

Abstract
Fanconi anemia (FA) is an inherited disease caused by mutations in at least 13 genes and characterized by genomic instability. In addition to displaying strikingly heterogenous clinical phenotypes, FA patients are exquisitely sensitive to treatments with crosslinking agents that create interstrand crosslinks (ICL). In contrast to bacteria and yeast, in which ICLs are repaired through replication-dependent and –independent mechanisms, it is thought that ICLs are repaired primarily during DNA replication in vertebrates (Moldovan and D’Andrea, 2009). However, recent data indicate that replication-independent ICL repair also operates in vertebrates. While the precise role of the FA pathway in ICL repair remains elusive, increasing evidence suggests that FA proteins function at different steps in the sensing, recognition and processing of ICLs, as well as in signaling from these very toxic lesions, which can be generated by a wide variety of cancer chemotherapeutic drugs. Here, we discuss some of the recent findings that have shed light on the role of the FA pathway in ICL repair with special emphasis on the implications of these findings for cancer therapy since disruption of FA genes have been associated with cancer predisposition.
Key terms: DNA repair, DNA replication, Crosslink, Checkpoint, Fanconi anemia
Introduction
Fanconi anemia (FA) is a rare inherited disease associated with genomic instability that has attracted the interest of a diverse audience from basic scientists studying the mechanisms of DNA repair to oncologists treating a variety of sporadic cancers. FA patients are characterized by developmental abnormalities, progressive bone marrow failure, and a predisposition to cancer, especially leukemias and carcinomas. Despite the diverse clinical phenotypes of the disease, a hallmark of cells derived from FA patients is a severe cellular hypersensitivity to crosslinking agents such as mitomycin C (MMC), cisplatin (CDDP), and diepoxybutane (DEB) (reviewed in (Moldovan and D’Andrea, 2009; Niedernhofer et al., 2005; Patel and Joenje, 2007; Thompson and Hinz, 2009; Wang, 2007)). This characteristic has led to a great deal of research implicating the FA pathway in crosslink repair and the maintenance of genomic stability. Furthermore, the FA pathway has also been associated with cancer susceptibility, primarily breast tumors, and with sensitivity and resistance to chemotherapeutic agents. Therefore, a better understanding of the mechanisms and roles of the FA pathway will not only help patients suffering from this rare disease, but also have an impact on cancer patients in the general population.
The FA pathway
Fanconi anemia results from mutations in one of 13 FA genes: Fanca, -b, -c, -d1, -d2, -e, -f, -g, -i, -j, -l, -m and -n (Table 1). FA-associated mutations are autosomal recessive apart from Fancb, which is X-linked. The proteins encoded by these genes make up the FA pathway. The FA pathway can be separated into three groups: the FA core complex, the FANCD2/FANCI (ID) complex, and FA proteins acting downstream. For more extensive reviews of the FA pathway, see (Kennedy and D’Andrea, 2005; Levitus et al., 2006; Mathew, 2006; Taniguchi and D’Andrea, 2006). The FA core complex consists of FANC-A, -B, -C, -E, -F, -G, -L, and -M, and accessory proteins FAAP24 and FAAP100. The downstream FA proteins consist of FANCD1, FANCJ, and FANCN, which are also known as BRCA2, BRIP1/BACH1, and PALB2, respectively. FANCD1/BRCA2 is a major breast and ovarian cancer susceptibility gene and plays an essential role in homology-directed repair (HDR) (Godthelp et al., 2006a; Godthelp et al., 2006b). FANCJ interacts directly with BRCA1 and is a member of the DEAH and XPD helicase families (Cantor et al., 2001; White, 2009). FANCN interacts with FANCD1/BRCA2 and is required for its recombination and checkpoint functions (Xia et al., 2006a; Xia et al., 2006b). This striking association between the FA pathway and breast cancer susceptibility appears to be restricted to this subset of FA genes (Garcia et al., 2009). The FA pathway is a unique example in which mutations from a large epistatic group of genes are associated with the same disorder. Recently, a homozygous mutation in RAD51C, a RAD51 paralog, was identified in a family with multiple congenital abnormalities characteristic of FA (Vaz et al., 2010). Based on the effect of RAD51C loss of function on RAD51 foci formation, RAD51C appears to operate downstream of the ID complex.
Table 1.
Fanconi proteins, functions and connections to cancer.
| FA Protein | Sub-complex | Role | Connection to cancer |
|---|---|---|---|
| FANCA | Core Complex | D2/I Ubiquitylation, Phosphorylated upon damage (ATR) | Inactivated by promoter methylation |
| FANCB | Core Complex | D2/I Ubiquitylation | |
| FANCC | Core Complex | D2/I Ubiquitylation | Inactivated by promoter methylation in AML and ALL |
| FANCE | Core Complex | D2/I Ubiquitylation, Phosphorylated upon damage (CHK1) | |
| FANCF | Core Complex | D2/I Ubiquitylation | Inactivated (methylation) in ovarian, lymphoid, bladder, cervical and lung cancer |
| FANCG | Core Complex | D2/I Ubiquitylation, phosphorylated in M | |
| FANCL | Core Complex | E3 Ligase for D2/I complex | Altered expression in splice variants |
| FANCM | Core Complex Localization to DNA | Helicase/translocase Phosphorylated in M | |
| FANCD2 | Ubiquitylated complex | Ub and Phosphorylated upon damage (ATM, ATR, CHK1) | |
| FANCI | Ubiquitylated complex | Ub and Phosphorylated upon damage (ATR) | |
| FANCD1/BRCA2 | Downstream foci | HR component | Breast, ovarian, prostate, pancreas Cancer Susceptibility, cisplatin sensitivity |
| FANCJ/BACH1 | Downstream foci | Helicase/translocase | Breast Cancer Susceptibility |
| FANCN/PALB2 | Downstream foci | FANCD1 binding | Breast Cancer Susceptibility. Inactivated by promoter methylation |
Modifications of FA proteins: Ubiquitylation
Central to the function and the regulation of the FA pathway is the ubiquitylation of the FANCD2/FANCI complex. The FA core complex monoubiquitylates FANCD2 and FANCI and activates the FA pathway (Ciccia et al., 2007; Dorsman et al., 2007; Garcia-Higuera et al., 2001; Ling et al., 2007; Meetei et al., 2003; Sims et al., 2007). While FANCL is the catalytic subunit (Meetei et al., 2003), all ten members of the FA core complex are required for its ubiquitin E3 ligase activity and UBE2T serves as the ubiquitin E2 ligase (Alpi et al., 2007; Alpi et al., 2008; Machida et al., 2006). FANCM, however, can be distinguished from the other components of the core complex since FANCD2 is still partially ubiquitylated and the core complex remains intact following FANCM inactivation (Kim et al., 2008). This has led to the proposal that FANCM-FAAP24 is a subunit targeting the core complex to DNA (Ali et al., 2009; Ciccia et al., 2007). In addition, FANCM and FAAP24 have been shown to interact with HCLK2 independently of the FA core complex and to function in ATR-mediated checkpoint signaling (Collis et al., 2008). Another accessory protein, FAAP100, has been shown to interact in a stable complex with FANCL and FANCB and is essential for FANCD2 monoubiquitylation and stability of the FA core complex (Ling et al., 2007). Monoubiquitylation of FANCD2 and FANCI is critical for activation of the FA pathway as it is required for the formation and the localization of the FANCI/D2 complex to nuclear foci (Dorsman et al., 2007; Sims et al., 2007; Smogorzewska et al., 2007; Wang, 2007). Functional grouping of FA proteins has been deduced from their roles in FANCD2/FANCI ubiquitylation (Figure 1). Components of the core complex are all required for ID modification and are therefore upstream. Inactivation of the downstream FA proteins does not affect ID ubiquitylation. The ID complex is formed by monoubiquitylated FANCD2 and FANCI whose monoubiquitylations are mutually dependent (Smogorzewska et al., 2007). The ID complex is formed and localized to chromatin during the S phase (Taniguchi et al., 2002a) and in response to DNA damaging agents including MMC, ionizing radiation, ultraviolet light, and hydroxyurea (Garcia-Higuera et al., 2001). On DNA, the ID complex forms foci with DNA repair proteins BRCA1 and RAD51, and FA downstream proteins (Garcia-Higuera et al., 2001; Taniguchi et al., 2002a; Wang et al., 2004). Whereas ubiquitylation of FANCD2 at K561 is essential for the activation of the FA pathway, ubiquitylation of FANCI at K523 is not (Ishiai et al., 2008).
Figure 1. FA pathway: subcomplexes, proteins and post-translational modifications.
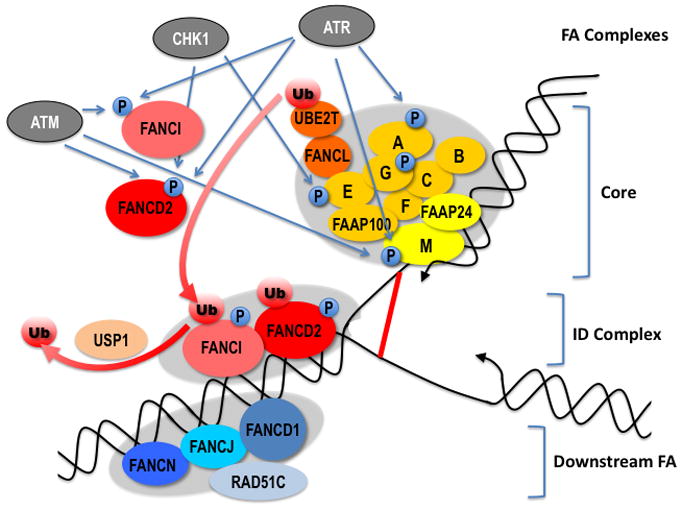
Hypothetical localization of the different FA subcomplexes at a replication fork encountering an interstrand crosslink (red line). The core complex together with the E2 ligase UBE2, mono-ubiquitylate FANCI and FANCD2 (ID complex), thus triggering its association to damaged chromatin. Several FA proteins are phosphorylated in response to DNA damage by the checkpoint kinases ATM, ATR and CHK1. In addition, FANCM and FANCG are phosphorylated in mitosis (See text for details). Note: a color version of the figure is available online.
