

Abstract
Inhibitors of the JAK family of non-receptor tyrosine kinases have demonstrated clinical efficacy in rheumatoid arthritis and other inflammatory disorders; however, the precise mechanisms by which JAK inhibition improves inflammatory immune responses remain unclear. Here we examined the mode of action of tofacitinib (CP-690,550) on JAK/STAT signaling pathways involved in adaptive and innate immune responses. To determine the extent of inhibition of specific JAK/STAT-dependent pathways, we analyzed cytokine stimulation of mouse and human T cells in vitro. We also investigated the consequences of CP-690,550 treatment on Th cell differentiation of naïve murine CD4+ T cells. CP-690,550 inhibited IL-4-dependent Th2 cell differentiation, and interestingly also interfered with Th17 cell differentiation. Expression of IL-23 receptor and of the Th17 cytokines IL-17A, IL-17F and IL-22 were blocked when naïve Th cells were stimulated with IL-6 and IL-23. In contrast, IL-17A-production was enhanced when Th17 cells were differentiated in the presence of TGF-β. Moreover, CP-690,550 also prevented activation of STAT1, induction of T-bet and subsequent generation of Th1 cells. In a model of established arthritis, CP-690,550 rapidly improved disease by inhibiting production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. Furthermore, efficacy in this disease model correlated with inhibition of both JAK1 and JAK3 signaling pathways. CP-690,550 also modulated innate responses to LPS in vivo through a mechanism likely involving inhibition of STAT1 signaling. Thus, CP-690,550 may improve autoimmune diseases and prevent transplant rejection by suppressing the differentiation of pathogenic Th1 and Th17 cells, as well as innate immune cell signaling.
Introduction
Cytokines are key mediators of development and homeostasis of hematopoietic cells, playing crucial roles in controlling both innate and adaptive immunity (1, 2). Type I and II cytokine receptors represent a structurally distinct class of integral membrane proteins that lack intrinsic enzymatic activity and associate with a family of cytoplasmic protein tyrosine kinases known as JAKs. Upon cytokine-induced activation, JAKs phosphorylate the cytoplasmic tail of the receptor, leading to recruitment of STATs, which are also phosphorylated by JAKs (3). Activated STATs dimerize, translocate to the nucleus and regulate expression of numerous genes (4). The vital role of JAK signaling is best illustrated by circumstances where these kinases are mutated or deleted (5, 6). For instance, while germline deletion of either JAK1 or JAK2 is lethal, mutation of JAK3 or TYK2 in humans and mice results in immunodeficiency (7, 8). TYK2 mainly transmits the signals derived from Type I IFNs and the IL-12 receptor β1 subunit sharing receptors for IL-12 and IL-23 (9), whereas JAK3 has a more discrete function and associates only with the IL-2 receptor γc-chain shared by the receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 (2). Deficiency of JAK1 leads to non-responsiveness to type I and type II IFNs, γc-cytokines and gp130 subunit-utilizing cytokines (10), whereas JAK2-deficient cells fail to respond to hormone-like cytokines such as erythropoietin, thrombopoietin or GM-CSF (11).
JAKs play a critical role in mediating inflammatory immune responses, and their pharmacological modulation represents a novel approach to the treatment of inflammatory immune-mediated diseases. Indeed, the JAK-STAT pathway has gained significant attention as a therapeutic target in inflammation, autoimmune disease, hematopoetic disorders, and transplant rejection (12, 13). Several small molecule JAK inhibitors have been developed and are currently under clinical investigation (14-17). Tofacitinib (CP-690,550, formerly tasocitinib) is a selective inhibitor of the JAK kinase family with nanomolar potency and a high degree of kinome selectivity (18-20). In cellular assays, it has demonstrated potent inhibition of γc-cytokine signaling by blocking IL-2 driven T cell proliferation and functional selectivity over JAK2-dependent GM-CSF-driven proliferation of HUO3 cells (18). More recently, CP-690,550 has been shown to potently inhibit both JAK3- and JAK1-dependent STAT activation with selectivity over JAK2-mediated pathways (21). Results from a phase II trial of oral CP-690,550 as monotherapy in patients with rheumatoid arthritis (RA) showed efficacy with 70 to 80% of patients achieving 20% improvement in the American College of Rheumatology criteria (ACR20) and an acceptable safety profile (22). CP-690,550 is currently being evaluated in phase III trials in RA and in other immune-mediated diseases including: psoriasis, Crohn's disease and organ transplant rejection (15, 23) (ClinicalTrials.gov identifier NCT00615199). Other JAK inhibitors being studied in the setting of autoimmune disease include the JAK3 inhibitors VX-509 and WYE-151650, the JAK1/JAK2 inhibitors INCB028050 and INCB018424 and the JAK3/Syk inhibitor R348 (12, 13, 17, 24, 25) (ClinicalTrials.gov identifiers NCT00902486, NCT00550043 and NCT00789126).
