluskey J, Rossjohn J. Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc Natl Acad Sci USA. 2004;101:7675–7680. doi: 10.1073/pnas.0402295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatzky KF, Liu W, Goldgraben K, Simmerling C, Smith SO, Constantinescu SN. Structural requirements of the extracellular to transmembrane domain junction for erythropoietin receptor function. J Biol Chem. 2005;280:14844–14854. doi: 10.1074/jbc.M411251200. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM. The safety on the TCR trigger. Cell. 2008;135:594–596. doi: 10.1016/j.cell.2008.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM, Garcia KC. Deconstructing the form and function of the TCR/CD3 complex. Immunity. 2006;24:133–139. doi: 10.1016/j.immuni.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227:150–160. doi: 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998a;391:703–707. doi: 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- Lanier LL, Corliss B, Wu J, Phillips JH. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity. 1998b;8:693–701. doi: 10.1016/s1074-7613(00)80574-9. [DOI] [PubMed] [Google Scholar]
- Lau TL, Dua V, Ulmer TS. Structure of the integrin alphaIIb transmembrane segment. J Biol Chem. 2008a;283:16162–16168. doi: 10.1074/jbc.M801748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau TL, Partridge AW, Ginsberg MH, Ulmer TS. Structure of the integrin beta3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry. 2008b;47:4008–4016. doi: 10.1021/bi800107a. [DOI] [PubMed] [Google Scholar]
- Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. 2010;2:a002485. doi: 10.1101/cshperspect.a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Takagi J, Springer TA. Association of the membrane proximal regions of the alpha and beta subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem. 2001;276:14642–14648. doi: 10.1074/jbc.M100600200. [DOI] [PubMed] [Google Scholar]
- Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2004;2:e153. doi: 10.1371/journal.pbio.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KR. Folding and stability of α-helical integral membrane proteins. Chem Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- Manolios N, Bonifacino JS, Klausner RD. Transmembrane helical interactions and the assembly of the T cell receptor complex. Science. 1990;249:274–277. doi: 10.1126/science.2142801. [DOI] [PubMed] [Google Scholar]
- Otomo T, Ito N, Kyogoku Y, Yamazaki T. NMR observation of selected segments in a larger protein: central-segment isotope labeling through intein-mediated ligation. Biochemistry. 1999;38:16040–16044. doi: 10.1021/bi991902j. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak RM, Chou JJ. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem Biophys Res Commun. 2010;401:58–63. doi: 10.1016/j.bbrc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Yu TY, Rice AJ, Walz T, Wagner G. Structural and functional characterization of the integral membrane protein VDAC-1 in lipid bilayer nanodiscs. J Am Chem Soc. 2009;131:17777–17779. doi: 10.1021/ja907918r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Etzkorn M, Wagner G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr Opin Struct Biol. 2010;20:471–479. doi: 10.1016/j.sbi.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samelson LE, Harford JB, Klausner RD. Identification of the components of the murine T cell antigen receptor complex. Cell. 1985;43:223–231. doi: 10.1016/0092-8674(85)90027-3. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Prosser RS. Bicelles: a model membrane system for all seasons? Struct Fold Des. 1998;6:1227–1234. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigalov AB, Aivazian DA, Uversky VN, Stern LJ. Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry. 2006;45:15731–15739. doi: 10.1021/bi061108f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LC, Clements CS, Beddoe T, Johnson D, Hoare HL, Lin J, Huyton T, Hopkins EJ, Reid HH, Wilce MC, et al. The heterodimeric assembly of the CD94-NKG2 receptor family and implications for human leukocyte antigen-E recognition. Immunity. 2007;27:900–911. doi: 10.1016/j.immuni.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Sussman JJ, Bonifacino JS, Lippincott-Schwartz J, Weissman AM, Saito T, Klausner RD, Ashwell JD. Failure to synthesize the T cell CD3-zeta chain: structure and function of a partial T cell receptor complex. Cell. 1988;52:85–95. doi: 10.1016/0092-8674(88)90533-8. [DOI] [PubMed] [Google Scholar]
- Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- Tomasello E, Olcese L, Vely F, Geourgeon C, Blery M, Moqrich A, Gautheret D, Djabali M, Mattei MG, Vivier E. Gene structure, expression pattern, and biological activity of mouse killer cell activating receptor-associated protein (KARAP)/DAP-12. J Biol Chem. 1998;273:34115–34119. doi: 10.1074/jbc.273.51.34115. [DOI] [PubMed] [Google Scholar]
- van Agthoven A, Terhorst C, Reinherz E, Schlossman S. Characterization of T cell surface glycoproteins T 1 and T 3 present on all human peripheral T lymphocytes and functionally mature thymocytes. Eur J Immunol. 1981;11:18–21. doi: 10.1002/eji.1830110105. [DOI] [PubMed] [Google Scholar]
- Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–597. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Vold RR, Prosser RS, Deese AJ. Isotropic solutions of phospholipid bicelles: a new membrane mimetic for high-resolution NMR studies of polypeptides. J Biomol NMR. 1997;9:329–335. doi: 10.1023/a:1018643312309. [DOI] [PubMed] [Google Scholar]
- Weljie AM, Hwang PM, Vogel HJ. Solution structures of the cytoplasmic tail complex from platelet integrin alpha IIb- and beta 3-subunits. Proc Natl Acad Sci USA. 2002;99:5878–5883. doi: 10.1073/pnas.092515799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA. Structure of an integrin with an alphaI domain, complement receptor type 4. Embo J. 2010;29:666–679. doi: 10.1038/emboj.2009.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ma YQ, Page RC, Misra S, Plow EF, Qin J. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc Natl Acad Sci USA. 2009;106:17729–17734. doi: 10.1073/pnas.0909589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol Biol Cell. 2002;13:2547–2557. doi: 10.1091/mbc.01-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Zhu J, Carman CV, Kim M, Shimaoka M, Springer TA, Luo BH. Requirement of alpha and beta subunit transmembrane helix separation for integrin outside-in signaling. Blood. 2007;110:2475–2483. doi: 10.1182/blood-2007-03-080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Luo BH, Barth P, Schonbrun J, Baker D, Springer TA. The structure of a receptor with two associating transmembrane domains on the cell surface: integrin alphaIIbbeta3. Mol Cell. 2009;34:234–249. doi: 10.1016/j.molcel.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]