The USP1/UAF1 complex deubiquitylates FANCD2 (Cohn et al., 2007; Nijman et al., 2005) and FANCI (Longerich et al., 2009). Deubiquitylation of FANCD2 is required for ICL repair (Oestergaard et al., 2007) and loss of USP1 is associated with ICL sensitivity (Kim et al., 2009). Notably, inactivation of USP1 in mice recapitulates several FA phenotypes reminiscent of the knock-out of FA genes (Kim et al., 2009). Proteasome inhibitors sensitize cells to DNA damage and a potential target for this sensitization is FA pathway activation (Jacquemont and Taniguchi, 2007). However, the specific protein target(s) of the proteasome has not been identified.
Modifications of FA proteins: phosphorylation
Several FA proteins are phosphorylated (Table 1; Figure 1). These modifications regulate the activity of the FA pathway following DNA damage and during the cell cycle. Whereas phosphorylation of FA proteins generally occurs in response to both DNA damage and replication stress, FANCA is specifically phosphorylated by ATR (ATM and Rad3-related) following DNA damage but not during DNA replication (Collins et al., 2009). FANCM is phosphorylated in response to DNA replication and DNA damage and this phosphorylation is regulated by the PIKKs ATM (Ataxia telangiectasia mutated) and ATR (Kim et al., 2008; Sobeck et al., 2009). The consequences of the DNA damage-dependent phosphorylation of FANCM are not known. FANCD2 is phosphorylated at multiple sites following DNA damage. Phosphorylation by ATR is critical for checkpoint signaling during S-phase (Andreassen et al., 2004) and for monoubiquitylation of FANCD2 (Ho et al., 2006). FANCD2 is also phosphorylated by ATM; however, this modification is independent of FANCD2 monoubiquitylation (Taniguchi et al., 2002b). FANCD2 and FANCE are phosphorylated by CHK1 and these modifications play a role in resistance to DNA crosslinking agents (Wang et al., 2007; Zhi et al., 2009). Finally, FANCG and FANCM are hyper-phosphorylated during mitosis (Kee et al., 2009; Mi et al., 2004). While the role of these cell cycle-dependent phosphorylations is not known, they could reflect a potential role for the FA pathway in mitosis. Notably, a genome-wide siRNA screen for genes that regulate mitosis progression showed that FANCC and FANCE down-regulation results in mitosis delay (Neumann et al., 2010). Furthermore, FANCD2 localizes to ultrafine chromosome bridges during anaphase, where it co-localizes with BLM (Neumann et al., 2010), suggesting a possible role for FANCD2 in preventing aneuploidy (Naim and Rosselli, 2009).
Crosslinking agents and DNA interstrand crosslinks
ICLs are particularly toxic DNA lesions because they involve both strands of DNA, blocking the essential processes that require translocation along the DNA; namely, DNA replication and transcription. In addition to this physical constraint on DNA, ICLs require repair of damage on both strands of the DNA. Comparative studies ranking in vitro and in vivo genotoxicity of large sets of compounds have ranked crosslinking agents among the most toxic (Lohman, 1999). DNA crosslinking agents cause gross-chromosomal aberrations including chromosome loss, deletions, and breaks, and the ability to yield chromosomal aberrations correlate with their cytotoxicity (Vogel et al., 1998; Vogel et al., 1996). Consequently, crosslinking agents are primarily clastogens rather than mutagens.
ICLs can be sensed by different mechanisms depending on the type of DNA transaction taking place on the crosslinked DNA molecule. In actively dividing cells, the replicative machinery will encounter an ICL during S-phase and the ICL will act as a physical barrier to replisome progression. This is thought to be a prevalent mechanism for sensing ICL lesions. In this setting, ICL repair involves the collision of a replication fork as a trigger to initiate repair. Upon sensing of an ICL during DNA replication, checkpoint activation, multistep repair and stabilization of the replisome must be tightly coordinated. ICL repair in vertebrates involves the generation of fragile DNA intermediates including DSBs, nicks and ssDNA gaps. If these DNA structures are left unrepaired or partially repaired, they may lead to replication fork collapse and aberrant recombination events such as chromosomal fusions that result in the radial chromosomes seen in FA patients.
It has been assumed that this replication-dependent mode of repair should be sufficient to cope with and repair ICL in a timely fashion. However, in non-proliferating cells such as post-mitotic differentiated cells or quiescent stem cells, endogenously generated ICLs need to be repaired in the absence of DNA replication. In this situation, if the ICL is positioned in an actively transcribed gene, a similar sensing process could take place following stalling of the RNA polymerase and transcription block could be the initiating event. However, much less is known about the consequences of blocked transcription by ICLs. Finally, it is thought that distorting ICLs (Table 2) can be recognized in the absence of collision with the replication or transcription machinery moving along DNA.
Table 2.
Properties of different classes of crosslinking agents. Nucleotides in bold characters show the crosslinked nucleotides in ICLs.
| Nitrogen mustards | Platinum compounds | Nitrosoureas | Mitomycin C | Psoralens | |
|---|---|---|---|---|---|
| Example of compounds | HN2, Melphalan Chlorambucyl | Cisplatin Carboplatin | BCNU Carmustine | MMC | Psolaren, 8-Methoxypsoralen |
| Crosslinked Sequences (ICL) | 5′-GNC-3′ 3′-CNG-5′ |
5′-GC-3′ 3′-CG-5′ |
5′-G-3′ 3′-C-3′ |
5′-CG-3′ 3′-GC-5′ |
5′-TA-3′ 3′-AT-5′ |
| Distortion | Variable | Major + bending | Unknown | Modest | Major, no bending |
| Fraction of ICLs | 1–5% | 2–5% | 3–8% | 5–13% | Up to 40% |
| Other major lesions | Mono-adducts | Intrastrand Crosslinks | Mono-adducts, Intrastrand crosslinks | Mono-adducts, ROS | Mono-adducts |
| Chemotherapeutic uses | Lymphoid tumors | Ovarian, Testicular, Lung | Glioblastoma | Gastrointestinal Breast, Bladder | Vitiligo, Psoriasis, Cutaneous T cell lymphoma |
| References | (Dy and Adjei, 2008; Rink and Hopkins, 1995) | (Coste et al., 1999; Huang et al., 1995; Kartalou and Essigmann, 2001) | (Colvin et al., 1976; Wiencke and Wiemels, 1995) | (Pagano, 2002; Rajski and Williams, 1998; Warren et al., 1998) | (Averbeck et al., 1988; Honig et al., 1994; Spielmann et al., 1995; Stern, 2007) |
Crosslinking agents and their relevance
ICLs are generated by bifunctional agents that fall in different classes. Nitrogen mustards (Bis(2-chloroethyl)methylamnine:HN2, chlorambucil, Melphalan) have been used for over 65 years and are still widely used compounds for cancer therapy (Gilman and Philips, 1946), in particular for lymphoid tumors (multiple myeloma, chronic lymphocytic leukemia (CLL)). They react with the N7 of guanine to form primarily mono-adducts to DNA. 1–5% of the lesions, however, are ICLs that are responsible for the cytotoxicity of these compounds (Rink and Hopkins, 1995).
Cisplatin (cisplatinum diammine chloride II, CDDP) and di- and trinuclear cis-DDP analogues were first identified as inhibitors of bacterial division (Rosenberg et al., 1965) and are used to treat a wide spectrum of solid tumors with the greatest impact seen in the treatment of testicular and ovarian cancers. As with nitrogen mustards, ICLs represent a small fraction of lesions induced by platinum drugs (1–5%), which are primarily intrastrand crosslinks (Brabec and Leng, 1993; Jones et al., 1991; Kartalou and Essigmann, 2001). These compounds also react with the N7 of guanine to form ICLs. While ICLs are thought to be the primary cause for cytotoxicity (Roberts and Friedlos, 1987), it is worth noting that intrastrand crosslinks are lesions that will trigger a checkpoint and activate DNA repair pathways overlapping with ICL repair. Notably, differences in cytotoxicity and efficacy between platinum compounds generating distinct intrastrand crosslinks have been reported. For example, trans-DDP, a less toxic analog of cis-DDP cannot form 5′-GG-3′ intrastrand crosslinks (Aris and Farrell, 2009).
Mitomycin C (MMC) is an antibiotic from Streptomyces that is widely used in cell-based studies of ICLs (Tomasz, 1995). MMC needs to be metabolized to active intermediates, which trigger 5–14 % of ICLs, among other adducts (Tomasz, 1995). MMC also generates reactive oxygen species (ROS), which contribute to its toxicity (Pagano, 2002). MMC is used to treat gastrointestinal, breast, lung and bladder cancers. Another class of antibiotic from Streptomyces are Pyrrolo[2,1-c][1,4]benzodiazepines (PBD) that form ICLs very efficiently (Clingen et al., 2005; Gregson et al., 2001).
Nitrosoureas (BCNU, carmustine) are chemotherapeutic agents that are metabolized to bifunctional molecules, which in turn generate less than 8% ICLs among other adducts (Wiencke and Wiemels, 1995). However, toxicity has limited the clinical use of nitrosoureas.
Finally, psoralen and its derivatives form inter-strand cross-links with DNA upon activation by UV irradiation with long-wavelength UV light. Psoralens are used for the treatment of psoriasis, vitiligo, and cutaneous T-cell lymphoma. They form the highest fraction of ICLs (up to 40%) among crosslinking agents (Averbeck et al., 1988; Dronkert and Kanaar, 2001; Honig et al., 1994; Warren et al., 1998).