Because CP-690,550 and the other inhibitors of this class target more than one JAK, their exact mode of action in the setting of inflammatory disease has not been resolved. Autoimmune diseases can be driven by CD4+ T cells that produce IFN-γ (Th1 cells), IL-17 (Th17 cells) or combinations of the two (26). The inflammatory response is supported by innate immune mechanisms that are also particularly relevant in autoimmunity (27). To begin to clarify the mechanism of JAK inhibition vis-à-vis the cognate cytokines that are blocked, we revisited the effects of CP-690,550 on adaptive and innate immune responses.
Materials and Methods
Mice
DBA/1J and C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME), and STAT1-deficient mice and littermate controls on a 129S6/SvEv background were from Taconic (Hudson, NY). Use of the animals in these studies was reviewed and approved by the Pfizer Institutional Animal Care and Use Committee or by the Institutional Animal Care and Use Committee of NIAMS. The animal care and use program at Pfizer is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. Experiments were performed according to the NIH guidelines for the use of live animals.
JAK inhibitor
CP-690,550 was prepared by Pfizer Research Laboratories or by the NIH Chemical Genomics Center and resuspended in 0.5% methylcellulose/0.025% Tween-20 (Sigma, St. Louis, MO) for in vivo studies or in DMSO for in vitro use.
T cell purification and differentiation
CD4+ T cells were purified by negative selection from spleens and lymph nodes of C57BL/6J, STAT1-deficient or wildtype 129S6/SvEv mice using magnetic cell separation technology (MACS, Miltenyi Biotec, Auburn, CA), and the naïve CD4+CD62L+CD44+CD25- population was sorted using a FACSAria II (BD Biosciences, San Jose, CA). Naïve T cells were activated by plate-bound anti-CD3/anti-CD28 (10 μg/ml) in fully supplemented RPMI 1640 medium containing 10% fetal calf serum, 2 mM glutamine, 100 IU/ml penicillin, 0.1 mg/ml streptomycin, HEPES buffer (all Invitrogen, Carlsbad, CA) and 2 μM β-mercaptoethanol (Sigma) or if indicated in serum-free medium (X-VIVO 20, Lonza, Walkersville, MD) for 3-4 days. During T cell activation cells were treated with the indicated concentrations of CP-690,550 dissolved in DMSO. Th1 cells were polarized in the presence of IL-12 (20 ng/ml) and anti-IL-4 (10 μg/ml), and Th2 cells in IL-4 (10 ng/ml) and anti-IFN-γ (10 μg/ml). For Th17 generation, CD4+ T cells were stimulated with the indicated combinations of IL-6 (20 ng/ml), TGF-β1 (0.5 ng/ml), IL-23 (50 ng/ml) or IL-1β (20 ng/ml, all R&D Systems, Minneapolis, MN) in the presence of anti-IFN-γ antibodies (10 μg/ml). TGF-β-signaling was neutralized by using anti-TGF-β1 and anti-TGF-β2 antibodies (5 μg/ml, R&D Systems). Following differentiation, cells were stimulated with PMA and ionomycin in the presence of brefeldin A for 4 h, fixed in 4% formyl saline and permeabilized with 0.1% saponin buffer prior to intracellular cytokine staining and flow cytometry using a FACSCalibur (BD Biosciences) with FlowJo software (Tree Star, Ashland, OR). The following antibodies were used: anti-IL-2-APC, -PE or -FITC, anti-IL-17A-PE, anti-IFN-γ-APC, -PE or -FITC, anti-IL13-PE (all BD Biosciences), anti-IL-17A-APC or -FITC, anti-IL-17F-AlexaFluor647, anti-IL-22-PE and anti-T-bet-AlexaFluor647 (all eBioscience, San Diego, CA). To study proliferation, T cells were labeled with 1 mM CFSE (Invitrogen) before culture.