The pathways of ICL repair have been primarily inferred through the use of these crosslinking agents. However, a variety of synthetic ICLs have also been used for in vitro studies and studies in cell-free extracts (see below). The development of systems utilizing DNA templates harboring single, defined ICL lesions offers significant insight into the mechanisms of ICL detection, signaling, and repair. This strategy is a significant advance over the use of crosslinking agents that generate only a minority of interstrand crosslinks and primarily generate intrastrand crosslinks and other forms of DNA damage that are capable of activating checkpoint signaling and activating repair mechanisms (Table 2).
It has been shown that ICLs can arise endogenously in cells (Niedernhofer et al., 2003). Proposed sources of endogenous ICLs include aldehydes such as malondialdehyde (Kozekov et al., 2003; Stone et al., 2008). These compounds, which act essentially as bifunctional alkylating agents, are products of lipid peroxidation. Another source of ICLs is following the action of psoralen-related furocoumarins, which might be found in plants (Kleiner et al., 2003). The extent to which endogenous ICLs form in cellular DNA and their possible biological effects remain unclear, however, it has been inferred that as many as 10 ICLs could be generated and repaired per cell each day (Lindahl and Barnes, 2000). Accumulation of ICL DNA lesions with time could contribute to the aging of cells (Grillari et al., 2007).
The strongest evidence supporting the idea that ICLs are the toxic lesions among the DNA adducts generated by crosslinking agents come from comparative studies between bifunctional compounds that are capable of forming ICLs and their chemically related monofunctional analogues, which are limited to the generation of monoadducts. Whereas monofunctional alkylating agents largely produce point mutations that are easily explained by the type of alkylation product formed, bifunctional crosslinking agents cause gross chromosomal alterations. For example, studies comparing chlorambucyl with its monofunctional analog show that while chlorambucyl, a nitrogen mustard that elicits ICL formation, treatment resulted in deletions, its monofunctional analog caused point mutations (Yaghi et al., 1998). Furthermore, ICL-generating agents are more effective at inducing chromosomal losses and sister chromatid exchanges than their monofunctional analogues (Bodell et al., 1985; Vogel et al., 1996).
Table 2 summarizes characteristics of some of the crosslinking agents used in cancer therapy and/or for experimental studies of ICLs. Notably, different crosslinking sources yield different ranges of aberrant DNA structures of which ICLs always compose the less abundant fraction. Moreover, ICLs have unique structural characteristics. In particular, the physico-chemical nature of an ICL influences the topology of the surrounding DNA helix, which in turn is thought to affect the manner in which the ICL is recognized and repaired (Noll et al., 2006; Rajski and Williams, 1998; Smeaton et al., 2008; Smeaton et al., 2009).
Model systems to study ICL repair
Some of the characteristics of the different strategies for repairing ICLs are outlined below. Please see (Dronkert and Kanaar, 2001; McVey, 2010; Niedernhofer et al., 2005; Patel and Joenje, 2007) for more complete reviews. ICL repair was first investigated in E. coli (Cole, 1973). There, ICL repair initiates by incisions on either side of the ICL by Uvr(A)2BC (Sladek et al., 1989; Van Houten et al., 1988). If homologous recombination is available, resection at one of the nicked positions elicits RecA loading and invasion of the undamaged template followed by DNA synthesis across the region containing the crosslink. Uvr(A)2BC then excises the unhooked crosslink and the resulting gap is filled. When HR is not available, error-prone translesion synthesis (via Pol II) occurs following Uvr(A)2BC incisions. ICL repair in yeast uses similar strategies but in addition, is tightly cell cycle-regulated (McHugh et al., 2001). Of note, models of ICL repair in S. cerevisiae are primarily based on genetic data and the sequence of events remains hypothetical. During S-phase, the collision of a replication fork with an ICL results in the generation of a DSB (McHugh et al., 2000). This DSB could arise as a consequence of nucleolytic cleavage following fork stalling by the ICL or could result from the fork encountering a single-stranded nick generated by a pre-existing incision in the proximity of the ICL. The ICL is then unhooked and following translesion synthesis, removed by the nucleotide excision repair (NER) pathway. Additionally, the HR pathway is required for repair of the DSB intermediate, as strongly suggested by the sensitivity of HR mutants to crosslinking agents (Henriques and Moustacchi, 1981). In contrast, cells in G1 lack moving replication forks to sense ICLs and lack replicated homologous sequences to use in a HR step in repair. In G1, ICL recognition is dependent on the NER machinery (Lehoczky et al., 2007) and is followed by translesion synthesis and a second set of incisions releasing the crosslinked nucleotide. The occurrence of ICL repair in G1 in yeast is supported by the mutagenic nature of repair and by the sensitivity of yeast harboring mutations in error-prone postreplication repair to crosslinking agents (Henriques and Moustacchi, 1981; McHugh et al., 2000). Notably, yeast use different sets of repair pathways to repair lesions induced by different crosslinking agents (Beljanski et al., 2004). This suggests that the structure of the crosslink influences, at least in part, the mode of repair.
ICL repair in higher eukaryotes is less understood. However, some fundamental similarities are thought to exist between yeast and higher eukaryotes. ICL repair is accompanied by DSB formation that require HR for repair. While HR- and replication-dependent ICL repair is thought to be the prominent mode of repair, replication-independent, HR-independent, and error-prone pathways of ICL repair have also been described (Wang et al., 2001; Zheng et al., 2003). In particular, it has been shown that psoralen crosslinks induced by UV laser irradiation are repaired in G1 cells by a XPC-dependent repair mechanism (Muniandy et al., 2009). Furthermore, it has been shown, using quantitative RT-PCR across repaired lesions, that a plasmid harboring a single ICL is repaired in G1-arrested mammalian cells ((Ben-Yehoyada et al., 2009) and see below).
A fundamental difference between yeast and other organisms is the requirement for FA proteins. The helicases FANCJ and FANCM appeared first in evolution and have homologs in bacteria and Archae (Meetei et al., 2005; White, 2009). Invertebrates have several FA genes with a FANCD2/I complex and downstream FA genes: FANCD1 and FANCJ in C. elegans and FANCD1 in Drosophila. While Drosophila has a FANCL homolog, C. elegans appears to be devoid of FA core complex proteins raising the question of the identity of the ubiquitin ligase responsible for FANCD2 ubiquitylation, which is observed in C. elegans (McVey, 2010).
Most studies on FA and ICL repair have been performed in human and mouse cells as well as in mouse models and in chicken lymphoblastic cells (DT40). In addition, cell-free extracts from Xenopus eggs have recently emerged as a powerful system to study ICL repair and signaling from ICLs.
FA proteins in the ICL damage response
FA proteins as sensors for DNA damage
The FA pathway has been implicated in the response to DNA damage, including but not limited to, ICL damage. Several FA proteins are modified following DNA damage, including the monoubiquitylation of FANCD2 and FANCI (Figure 1). In addition to their role downstream of checkpoint kinases, it has been speculated that FA proteins could also play a role in the early steps of the DNA damage response, i.e. as sensors for DNA damage.
The FA pathway has been shown to be an early responder to ICL damage. FA cells display a defect in the initial ICL incision step, generally attributed to XPF/ERCC1 activity (Fujiwara, 1982; Kumaresan and Lambert, 2000; Kumaresan et al., 2007), although XPF could have additional functions (reviewed in (Bergstralh and Sekelsky, 2008; Hlavin et al., 2010)). This has implicated the FA pathway in the sensing of ICLs and/or the recruitment of XPF/ERCC1. This idea was recently supported by experiments in Xenopus cell-free extracts showing that FANCD2 depletion resulted in an incision defect and subsequent delay in translesion synthesis (Knipscheer et al., 2009).
Experiments using small plasmid templates harboring a single ICL have considerably strengthened the idea that FA proteins could play a direct role in sensing ICLs. Shen et al. employed a substrate harboring an EBV replication start site and a psoralen-induced ICL and found that FA proteins are enriched at the ICL site (Shen et al., 2009). Importantly, in these experimental conditions, FA core complex components and FANCD2/FANCI are recruited to the ICL in a replication-independent manner, implicating the FA pathway in an early step in the sensing of an ICL. In contrast, the recruitment of the downstream FA proteins FANCD1, FANCJ and FANCN requires replication but not the FA core complex, suggesting that the FA core and FANCD2/FANCI proteins are recruited directly to ICL and the BRCA-related FA proteins are recruited to the stalled replication fork at the ICL (Shen et al., 2009). The differential recruitment of FA proteins could reflect the involvement of the FA pathway in distinct ICL repair pathways and is consistent with a sensing role for FA proteins in replication-independent ICL repair and a role of the “downstream” FA proteins in HDR, which takes place in replication-dependent repair of ICLs. Consistent with this, using cell-free extracts from Xenopus, it was shown that FA proteins are recruited to a plasmid harboring a single distorting ICL regardless of whether DNA replication is taking place or not (Ben-Yehoyada et al., 2009). It was also demonstrated that the recruitment of FA proteins was independent of the RPA- and ATR-dependent checkpoint (Ben-Yehoyada et al., 2009). These data implicate the FA pathway in an early step in the sensing of an ICL, upstream of checkpoint signaling. This may explain why FA cells are so exquisitely sensitive to ICLs since they are unable to properly recognize ICLs and therefore unable to activate the subsequent necessary steps to remove the ICL.
FA proteins and ICL repair
The hypersensitivity to crosslinking agents shared by all FA patients indicates that the FA pathway plays an essential role in either sensing, signaling from, or repairing lesions generated by these agents. However, the exact role of the FA proteins in the response to ICLs still remains largely elusive. The FA pathway could participate in both replication-dependent and –independent pathways of ICL repair outlined above. When FA cells are exposed to crosslinking agents, they accumulate chromosomal breaks and radial chromosomes (Auerbach, 1993), an outcome that indicates a defect in the cellular response to ICLs.
After recognizing the ICL and signaling cell cycle arrest to allow its repair, the FA pathway may then function to coordinate the repair of the ICL. Disruption of the FA core complex and the ID complex has been shown to decrease ICL repair efficiency (Ben-Yehoyada et al., 2009; Shen et al., 2009). The mechanisms of ICL repair are believed to involve HDR, translesion synthesis (TLS), and part of the NER machinery. Whereas TLS and nuclease(s) are required for both replication-dependent and –independent repair, homology-dependent repair is thought to be required only in replication-dependent repair when sequences homologous to the lesion are available. However, it is also conceivable that replication-independent, HDR-dependent ICL repair could operate in G2 phase of the cell cycle. Notably, the FA pathway has been associated with proteins involved in HDR, TLS and NER. The exact role of FA proteins in HDR is unclear. While FANCD1 cells show a marked defect in HDR (Moynahan et al., 2001; Nakanishi et al., 2005; Stark et al., 2004), other FA cells show a milder HDR defect (Hirano et al., 2005; Nakanishi et al., 2005; Niedzwiedz et al., 2004; Yamamoto et al., 2005; Yamamoto et al., 2003; Yang et al., 2005). However, the link between the FA pathway and the BRCA pathway is well established since FANCD1 is identical to BRCA2 and several FA pathway proteins interact directly with BRCA1 or BRCA2. Therefore, it is possible that while the FA pathway does not play a major role in all HDR, it plays a specific role in the recruitment of repair proteins and the coordination of HDR repair in the context of ICL damage. Indeed, in vitro studies suggest that FANCD1/BRCA2 play a significant role in ICL repair (Cipak et al., 2006). FANCD2 also associates with the Mre11-Rad50-Nbs1 (MRN) complex, which is essential for DNA resection at DSBs, the first step of all homology-dependent repair processes (Roques et al., 2009). MRN is required for FANCD2 foci formation at sites of damage: ssDNA gaps and DSBs (Roques et al., 2009).
In response to crosslinking agents, FANCD2 has been shown to colocalize with NER component XPF (Sridharan et al., 2003) and XPF affects the stability of ubiquitylated FANCD2 (Bhagwat et al., 2009). FANCD2 has also been shown to colocalize with TLS polymerase Rev1 after replication arrest (Niedzwiedz et al., 2004) and FA core complex components FANCA and FANCG have been shown to be required for efficient Rev1 foci formation (Mirchandani et al., 2008). Playing an ICL-specific role in HDR and a role upstream of TLS and NER, the FA pathway helps to coordinate and regulate these repair mechanisms for the efficient and proper removal of ICL damage. Consistent with these ideas, it was shown that inactivation of FANCD2 affected both nucleolytic incision and translesion synthesis (Knipscheer et al., 2009).
Recent experiments have examined the role of the FA pathway in ICL repair using DNA substrates containing a single, site-specific ICL in cell-free extracts from Xenopus (Ben-Yehoyada et al., 2009; Raschle et al., 2008). In addition, the behavior of ICL-containing DNA templates were monitored in mammalian cells upon transfection (Ben-Yehoyada et al., 2009; Shen et al., 2009).
One of the Xenopus studies reported a system in which ICL repair is exclusively replication-dependent (Knipscheer et al., 2009; Raschle et al., 2008). This study describes in detail the initial steps in ICL processing in a replication-dependent system using primarily a nitrogen mustard-like ICL adduct that minimally distorts the DNA helix (Raschle et al., 2008). Two replication forks converge on the ICL with the leading strand stalling initially 20–24 nucleotides from the ICL followed by the advancement of one leading strand to within 1 nucleotide of the crosslink. Notably, these findings suggest that translesion synthesis precedes ICL unhooking. This model also departs from earlier models invoking the collision of a single replication fork with an ICL (Niedernhofer et al., 2005). On a small plasmid DNA template, convergence of 2 forks on the ICL is the rule. However, on a chromosome, it is not clear how often this phenomenon will take place. The distance between origins of replication ranges from 150 to 500 kb in somatic cells and given that ICLs activate a checkpoint that will decrease the density of active origins, it is reasonable to think that a significant fraction of ICLs will be met by a single replication fork. Therefore these two modes of replication-dependent ICL repair might co-exist.
Using a DNA substrate containing a MMC-like ICL adduct that significantly distorts the DNA helix, another study found that ICL repair can proceed through replication-dependent and -independent mechanisms (Ben-Yehoyada et al., 2009). Repair is associated with a significant amount of DNA synthesis (Ben-Yehoyada et al., 2009). The observation that ICL repair could take place in the absence of DNA replication in Xenopus extracts and upon transfection of an ICL-containing plasmid in G1-arrested mammalian cells (Ben-Yehoyada et al., 2009) is consistent with accumulating evidence for ICL repair in G1, discussed above (Hlavin et al., 2010; Muniandy et al., 2010; Sarkar et al., 2006; Shen and Li, 2010). Some of the differences reported in Xenopus could stem from the different types of ICLs used. As mentioned above, yeasts use different repair pathways to repair ICLs generated by different crosslinking agents (Beljanski et al., 2004). Similarly, it has been shown that in mammalian cell extracts, the level of unhooking of a 5′-GC-3′ ICL (distorting) was 10-fold greater than for a 5′-CG-3′ ICL (non-distorting). These results demonstrate that helix distortion influences significantly ICL recognition and subsequent repair (Smeaton et al., 2008).
The development of site-specific ICL template systems offers great promise for the future of ICL research as these substrates provide the opportunity to examine ICL processes specifically in the absence of other forms of DNA damage. The use of these substrates will help delineate the molecular steps involved in ICL processing and the proteins recruited. The site-specific nature of these ICL substrates allows for the determination of repair efficiency and analysis of repair intermediates and repair errors. In addition, the ability to introduce site-specific ICL adducts into mammalian cells allows for in vivo studies of ICL repair and signaling. This should also provide a prognostic tool to assess the ability of cancer patients’ cells to repair ICLs and subsequently to predict the patients’ response to crosslinking drugs. Likewise, this tool may be used to assess the efficacy of therapeutics targeting crosslink repair.
FA proteins and checkpoint activation
The FA pathway has been connected to checkpoint activity in multiple ways. The complexity of these interactions is seen by the fact that FA is involved upstream and downstream of checkpoint kinase signaling. Studies in Xenopus cell-free extracts using a DNA substrate containing a 5′-GC-3′ ICL adduct that distorts the DNA helix, found that the ICL activates robust replication-dependent and -independent checkpoints. Following recognizing ICL damage, the FA pathway plays an essential role in the induction of an ATR/Chk1-dependent checkpoint (Ben-Yehoyada et al., 2009). This is consistent with the replication-independent specific association of FA core and ID complexes to a psoralen ICL (Shen et al., 2009). Depletion of a FA core protein (FANCL) or of FANCD2 impairs checkpoint signaling. Checkpoint activity was measured by the ability of ICL-containing DNA to inhibit the replication of an undamaged plasmid in trans. Inhibition of the FA pathway also inhibited phosphorylation of CHK1. Consistent with this finding, depletion of FA proteins prevented the recruitment of RPA and ATR to the ICL. This establishes that the FA pathway operates upstream of the RPA-ATR-CHK1 signaling pathway. Importantly, recruitment of RPA and ATR to plasmids harboring UV lesions is not affected by FA pathway inhibition, thus identifying an ICL-specific signaling function for the FA pathway (Ben-Yehoyada et al., 2009). The generation of ssDNA-RPA intermediates upon encountering an ICL with a progressing replication fork can be envisioned as a consequence of fork stalling and processing of the ICL (Figure 2). In contrast, the mechanism responsible for generating ssDNA-RPA in the absence of DNA replication is unknown (Figure 2). In particular, it remains to be determined whether the recruitment of FA proteins is a prerequisite for unhooking of the ICL and/or whether incision(s) at the ICL is required for checkpoint activation.
Figure 2. Cell cycle regulation of signaling from crosslink and crosslink repair.
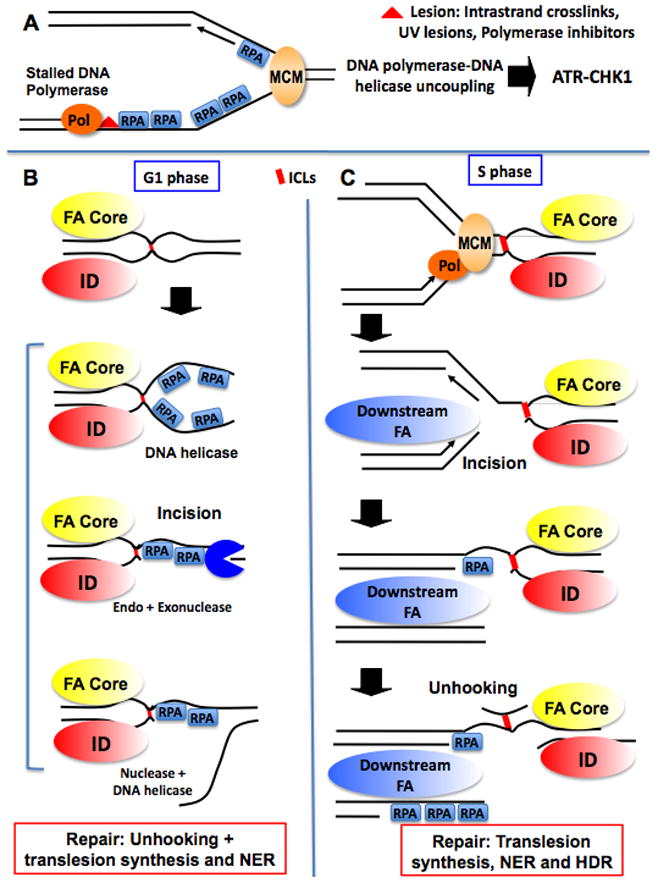
A. Canonical replication checkpoint activated by a lesion on one strand of the DNA. The lesion results in DNA polymerase stalling that becomes uncoupled from the MCM DNA helicase. The resulting ssDNA-RPA intermediates activate the ATR-CHK1 checkpoint pathway (Byun et al., 2005). This checkpoint is activated in response to UV lesions, DNA polymerase inhibition but also intra-strand crosslink generated by crosslinkers such as cisplatin.