Mouse arthritis model and LPS model
DBA/1J mice were immunized subcutaneously with 50 μg of chicken type II collagen (Western Institute for Biomedical Research, Salt Lake City, UT) emulsified in complete Freund's adjuvant (Sigma), and boosted 21 days later with 50 μg of the same antigen in incomplete Freund's adjuvant (Sigma). In therapeutic efficacy studies, disease was monitored beginning on day 45 and severity scored on a scale of 0-3 for each paw, as previously described (28). Mice were sorted into groups of equivalent severity, and dosed orally b.i.d. with vehicle or 50 mg/kg CP-690,550 beginning on day 48. For protein and gene expression analysis, groups of mice were bled by cardiac puncture, euthanized and both hind paws were excised and flash frozen 4 hours post-treatment on days 48, 49, 52 and 55. For histopathology and immunohistochemistry (IHC) analysis, fore and rear paws were harvested 4 and 12 hours post-treatment on day 48, and 4 hours post-treatment on days 49 and 55, and fixed in 10% neutral buffered formalin. In studies used to correlate JAK inhibition with efficacy, mice were orally administered vehicle or varying b.i.d. doses of CP-690,550 on days 22 through 56, disease was monitored beginning on day 42, and severity scored, as described. Efficacy was determined using the area under the curve of disease severity time-course for each dose.
DBA/1J or C57BL/6J mice were orally administered vehicle or a 5 mg/kg dose of CP-690,550, and one hour later injected i.p. with 10 μg of Salmonella typhosa LPS (Sigma). Plasma was collected before LPS administration, as well as, 1, 2 and 6 hours after LPS injection.
Plasma cytokine determination
Plasma cytokines were detected using Luminex-based murine multiplex assays (Millipore, St. Charles, MO) and Luminex 200 instrumentation (Luminex Corporation, Austin, TX) or with Meso Scale Discovery technology (Gaithersburg, MD). Serum amyloid A (SAA) was detected using a murine SAA ELISA (Invitrogen) following the manufacturer's protocol.
Knockdown of JAK1 and JAK3 with siRNA
Human CD4+ T cells were purified by negative selection from PBMC using magnetic cell separation technology. Cells were transfected with 1.4 μM SMARTpool siRNA for human JAK1 or JAK3 (Dharmacon, Lafayette, CO), or with a scrambled control using Nucleofector technology (Lonza). Transfected cells were stimulated with 100 ng/ml IL-6 or IL-7 for 15 minutes, and STAT phosphorylation was assessed by intracellular flow cytometry, as described below.
RNA isolation and gene expression
For cultured T cells, total RNA was isolated using the mirVana miRNA isolation kit (Applied Biosystems, Foster City, CA). Frozen paw tissue was powdered in a freezer mill and total RNA was prepared from each sample using TRIzol (Invitrogen). Relative gene expression levels were determined by quantitative RT-PCR using Taqman Gene Expression primer probe sets and ABI PRISM 7700 or 7900 Taqman systems (Applied Biosystems). The comparative threshold cycle method and internal controls (cyclophillin or β-actin) were used to normalize expression of target genes.
Western blotting
Freshly isolated CD4+ T cells were activated with plate-bound anti-CD3/CD28 and expanded with IL-2. Cells were washed and rested in fresh medium in the presence or absence of CP-690,550 for 30 minutes before addition of the indicated cytokines (100 ng/ml). Following stimulation, cells were lysed in Triton lysis buffer containing protease inhibitors. Equal amounts of total protein were separated by PAGE, transferred to nitrocellulose and blotted with antibodies recognizing actin (Millipore), pAKT and specific pSTATs (Cell Signaling Technology, MA or Invitrogen). IRDye800- (Rockland, Gilbertsville, PA) and AlexaFluor680- (Invitrogen) labeled secondary antibodies were used for detection and specific bands were visualized using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE).