B. Checkpoint activation and ICL repair in G1. In G1, the core and ID complexes associated to ICL damage and a checkpoint is activated. It is not known whether ssDNA-RPA accumulates prior to, or as a consequence of an incision in the proximity of the crosslink. Several hypothetical modes of ssDNA-RPA are presented. For clarity, accumulation of ssDNA-RPA is shown only on one side of the ICL but could take place on both sides. Repair will proceed following unhooking, via translesion synthesis and NER.
C. Checkpoint activation and ICL repair in S phase. In S-phase, FANCD1/FANCJ/FANCN (Downstream FA) associate to the fork stalled at the ICL independently of the core and ID complexes. Hypothetically, ssDNA-RPA could be generated following the incision and/or unhooking of the ICL on the lagging strand. In addition, the resected DSB generated following incision could be an additional source of ssDNA-RPA to activate a checkpoint. Note that the MCM helicase is thought to be loaded around dsDNA and therefore unable to unwind DNA passed the ICL. Therefore, the mechanism of checkpoint activation at an ICL is presumably distinct from the polymerase-helicase uncoupling described in A. Repair will proceed following unhooking, via translesion synthesis, NER and HDR. Note: a color version of the figure is available online.
FANCM is involved in the ATR-mediated checkpoint response, independently of the FA core complex (Collis et al., 2008). In the absence of FANCM, TOPBP1 is not retained on chromatin and phosphorylation of ATR downstream targets is defective (Luke-Glaser et al., 2010; Schwab et al., 2010). Interestingly, FANCM appears to be central to a feedback loop in checkpoint activation as CHK1 prevents FANCM proteasome-dependent degradation upon DNA damage.
FA proteins are also required downstream of checkpoint kinases for the proper execution of checkpoint signaling. This was first demonstrated by showing that phosphorylation of FANCD2 by ATM following IR was required for the activation of an S-phase checkpoint (Taniguchi et al., 2002b). Similarly, it was shown that the FA core complex and FANCD2 regulate the ATR-dependent intra-S-phase checkpoint in response to distorting ICLs induced by psoralen (Pichierri and Rosselli, 2004a, b). In contrast, other studies suggested that the FA pathway does not regulate the intra-S-phase checkpoint following treatment with MMC that generate non-distorting ICLs (Andreassen et al., 2004). These differences could reflect distinct responses from cells deficient in different FA genes and/or different responses to different crosslinking agents yielding ICL lesions with different properties (Table 2).
Checkpoint pathways also regulate the FA pathway and in particular, FANCD2 activation. ATR is required for the efficient ubiquitylation of FANCD2 (Andreassen et al., 2004). Both ATR and BRCA1 were shown to facilitate FANCD2 ubiquitylation in response to rereplication (Zhu and Dutta, 2006b). The FA pathway appears to participate in a feedback loop in response to rereplication since the FA core complex, but not FANCD2, is required for checkpoint activation in rereplicating cells (Zhu and Dutta, 2006a).
The FA pathway and DNA replication
The FA pathway is not only activated following treatment with crosslinking agents, but is also responsive to treatments that affect DNA replication (such as the induction of replication stress). The FA pathway is also activated during normal S-phase and the ID complex binds to chromatin during S phase, in the absence of exogenous DNA damage (Mi and Kupfer, 2005; Sobeck et al., 2006). Disruption of the FA pathway leads to the accumulation of DSBs in the absence of exogenous damage (Sobeck et al., 2006), suggesting that the FA pathway plays a role in preventing the accumulation of damage during unperturbed DNA replication. The nature of the DNA structure(s) that are recognized and processed by the FA pathway during DNA replication remains elusive. In cell-free extracts, the FA pathway, as seen by FANCD2 ubiquitylation, is activated by various structures (Sobeck et al., 2007).
The FA core complex interacts with BLM, Topoisomerase IIα, and RPA in the BRAFT complex (Meetei et al., 2003b) and is required for ICL-dependent BLM phosphorylation and recruitment to nuclear foci (Pichierri et al., 2004). Both ATR and BLM have been shown to function at stalled replication forks to stabilize the forks during repair processes and prevent the collapse of the replication fork. Disruption of the FA pathway increases the expression of fragile sites as well as the number of breaks and gaps at known fragile sites (Howlett et al., 2005). FANCD2, FANCI and BLM proteins associate with ultrafine bridges (UFB) during mitosis. These structures are thought to reflect improper replication of fragile site loci (Chan et al., 2009). Moreover, FANCJ and FANCM are enzymes that can process DNA structures arising at replication forks (Schwab et al., 2010; Sommers et al., 2009; Wu et al., 2008). ATM and ATR can phosphorylate a number of FA proteins and many of these phosphorylations are required for efficient activation of the FA pathway during DNA replication (Ho et al., 2006; Matsuoka et al., 2007; Qiao et al., 2004; Wang et al., 2007; Yamashita et al., 1998).
FA proteins can bind specifically at the DNA replication fork where they are poised to detect and respond to DNA damage and stabilize the replication fork (Wang et al., 2008). Disruption of the FA pathway following inactivation of FANCL leads to a defect in replication restart at collapsed replication forks after treatment with MMC or camptothecin (CPT), a topoisomerase I inhibitor (Wang et al., 2008). Similarly, cells deficient for FANCM are defective in DNA replication restart following CPT treatment (Luke-Glaser et al., 2010; Schwab et al., 2010). While MMC generates ICLs (among other DNA lesions) and CPT ultimately leads to the generation of DSBs, both treatments lead to the formation of DNA lesions blocking the replication fork. The requirement for the FA pathway in replication restart after both treatments indicates that the FA pathway functions to stabilize the replication fork when it encounters a physical lesion blocking the fork and/or to coordinate the reassembly of the replication fork and the restart of replication after the removal of the lesion.
FA proteins in cancer
The FA pathway is connected to cancer in many ways. First, Fanconi anemia is associated with cancer predisposition. Second, because the FA pathway is critical for ICL sensing and repair, its integrity determines the sensitivity or resistance to chemotherapeutic drugs that generate ICLs.
The relationship between Fanconi anemia and cancer predisposition has been well established. Almost 25% of FA patients develop malignancies, especially myelodysplastic syndrome, acute myelocytic leukemia, and squamous cell carcinomas of the head and neck (Kutler et al., 2003). However, more recently, FA proteins have been implicated in familial and sporadic cancers outside the FA patient population. FANCD1, FANCJ and FANCN have been implicated in breast cancer susceptibility as truncating mutations in each of these genes have been identified (Erkko et al., 2007; Rahman et al., 2007; Reid et al., 2007; Seal et al., 2006; Turnbull and Rahman, 2008; Xia et al., 2007). FANCD1 mutations have also been associated with ovarian, prostate, stomach and pancreatic cancers (Friedenson, 2005) and FANCN/PALB2 truncations have been implicated in prostate cancer (Erkko et al., 2007). RAD51C, which was recently implicated in a FA-like disorder (Vaz et al., 2010), is also involved in breast and ovarian cancer susceptibility (Meindl et al., 2010). Monoallelic, dominant mutations were observed in families with both breast and ovarian cancer, reminiscent of families with BRCA2 deficiencies (Levy-Lahad, 2010; Meindl et al., 2010). Promoter methylation has also been identified as a method of FA inactivation. FANCF promoter methylation was first found to be associated with ovarian cancer by Taniguchi et al. (Taniguchi et al., 2003) and this finding has been confirmed by several other groups (Dhillon et al., 2004; Wang et al., 2006). In addition to ovarian cancer, FANCF promoter methylation has been implicated in acute myeloid leukemia (Tischkowitz et al., 2003), multiple myeloma (Chen et al., 2005; Hazlehurst et al., 2003), bladder cancer (Neveling et al., 2007), cervical cancer (Narayan et al., 2004), head and neck carcinomas and non-small cell lung cancers (Marsit et al., 2004). Promoter methylation of other FA core complex members, FANCC, FANCG, and FANCL, have also been implicated in pancreatic cancer (Couch et al., 2005; Rogers et al., 2004; van der Heijden et al., 2003), breast cancer (Sinha et al., 2008) and leukemia (Hess et al., 2008). Promoter methylation of PALB2 has been reported in breast tumors (Potapova et al., 2008). In addition, altered expression of FANCL due to a splice variant was found in Calu-6 lung cancer cells (Zhang et al., 2006), and a FANCD2 SNP was found to be associated with sporadic breast cancer in the Spanish population (Barroso et al., 2006). Genetic inactivation and epigenetic silencing of the FA pathway in sporadic cancers underscore the critical tumor suppressor function of this pathway.
The response of cancer cells, either sensitivity or resistance, to DNA damaging drugs such as the crosslinking agent cisplatin can vary depending on the status of certain DNA repair pathways. For example, cisplatin sensitivity in testicular germ cells and non-small cell lung cancer cells is associated with decreased expression of nucleotide excision repair proteins XPA and ERCC1, respectively (Fujii et al., 2008; Koberle et al., 1999; Vilmar and Sorensen, 2008; Welsh et al., 2004). In addition, secondary mutations in BRCA1 mediate cisplatin resistance in BRCA1-mutated ovarian tumors (Swisher et al., 2008). Therefore, the status of and modulation of DNA repair pathways can play a role in tailoring chemotherapy regimes and re-sensitizing cisplatin-resistant cancers to treatment, respectively.