STAT phosphorylation in Whole Blood
Heparinized blood from normal human donors was pre-incubated with CP-690,550 for 1 h prior to cytokine stimulation. Non-immunized DBA/1J mice were orally administered varying doses of CP-690,550 and blood was collected after 1 h. Alternatively, mice immunized to develop collagen-induced arthritis (CIA) were orally administered varying doses of CP-690,550 twice daily (b.i.d.) on days 22 through 56 and blood was collected 1 hour after the final dose. Whole blood leukocytes were surface labeled with FITC- and PE-labeled lineage-specific antibodies (BD Biosciences or eBioscience) in advance of cytokine stimulation. CD3 was used to identify human T cells, CD3 and CD8 for mouse T cell subsets, and CD11b and F4/80 for mouse monocytes. Blood was stimulated with or without cytokine (100 ng/ml IL-2, IL-4, IL-6, IL-7, IL-15 or IL-21, or 20 ng/ml GM-CSF) for 15-20 minutes, and activation was stopped by the addition of Lyse/Fix Buffer (BD Biosciences) following the manufacturer's protocol. Cells were washed, permeabilized in ice-cold Perm Buffer III (BD Biosciences) for 20-30 minutes and stained intracellularly with AlexaFluor647-conjugated pSTAT-specific mAb (BD Biosciences). Flow cytometric analysis was performed on a FACSCalibur. In human, STAT phosphorylation was assessed in CD3+ T cells; however, in mice, IL-15- and IL-6-driven STAT activation was examined within CD8+ T cells because that sub-population reproducibly yielded greater signal for quantitative evaluation of inhibition. AlexaFluor 647 geometric mean channel fluorescence derived from gated populations was used to determine percent of control stimulation by comparison of compound-treated and vehicle treated animals. Plasma from each sample was collected and CP-690,550 concentration determined by liquid chromatography/mass spectroscopy.
Histopathology and immunohistochemistry
Fixed paws were decalcified in Immunocal (Decal Chemical Corporation, Tallman, NY) for 7 days and paraffin-embedded. To assess general inflammation, 4 μm sections were stained with H&E, independently examined by two board-certified veterinary pathologists (TPL and ZAR) and scored semi-quantitatively, as previously described (29). For monocyte/macrophage IHC, tissue sections were treated with proteinase K (Dako, Carpinteria, CA) and blocked. Incubation with anti-F4/80 mAb (eBioscience) was followed by HRP-conjugated rat on mouse micro polymer (Biocare Medical, Concord, CA), diaminobenzidine (DAB) detection (Dako), and light hematoxylin counterstaining. Matched rat IgG was used as a negative control. T cell IHC utilized Borg high pH retrieval (Biocare Medical) followed by incubation with a rabbit anti-CD3 antibody (Accurate Chemical, Westbury, NY). HRP-conjugated secondary antibody incubation was followed by detection with DAB and hematoxylin counterstaining. Macrophage and T cell infiltration were scored semi-quantitatively, as previously described (30).
Results
CP-690,550 disrupts γc-chain cytokine signaling in CD4+ Th cells
CP-690,550 was originally designed as a JAK3 inhibitor and therefore was expected to interfere with γc-chain cytokine signaling. As shown in Fig. 1A, IL-2 induced the phosphorylation of STAT5 and AKT, and CP-690,550 inhibited both events very effectively. While it is well established that STATs are JAK substrates, the ability of CP-690,550 to inhibit AKT phosphorylation argues that this pathway is also downstream of JAKs. The CP-690,550 related compound PF-956980 has also been shown to inhibit IL-7-mediated AKT phosphorylation in human thymocytes (31). These results indicate that JAK inhibition interferes with both of the major pathways emanating from cytokine receptors.
Figure 1.
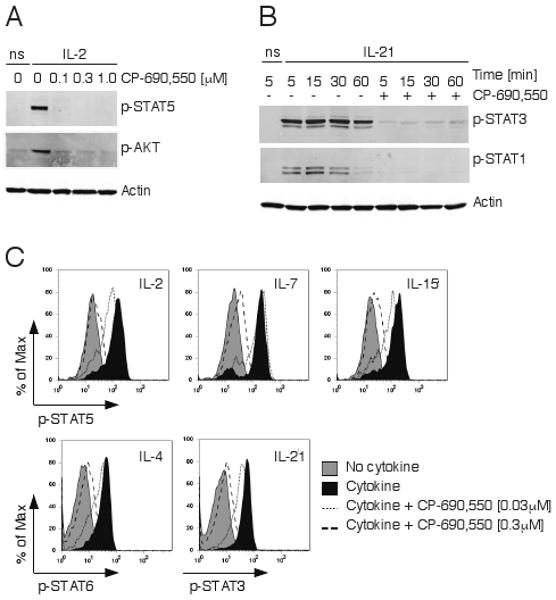
CP-690,550 inhibits γc-chain cytokine signaling in T cells. Equal numbers of mouse CD4+ T cells were pre-incubated with the indicated concentrations of CP-690,550 before stimulation with or without IL-2 for 15 minutes (ns, no cytokine stimulation). Activation of STAT5 and AKT were determined in cell lysates by immunoblotting with phospho-specific antibodies (A). Mouse CD4+ T cells were pre-incubated with or without 0.3 μM CP-690,550 and stimulated with IL-21 for the indicated time periods (ns, no cytokine stimulation). Activation of STAT3 and STAT1 was determined as in A (B). Human whole blood was pre-incubated with or without CP-690,550 before stimulation with the indicated cytokines for 15 minutes. The extent of specific STAT phosphorylation within the CD3+ T cell population was assessed by intracellular flow cytometry using phospho-specific antibodies. Results are representative of 8 separate experiments (C).
IL-21 is a critical immunoregulatory cytokine with important actions on T cells, B cells and NK cells (32), and it too uses the common γ chain (33). As expected, CP-690,550 interfered with IL-21 signaling in mouse CD4+ T cells, as shown by the inhibition of STAT3 and STAT1 phosphorylation (Fig. 1B). We also investigated the ability of CP-690,550 to inhibit γc-cytokine signaling in human T cells, and as depicted in Fig. 1C the inhibitor blocked STAT phosphorylation induced by IL-2, IL-4, IL-7, IL-15 and IL-21 with similar potencies. These results confirmed that CP-690,550 clearly affects signaling pathways downstream of JAK3-dependent γc-cytokine receptors in both mouse and human T cells. Since evidence from kinase binding assays have shown that CP-690,550 can also affect JAK members other than JAK3 (19), we next asked if the inhibitor also interfered with non-γc cytokine receptor signaling in T cells.
CP-690,550 disrupts non-γc chain cytokine signaling
To investigate the effects of CP-690,550 on JAK3-independent cytokine receptor signaling we stimulated CD4+ T cells with IL-6, a key inflammatory mediator in CIA and RA (34, 35). IL-6 signaling involves JAK1 in conjunction with JAK2 (2, 10, 11) and in human cells, TYK2 is also a contributor (36, 37). As depicted in Fig. 2A, CP-690,550 inhibited IL-6-driven phosphorylation of STAT3 and STAT1 in mouse CD4+ T cells. Similar results were obtained in human whole blood T cells (Fig. 2B). In both cases, STAT1 phosphorylation was more sensitive to tasocitinb than STAT3 phosphorylation. To further examine the role that particular JAK family members might play in STAT activation, we used RNA interference (Fig. 2C). Interestingly, inhibiting JAK1 expression in human CD4+ T cells completely suppressed IL-6-mediated STAT1 phosphorylation but had only a partial effect on STAT3 phosphorylation. In these studies, JAK1 inhibition corresponded to greater than 75% reduction in transcript copy number compared to control siRNA, as assessed by quantitative RT-PCR. JAK1 siRNA also abrogated IL-7-dependent STAT5 phosphorylation. As a control, JAK3 siRNA was included and while it had no effect on IL-6 signaling, JAK3 knockdown completely suppressed IL-7-mediated STAT5 phosphorylation, as would be expected. These data suggest that at least in T cells, both IL-6 activation of STAT1, as well as γc-cytokine activated STAT pathways are critically dependent on JAK1. Using the same approach we were unsuccessful in our attempts to suppress JAK2 or TYK2 expression using RNAi.
Figure 2.