Like cells from FA patients, many of the cancer cells lines and primary tumor samples with a disruption in the FA pathway show crosslinker sensitivity. Inactivation of the FA pathway has been associated with increased sensitivity to crosslinking agents such as cisplatin, MMC, and melphalan in ovarian (Taniguchi et al., 2003), pancreatic (Sakai et al., 2008; van der Heijden et al., 2005; van der Heijden et al., 2004), bladder (Neveling et al., 2007), multiple myeloma (Yarde et al., 2009) and lung cancers (Zhang et al., 2006). In addition, defects in the FA pathway have been associated with sensitivity to alkylating agents in glioma cells (Chen et al., 2007). The FA pathway defect was found to be directly responsible for chemosensitivity since restoration of an intact FA pathway abolished the sensitivity and led to chemoresistance (Taniguchi et al., 2003; van der Heijden et al., 2005). Several studies have also shown that the targeted disruption of the FA pathway by deletion of FANCC or FANCG, the introduction of dominant-negative FANCA, siRNA targeting of FANCF or FANCD2, or inhibition by the small molecule inhibitor curcumin directly leads to chemosensitization to cisplatin, melphalan, alkylating agents, and PARP inhibitors (Chen et al., 2007; Chen et al., 2005; Ferrer et al., 2004; Gallmeier et al., 2006; Sakai et al., 2008). Finally, inactivation of BRCA2/FANCD1 has been associated with sensitivity to cisplatin in ovarian carcinoma. The connection between FA pathway disruption and chemosensitivity identifies the modulation of the FA pathway as a means of chemosensitization. Likewise, identifying FA pathway defects in tumors can lead to the generation of targeted cancer therapy regimes that take advantage of this tumor-specific defect.
While FA pathway disruption leads to cisplatin sensitivity, reactivation of the pathway functions in the development of cisplatin resistance. Strikingly, de novo resistance of BRCA2-deficient ovarian tumor caused by reversion of BRCA2 point mutations have been described in patients and in cells in culture (Sakai et al., 2009; Sakai et al., 2008). In vitro incubation of cisplatin-sensitive cells containing a FANCD1/BRCA2 mutation or FANCF promoter hypermethylation also resulted in acquired cisplatin-resistance mediated by the restoration of the FA pathway (Sakai et al., 2008; Taniguchi et al., 2003). In the case of FANCF promoter methylation, treatment with demethylating agents restores cisplatin resistance (Taniguchi et al., 2003). The processes of FA inactivation and reactivation shed light on the mechanisms of cisplatin sensitivity and resistance and also suggest new modalities of cancer therapy through inhibition of the FA pathway. A cell may initially take advantage of an inactivated FA pathway to generate chromosomal instability and accelerate mutagenesis, during which time it is sensitive to cisplatin treatment. However, cisplatin treatment may select for revertant cells that have reactivated the FA pathway and are now cisplatin-resistant. Therefore, targeting the FA pathway may serve to re-sensitize these cells to cisplatin and can be used in combination with cisplatin to improve the efficacy of this treatment.
Screens have identified genes that, when deregulated, sensitize FA cells. Of note, both major branches of the DNA damage checkpoint pathways, i.e. ATM and ATR-CHK1, sensitize FA cells when downregulated (Chen et al., 2009; Kennedy et al., 2007). Similarly, a small molecule was described that renders FA cells hypersensitive to cross-linking agents (Gallmeier et al., 2007). Several groups have also started the search for FA pathway inhibitors. Chirnomas et al. reported an initial finding of 4 FA/BRCA pathway inhibitors – wortmannin, H-9, alsterpaullone, and curcumin – from a high-throughput screen using human cells (Chirnomas et al., 2006). Landais et al. have developed a Xenopus cell-free extract based assay for FA inhibitors (Landais et al., 2009; Landais et al., 2008). Jacquemont and Taniguchi have shown that proteosomal inhibitors also inhibit the FA pathway (Jacquemont and Taniguchi, 2007) and Burkitt and Ljungman have shown that phenylbutyrate inhibits the FA pathway (Burkitt and Ljungman, 2008). The specific inhibition of the FA pathway offers a promising future for cancer therapies, allowing us to improve the efficacies of our current regimens and overcome the development of resistance.
Conclusions
The FA pathway plays a central role in ICL repair and the maintenance of genomic integrity. Functioning upstream of checkpoint signaling and repair mechanisms, the FA pathway acts as a sentinel in the detection of ICLs. Once an ICL is encountered, the FA pathway is activated and signals downstream to activate an ATR/Chk1-dependent checkpoint. The FA pathway also functions to coordinate HDR, TLS, and NER repair proteins in the repair of ICLs. During ICL recognition and repair, the FA pathway stabilizes the replication fork so that replication can recommence once the damage is repaired. In the absence of an intact FA pathway, cells are sensitive to spontaneous and DNA damage-induced chromosomal breaks.
Disruption of the FA pathway has been found in a number of cancers and may play a role in carcinogenesis by contributing to genomic instability. Inactivation of the FA pathway by promoter methylation and truncating mutations of FA proteins disrupts genomic integrity and contributes to cisplatin sensitivity in many cancers. However, cisplatin resistance may develop after initial treatments with cisplatin as the FA pathway becomes reactivated. The essential role of the FA pathway in cisplatin sensitivity and resistance identifies the FA pathway as a specific target for cancer therapy. Assessing the status of FA pathway function can identify the specific tumors susceptible to crosslinking agents. In addition, targeted disruption of the FA pathway can restore cisplatin sensitivity to resistant tumors, and can also be used as combination therapy with a variety of chemotherapeutic agents including crosslinking agents, ionizing radiation, alkylating agents, and PARP inhibitors to increase the efficacy of these treatments.
The development of tailored chemotherapies and combination therapy with specific chemosensitizers that target DNA repair pathways hold great promise for the future of cancer treatment. However, it will be important to target these chemosensitizers to tumor cells to avoid the disruption of key DNA repair pathways in adjacent normal cells. In addition, the inhibitors should target key downstream proteins so as to avoid the development of compensating mechanisms leading to chemoresistance. The initial findings of small molecule inhibitors of the FA pathway and their implication in restoring cisplatin sensitivity identify the FA pathway as a DNA repair pathway that can be specifically targeted in this manner. As greater understanding of the roles and mechanisms of the FA pathway is achieved and specific inhibitors of this pathway are identified, targeted modulation of the FA pathway may play an important role in many chemotherapeutic regimes.
Footnotes
Declaration of Interest section
The authors acknowledge funding from the National Institute of Health (GM 077495 and CA92245 to J. G.)
References
- Ali AM, Singh TR, Meetei AR. FANCM-FAAP24 and FANCJ: FA proteins that metabolize DNA. Mutation research. 2009;668:20–26. doi: 10.1016/j.mrfmmm.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpi A, Langevin F, Mosedale G, Machida YJ, Dutta A, Patel KJ. UBE2T, the Fanconi anemia core complex, and FANCD2 are recruited independently to chromatin: a basis for the regulation of FANCD2 monoubiquitination. Mol Cell Biol. 2007;27:8421–8430. doi: 10.1128/MCB.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes & development. 2004;18:1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris SM, Farrell NP. Towards Antitumor Active trans-Platinum Compounds. Eur J Inorg Chem. 2009;2009:1293. doi: 10.1002/ejic.200801118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Experimental hematology. 1993;21:731–733. [PubMed] [Google Scholar]
- Averbeck D, Papadopoulo D, Moustacchi E. Repair of 4,5′,8-trimethylpsoralen plus light-induced DNA damage in normal and Fanconi’s anemia cell lines. Cancer research. 1988;48:2015–2020. [PubMed] [Google Scholar]
- Barroso E, Milne RL, Fernandez LP, Zamora P, Arias JI, Benitez J, Ribas G. FANCD2 associated with sporadic breast cancer risk. Carcinogenesis. 2006;27:1930–1937. doi: 10.1093/carcin/bgl062. [DOI] [PubMed] [Google Scholar]
- Beljanski V, Marzilli LG, Doetsch PW. DNA damage-processing pathways involved in the eukaryotic cellular response to anticancer DNA cross-linking drugs. Mol Pharmacol. 2004;65:1496–1506. doi: 10.1124/mol.65.6.1496. [DOI] [PubMed] [Google Scholar]
- Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J. Checkpoint signaling from a single DNA interstrand crosslink. Mol Cell. 2009;35:704–715. doi: 10.1016/j.molcel.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh DT, Sekelsky J. Interstrand crosslink repair: can XPF-ERCC1 be let off the hook? Trends Genet. 2008;24:70–76. doi: 10.1016/j.tig.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, D’Andrea A, Niedernhofer LJ, McHugh PJ. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol. 2009;29:6427–6437. doi: 10.1128/MCB.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodell WJ, Aida T, Rasmussen J. Comparison of sister-chromatid exchange induction caused by nitrosoureas that alkylate or alkylate and crosslink DNA. Mutation research. 1985;149:95–100. doi: 10.1016/0027-5107(85)90013-2. [DOI] [PubMed] [Google Scholar]
- Brabec V, Leng M. DNA interstrand cross-links of trans-diamminedichloroplatinum(II) are preferentially formed between guanine and complementary cytosine residues. Proc Natl Acad Sci U S A. 1993;90:5345–5349. doi: 10.1073/pnas.90.11.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkitt K, Ljungman M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Molecular cancer. 2008;7:24. doi: 10.1186/1476-4598-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood. 2005;106:698–705. doi: 10.1182/blood-2004-11-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani el H, Joenje H, McDonald N, de Winter JP, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Cipak L, Watanabe N, Bessho T. The role of BRCA2 in replication-coupled DNA interstrand cross-link repair in vitro. Nature structural & molecular biology. 2006;13:729–733. doi: 10.1038/nsmb1120. [DOI] [PubMed] [Google Scholar]
- Clingen PH, De Silva IU, McHugh PJ, Ghadessy FJ, Tilby MJ, Thurston DE, Hartley JA. The XPF-ERCC1 endonuclease and homologous recombination contribute to the repair of minor groove DNA interstrand crosslinks in mammalian cells produced by the pyrrolo[2,1-c][1,4]benzodiazepine dimer SJG-136. Nucleic Acids Res. 2005;33:3283–3291. doi: 10.1093/nar/gki639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D’Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28:786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Cole RS. Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci U S A. 1973;70:1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, Kupfer GM. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;113:2181–2190. doi: 10.1182/blood-2008-05-154294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, Boulton SJ. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell. 2008;32:313–324. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Couch FJ, Johnson MR, Rabe K, Boardman L, McWilliams R, de Andrade M, Petersen G. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer research. 2005;65:383–386. [PubMed] [Google Scholar]
- de Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutation research. 2009;668:11–19. doi: 10.1016/j.mrfmmm.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol. 2007;29:211–218. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutation research. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, Kallioniemi A, Pylkas K, Karppinen SM, Rapakko K, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007 doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed. 2005;7:60. [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Toyooka S, Ichimura K, Fujiwara Y, Hotta K, Soh J, Suehisa H, Kobayashi N, Aoe M, Yoshino T, et al. ERCC1 protein expression predicts the response of cisplatin-based neoadjuvant chemotherapy in non-small-cell lung cancer. Lung cancer (Amsterdam, Netherlands) 2008;59:377–384. doi: 10.1016/j.lungcan.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y. Defective repair of mitomycin C crosslinks in Fanconi’s anemia and loss in confluent normal human and xeroderma pigmentosum cells. Biochimica et biophysica acta. 1982;699:217–225. doi: 10.1016/0167-4781(82)90110-5. [DOI] [PubMed] [Google Scholar]
- Garcia MJ, Fernandez V, Osorio A, Barroso A, Fernandez F, Urioste M, Benitez J. Mutational analysis of FANCL, FANCM and the recently identified FANCI suggests that among the 13 known Fanconi Anemia genes, only FANCD1/BRCA2 plays a major role in high-risk breast cancer predisposition. Carcinogenesis. 2009;30:1898–1902. doi: 10.1093/carcin/bgp218. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gilman A, Philips FS. The Biological Actions and Therapeutic Applications of the B-Chloroethyl Amines and Sulfides. Science (New York, NY. 1946;103:409–436. doi: 10.1126/science.103.2675.409. [DOI] [PubMed] [Google Scholar]
- Godthelp BC, van Buul PP, Jaspers NG, Elghalbzouri-Maghrani E, van Duijn-Goedhart A, Arwert F, Joenje H, Zdzienicka MZ. Cellular characterization of cells from the Fanconi anemia complementation group, FA-D1/BRCA2. Mutat Res. 2006a;601:191–201. doi: 10.1016/j.mrfmmm.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Godthelp BC, Wiegant WW, Waisfisz Q, Medhurst AL, Arwert F, Joenje H, Zdzienicka MZ. Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/BRCA2. Mutat Res. 2006b;594:39–48. doi: 10.1016/j.mrfmmm.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Gregson SJ, Howard PW, Hartley JA, Brooks NA, Adams LJ, Jenkins TC, Kelland LR, Thurston DE. Design, synthesis, and evaluation of a novel pyrrolobenzodiazepine DNA-interactive agent with highly efficient cross-linking ability and potent cytotoxicity. J Med Chem. 2001;44:737–748. doi: 10.1021/jm001064n. [DOI] [PubMed] [Google Scholar]
- Grillari J, Katinger H, Voglauer R. Contributions of DNA interstrand cross-links to aging of cells and organisms. Nucleic Acids Res. 2007;35:7566–7576. doi: 10.1093/nar/gkm1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlehurst LA, Enkemann SA, Beam CA, Argilagos RF, Painter J, Shain KH, Saporta S, Boulware D, Moscinski L, Alsina M, et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer research. 2003;63:7900–7906. [PubMed] [Google Scholar]
- Henriques JA, Moustacchi E. Interactions between mutations for sensitivity to psoralen photoaddition (pso) and to radiation (rad) in Saccharomyces cerevisiae. J Bacteriol. 1981;148:248–256. doi: 10.1128/jb.148.1.248-256.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess CJ, Ameziane N, Schuurhuis GJ, Errami A, Denkers F, Kaspers GJ, Cloos J, Joenje H, Reinhardt D, Ossenkoppele GJ, et al. Hypermethylation of the FANCC and FANCL promoter regions in sporadic acute leukaemia. Cell Oncol. 2008;30:299–306. doi: 10.3233/CLO-2008-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Yamamoto K, Ishiai M, Yamazoe M, Seki M, Matsushita N, Ohzeki M, Yamashita YM, Arakawa H, Buerstedde JM, et al. Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. Embo J. 2005;24:418–427. doi: 10.1038/sj.emboj.7600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavin EM, Smeaton MB, Noronha AM, Wilds CJ, Miller PS. Cross-Link Structure Affects Replication-Independent DNA Interstrand Cross-Link Repair in Mammalian Cells. Biochemistry. 2010 doi: 10.1021/bi902169q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GP, Margossian S, Taniguchi T, D’Andrea AD. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol. 2006;26:7005–7015. doi: 10.1128/MCB.02018-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig B, Morison WL, Karp D. Photochemotherapy beyond psoriasis. J Am Acad Dermatol. 1994;31:775–790. doi: 10.1016/s0190-9622(94)70240-3. [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nature structural & molecular biology. 2008;15:1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer research. 2007;67:7395–7405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- Jones JC, Zhen WP, Reed E, Parker RJ, Sancar A, Bohr VA. Gene-specific formation and repair of cisplatin intrastrand adducts and interstrand cross-links in Chinese hamster ovary cells. J Biol Chem. 1991;266:7101–7107. [PubMed] [Google Scholar]
- Kartalou M, Essigmann JM. Recognition of cisplatin adducts by cellular proteins. Mutation research. 2001;478:1–21. doi: 10.1016/s0027-5107(01)00142-7. [DOI] [PubMed] [Google Scholar]
- Kee Y, Kim JM, D’Andrea AD. Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes & development. 2009;23:555–560. doi: 10.1101/gad.1761309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes & development. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kee Y, Gurtan A, D’Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D’Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009;16:314–320. doi: 10.1016/j.devcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner HE, Reed MJ, DiGiovanni J. Naturally occurring coumarins inhibit human cytochromes P450 and block benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene DNA adduct formation in MCF-7 cells. Chem Res Toxicol. 2003;16:415–422. doi: 10.1021/tx025636d. [DOI] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science (New York, NY) 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koberle B, Masters JR, Hartley JA, Wood RD. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol. 1999;9:273–276. doi: 10.1016/s0960-9822(99)80118-3. [DOI] [PubMed] [Google Scholar]
- Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. DNA interchain cross-links formed by acrolein and crotonaldehyde. J Am Chem Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- Kumaresan KR, Lambert MW. Fanconi anemia, complementation group A, cells are defective in ability to produce incisions at sites of psoralen interstrand cross-links. Carcinogenesis. 2000;21:741–751. doi: 10.1093/carcin/21.4.741. [DOI] [PubMed] [Google Scholar]
- Kumaresan KR, Sridharan DM, McMahon LW, Lambert MW. Deficiency in incisions produced by XPF at the site of a DNA interstrand cross-link in Fanconi anemia cells. Biochemistry. 2007;46:14359–14368. doi: 10.1021/bi7015958. [DOI] [PubMed] [Google Scholar]
- Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101:1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- Lehoczky P, McHugh PJ, Chovanec M. DNA interstrand cross-link repair in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2007;31:109–133. doi: 10.1111/j.1574-6976.2006.00046.x. [DOI] [PubMed] [Google Scholar]
- Levitus M, Joenje H, de Winter JP. The Fanconi anemia pathway of genomic maintenance. Cell Oncol. 2006;28:3–29. doi: 10.1155/2006/974975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. The EMBO journal. 2007;26:2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman PH. Qualitative and quantitative procedures for health risk assessment. Mutation research. 1999;428:237–254. doi: 10.1016/s1383-5742(99)00051-4. [DOI] [PubMed] [Google Scholar]
- Longerich S, San Filippo J, Liu D, Sung P. FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem. 2009;284:23182–23186. doi: 10.1074/jbc.C109.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke-Glaser S, Luke B, Grossi S, Constantinou A. FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. The EMBO journal. 2010;29:795–805. doi: 10.1038/emboj.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D’Andrea AD, Dutta A. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006;23:589–596. doi: 10.1016/j.molcel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25:5875–5884. doi: 10.1038/sj.onc.1209878. [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:3425–3433. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2:483–490. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- McVey M. Strategies for DNA interstrand crosslink repair: Insights from worms, flies, frogs, and slime molds. Environ Mol Mutagen. 2010 doi: 10.1002/em.20551. [DOI] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J, Kupfer GM. The Fanconi anemia core complex associates with chromatin during S phase. Blood. 2005;105:759–766. doi: 10.1182/blood-2004-01-0001. [DOI] [PubMed] [Google Scholar]