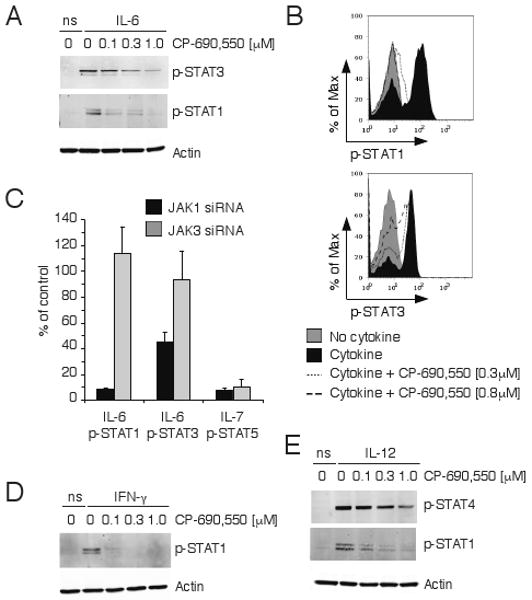
Inhibition of JAK-mediated IL-6, IFN-γ and IL-12 signaling by CP-690,550. Mouse CD4+ T cells (A) or human whole blood CD3+ T cells (B) were pre-incubated with the indicated concentrations of CP-690,550 and stimulated with IL-6 for 15 minutes (ns, no cytokine stimulation). Activation of STAT3 or STAT1 was determined by phospho-specific antibodies and immunoblotting (A) or flow cytometry (B). Cytokine-induced STAT activation following JAK1 or JAK3 siRNA transfection of human CD4+ T cells was assessed by flow cytometry and represents the percentage of STAT phosphorylation compared to control siRNA-transfected cells (C). Data represents the mean ± SEM from 3 separate experiments. Mouse CD4+ T cells were pre-incubated with CP-690,550 at the indicated concentrations before stimulation with IFN-γ for 30 minutes (D) or IL-12 for 15 minutes (E). STAT activation was determined as in A (ns, no cytokine stimulation).
Since CP-690,550 blocked IL-6 signaling, we next studied the effects of JAK inhibition on IFN-γ, a cytokine that also utilizes JAK1 and JAK2. The inhibitor potently blocked the IFN-γ-mediated phosphorylation of STAT1 in CD4+ T cells (Fig. 2D), further confirming that this inhibitor targets not only JAK3, but also JAK1 and/or JAK2. We then considered the possibility that CP-690,550 might also interfere with IL-12 signaling, which is dependent on JAK2 and TYK2. As shown in Fig. 2E, CP-690,550 blocked IL-12-driven phosphorylation of STAT1, but showed only modest suppression of STAT4 activation. Thus, CP-690,550 evidently interferes with multiple JAKs and hence multiple cytokine signaling pathways in T cells. Given these effects on lineage-promoting cytokine signaling, we considered the possibility that this JAK inhibitor would also affect the differentiation of Th cells to various fates.
CP-690,550 blocks the differentiation of Th2 and Th1 cells
Differentiation of Th2 cells is mediated by IL-4, a γc-cytokine that signals through JAK3 and JAK1. We therefore expected that CP-690,550 would effectively antagonize Th2 specification. Activating naïve Th cells with IL-4 and anti-CD3/anti-CD28 mAb, effectively generated GATA3 in cells that expressed high levels of IL-13 (Fig. 3, A and B). As expected, the expression of GATA3 and Th2 cytokines were inhibited by CP-690,550. However, TCR-mediated proliferation of Th cells was not affected. IL-2 production was enhanced, consistent with the recognized ability of IL-2 to limit its own production in Th2 and Th1 cells by activating STAT5 (38). It should be noted that adding a strong TCR stimulus (i.e. anti-CD3/anti-CD28) generates cells that produce little IL-4 protein (39). IL-4 mRNA expression was readily detectable in Th2 cells and abolished in the presence of CP-690,550 (data not shown). These findings are consistent with the efficacy of CP-690,550 in preclinical models of Th2-mediated allergic disease (40).
Figure 3.

CP-690,550 inhibits Th2 and Th1 differentiation. Naïve mouse CD4+ T cells were stimulated with anti-CD3/anti-CD28 antibodies in the presence or absence of the indicated concentrations of CP-690,550, using either Th2 polarizing conditions (IL-4 and anti-IFN-γ) or Th1 polarizing conditions (IL-12 and anti-IL-4). On day 3, the expression of lineage-associated transcription factors GATA3 and T-bet was determined by quantitative RT-PCR. Results were normalized using β-actin transcripts and represent relative expression ± SEM, *P<0.001, (A). After 3 days in culture, cells were activated with PMA/ionomycin. Th cell differentiation and proliferation were assessed by intracellular cytokine staining and CFSE dilution. Th2 cell polarization was evaluated by IL-13 expression (B), and Th1 cell polarization by IFN-γ expression (C). Data are representative of 3 separate experiments. Naïve CD4+ T cells from wild type (WT) or STAT1-deficient (STAT1 KO) mice were activated with IL-12 and anti-IL-4 for 3 days in the presence or absence of either CP-690,550 or anti-IFN-γ neutralizing antibodies. Intracellular staining of PMA/ionomycin activated cells shows IFN-γ and T-bet expression (D). Data are representative of 3-4 separate experiments with similar results.