- Mi J, Qiao F, Wilson JB, High AA, Schroeder MJ, Stukenberg PT, Moss A, Shabanowitz J, Hunt DF, Jones NJ, et al. FANCG is phosphorylated at serines 383 and 387 during mitosis. Mol Cell Biol. 2004;24:8576–8585. doi: 10.1128/MCB.24.19.8576-8585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirchandani KD, McCaffrey RM, D’Andrea AD. The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair (Amst) 2008;7:902–911. doi: 10.1016/j.dnarep.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol. 2010;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM. Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J Biol Chem. 2009;284:27908–27917. doi: 10.1074/jbc.M109.029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveling K, Kalb R, Florl AR, Herterich S, Friedl R, Hoehn H, Hader C, Hartmann FH, Nanda I, Steinlein C, et al. Disruption of the FA/BRCA pathway in bladder cancer. Cytogenetic and genome research. 2007;118:166–176. doi: 10.1159/000108297. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003;278:31426–31433. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28:798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano G. Redox-modulated xenobiotic action and ROS formation: a mirror or a window? Hum Exp Toxicol. 2002;21:77–81. doi: 10.1191/0960327102ht214oa. [DOI] [PubMed] [Google Scholar]
- Patel KJ, Joenje H. Fanconi anemia and DNA replication repair. DNA repair. 2007;6:885–890. doi: 10.1016/j.dnarep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. Embo J. 2004;23:3154–3163. doi: 10.1038/sj.emboj.7600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Rosselli F. Fanconi anemia proteins and the s phase checkpoint. Cell Cycle. 2004a;3:698–700. [PubMed] [Google Scholar]
- Pichierri P, Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. The EMBO journal. 2004b;23:1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P. Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer research. 2008;68:998–1002. doi: 10.1158/0008-5472.CAN-07-2418. [DOI] [PubMed] [Google Scholar]
- Rajski SR, Williams RM. DNA Cross-Linking Agents as Antitumor Drugs. Chem Rev. 1998;98:2723–2796. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- Raschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink SM, Hopkins PB. A mechlorethamine-induced DNA interstrand cross-link bends duplex DNA. Biochemistry. 1995;34:1439–1445. doi: 10.1021/bi00004a039. [DOI] [PubMed] [Google Scholar]
- Roberts JJ, Friedlos F. Quantitative estimation of cisplatin-induced DNA interstrand cross-links and their repair in mammalian cells: relationship to toxicity. Pharmacol Ther. 1987;34:215–246. doi: 10.1016/0163-7258(87)90012-x. [DOI] [PubMed] [Google Scholar]
- Rogers CD, van der Heijden MS, Brune K, Yeo CJ, Hruban RH, Kern SE, Goggins M. The genetics of FANCC and FANCG in familial pancreatic cancer. Cancer biology & therapy. 2004;3:167–169. doi: 10.4161/cbt.3.2.609. [DOI] [PubMed] [Google Scholar]
- Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, et al. MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. The EMBO journal. 2009;28:2400–2413. doi: 10.1038/emboj.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg B, Vancamp L, Krigas T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. The EMBO journal. 2006;25:1285–1294. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. The EMBO journal. 2010;29:806–818. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell. 2009;35:716–723. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Li L. Mutagenic repair of DNA interstrand crosslinks. Environ Mol Mutagen. 2010 doi: 10.1002/em.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nature structural & molecular biology. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Sladek FM, Munn MM, Rupp WD, Howard-Flanders P. In vitro repair of psoralen-DNA cross-links by RecA, UvrABC, and the 5′-exonuclease of DNA polymerase I. J Biol Chem. 1989;264:6755–6765. [PubMed] [Google Scholar]
- Smeaton MB, Hlavin EM, McGregor Mason T, Noronha AM, Wilds CJ, Miller PS. Distortion-dependent unhooking of interstrand cross-links in mammalian cell extracts. Biochemistry. 2008;47:9920–9930. doi: 10.1021/bi800925e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeaton MB, Hlavin EM, Noronha AM, Murphy SP, Wilds CJ, Miller PS. Effect of cross-link structure on DNA interstrand cross-link repair synthesis. Chem Res Toxicol. 2009;22:1285–1297. doi: 10.1021/tx9000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, Wallisch M, Akkari Y, Olson S, Wang W, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol Cell Biol. 2006;26:425–437. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME. The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways. J Biol Chem. 2009;284:25560–25568. doi: 10.1074/jbc.M109.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan D, Brown M, Lambert WC, McMahon LW, Lambert MW. Nonerythroid alphaII spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links. J Cell Sci. 2003;116:823–835. doi: 10.1242/jcs.00294. [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, et al. Interstrand DNA cross-links induced by alpha,beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res. 2008;41:793–804. doi: 10.1021/ar700246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer research. 2008;68:2581–2586. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002a;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D’Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002b;109:459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D’Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutation research. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkowitz M, Ameziane N, Waisfisz Q, De Winter JP, Harris R, Taniguchi T, D’Andrea A, Hodgson SV, Mathew CG, Joenje H. Bi-allelic silencing of the Fanconi anaemia gene FANCF in acute myeloid leukaemia. Br J Haematol. 2003;123:469–471. doi: 10.1046/j.1365-2141.2003.04640.x. [DOI] [PubMed] [Google Scholar]
- Tomasz M. Mitomycin C: small, fast and deadly (but very selective) Chem Biol. 1995;2:575–579. doi: 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]
- van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer research. 2003;63:2585–2588. [PubMed] [Google Scholar]
- Van Houten B, Gamper H, Hearst JE, Sancar A. Analysis of sequential steps of nucleotide excision repair in Escherichia coli using synthetic substrates containing single psoralen adducts. J Biol Chem. 1988;263:16553–16560. [PubMed] [Google Scholar]
- Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- Vilmar A, Sorensen JB. Excision repair cross-complementation group 1 (ERCC1) in platinum-based treatment of non-small cell lung cancer with special emphasis on carboplatin: A review of current literature. Lung cancer (Amsterdam, Netherlands) 2008 doi: 10.1016/j.lungcan.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Vogel EW, Barbin A, Nivard MJ, Stack HF, Waters MD, Lohman PH. Heritable and cancer risks of exposures to anticancer drugs: inter-species comparisons of covalent deoxyribonucleic acid-binding agents. Mutation research. 1998;400:509–540. doi: 10.1016/s0027-5107(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Vogel EW, Nivard MJ, Ballering LA, Bartsch H, Barbin A, Nair J, Comendador MA, Sierra LM, Aguirrezabalaga I, Tosal L, et al. DNA damage and repair in mutagenesis and carcinogenesis: implications of structure-activity relationships for cross-species extrapolation. Mutation research. 1996;353:177–218. doi: 10.1016/0027-5107(96)00032-2. [DOI] [PubMed] [Google Scholar]
- Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteins stabilize replication forks. DNA Repair (Amst) 2008 doi: 10.1016/j.dnarep.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24:5850–5862. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D’Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2007;27:3098–3108. doi: 10.1128/MCB.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol. 2001;21:713–720. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren AJ, Maccubbin AE, Hamilton JW. Detection of mitomycin C-DNA adducts in vivo by 32P-postlabeling: time course for formation and removal of adducts and biochemical modulation. Cancer research. 1998;58:453–461. [PubMed] [Google Scholar]
- Welsh C, Day R, McGurk C, Masters JR, Wood RD, Koberle B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. International journal of cancer. 2004;110:352–361. doi: 10.1002/ijc.20134. [DOI] [PubMed] [Google Scholar]
- White MF. Structure, function and evolution of the XPD family of iron-sulfur-containing 5′-->3′ DNA helicases. Biochem Soc Trans. 2009;37:547–551. doi: 10.1042/BST0370547. [DOI] [PubMed] [Google Scholar]
- Wiencke JK, Wiemels J. Genotoxicity of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) Mutation research. 1995;339:91–119. doi: 10.1016/0165-1110(95)90005-5. [DOI] [PubMed] [Google Scholar]
- Xia B, Dorsman J, De Vries Y, Ameziane N, Rooimans MA, Oostra AB, Sheng Q, Steltenpool J, Gluckman E, Llera J, et al. PALB2, a New Member of the FA/BRCA Pathway. Paper presented at: Eighteenth Annual Fanconi Anemia Research Fund Scientific Symposium.2006a. [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006b;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Yaghi BM, Turner PM, Denny WA, Turner PR, O’Connor CJ, Ferguson LR. Comparative mutational spectra of the nitrogen mustard chlorambucil and its half-mustard analogue in Chinese hamster AS52 cells. Mutation research. 1998;401:153–164. doi: 10.1016/s0027-5107(98)00005-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hirano S, Ishiai M, Morishima K, Kitao H, Namikoshi K, Kimura M, Matsushita N, Arakawa H, Buerstedde JM, et al. Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol Cell Biol. 2005;25:34–43. doi: 10.1128/MCB.25.1.34-43.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH, Takata M. Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol Cell Biol. 2003;23:5421–5430. doi: 10.1128/MCB.23.15.5421-5430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Herceg Z, Nakanishi K, Demuth I, Piccoli C, Michelon J, Hildebrand G, Jasin M, Digweed M, Wang ZQ. The Fanconi anemia group A protein modulates homologous repair of DNA double-strand breaks in mammalian cells. Carcinogenesis. 2005;26:1731–1740. doi: 10.1093/carcin/bgi134. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang X, Lin CJ, Couch FJ, Fei P. Altered expression of FANCL confers mitomycin C sensitivity in Calu-6 lung cancer cells. Cancer biology & therapy. 2006;5:1632–1636. doi: 10.4161/cbt.5.12.3351. [DOI] [PubMed] [Google Scholar]
- Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi G, Wilson JB, Chen X, Krause DS, Xiao Y, Jones NJ, Kupfer GM. Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for fanconi anemia pathway function and BRCA2 interaction. Cancer research. 2009;69:8775–8783. doi: 10.1158/0008-5472.CAN-09-2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Dutta A. Activation of fanconi anemia pathway in cells with re-replicated DNA. Cell Cycle. 2006a;5:2306–2309. doi: 10.4161/cc.5.20.3364. [DOI] [PubMed] [Google Scholar]
- Zhu W, Dutta A. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol. 2006b;26:4601–4611. doi: 10.1128/MCB.02141-05. [DOI] [PMC free article] [PubMed] [Google Scholar]