Whereas Th2 cells are non-pathogenic in experimental autoimmune models like CIA, IFN-γ-producing Th1 cells and IL-17-producing Th17 cells have been reported to be responsible for destructive arthritis in mice and humans (41). Therefore we next studied the effects of CP-690,550 on Th1 cell differentiation. The inhibitor potently suppressed the expression of T-bet and the differentiation of IFN-γ-producing Th1 cells without suppressing cell proliferation (Fig. 3, A and C). As in Th2 cells, CP-690,550 also enhanced IL-2 production in Th1 cells. Th1 specification is initiated by IL-12 and STAT4 activation; however, IFN-γ amplifies T-bet and IFN-γ expression in Th1 cells through STAT1 activation (42). Of note, the inhibition of T-bet and IFN-γ expression by CP-690,550 was inhibited to the same extent as seen with IFN-γ neutralizing antibody or STAT1-deficient T cells (Fig. 3D). In view of this data and that presented in Fig. 2, D and E, we would argue that the primary mechanism by which CP-690,550 appears to inhibit Th1 differentiation is through inhibition of IFN-γ- and IL-12-mediated STAT1 signaling.
CP-690,550 inhibits the generation of IL-23-dependent Th17 cells
Although Th1 cells were originally thought to be the major mediators of immunopathogenesis, it is now increasingly recognized that Th17 cells are also important drivers of autoimmunity (41, 43, 44). Extensive work indicates that cells which selectively produce IL-17A, and not other cytokines, can arise from naïve CD4+ T cells in response to specific cytokine stimulation (45). IL-23 was initially thought to be important for driving Th17 differentiation; however, it was later argued that IL-6 in conjunction with TGF-β were responsible for the initial specification of mouse Th17 cells (46-48). In human cells, the requirement for TGF-β has been less clear, and lately the necessity for TGF-β in the mouse has been called into question (49, 50). We have recently shown that Th17 cells can be generated from naïve T cells in the absence of TGF-β signaling when using IL-23, IL-1β and IL-6 (51). Such IL-23-induced Th17 cells express a distinct repertoire of transcription factors, receptors and mediators and are more pathogenic in vivo. Since IL-2 and IFN-γ inhibit Th17 differentiation (45, 52) it was difficult to predict what the effect of CP-690,550 treatment might be on this lineage. We first polarized cells in the conventional manner, using TGF-β1 and IL-6. Under these conditions addition of CP-690,550 enhanced IL-17A and IL-2 production (Fig. 4A, upper row), consistent with the ability of the inhibitor to block feedback inhibition mediated by IL-2 (38, 52). Addition of anti-IL-2 had a similar effect to that of the inhibitor (data not shown). Interestingly, the IL-17A-inducing effect of CP-690,550 on Th17 differentiation was strictly dependent on the presence of TGF-β1, as neutralizing the biologic activity of this cytokine abolished IL-17A production (Fig. 4 A and B).
Figure 4.

Modulation of Th17 differentiation by CP-690,550. Sorted naïve CD4+ T cells were activated for 3 days with anti-CD3/anti-CD28 antibodies in the presence of IL-6 in combination with either TGF-β1, IL-23 or TGF-β1 and IL-23. CP-690,550 was added to the cultures at the indicated concentrations. Expression of IL-17A and IL-2 was determined by intracellular cytokine staining after stimulation with PMA/ionomycin. The effect of CP-690,550 on IL-17A expression was also studied in conditions, where TGF-β1 signaling was blocked by neutralizing antibodies. Representative flow cytometry plots (A) and summarized data of 3-4 separate experiments are shown (B). Expression of Rorc, Ahr and Il23r in cells activated as in A was determined by quantitative RT-PCR. Results were normalized using β-actin transcripts and represent fold increase of expression (mean ± SEM) compared to Th0 conditions (C).
IL-17A-producing cells generated by TGF-β1 and IL-6 can produce the anti-inflammatory cytokine IL-10 and are less pathogenic upon adoptive transfer than those generated in the presence of IL-23 (53). We have also recently shown that IL-17A-producing cells generated in the absence of TGF-β1 are more pathogenic than those generated in the presence of this immunoregulatory cytokine (51). We therefore examined the effects of CP-690,550 on Th17 cells induced in the absence of TGF-β, and found that in sharp contrast to its effects on IL-17A production induced by IL-6 and TGF-β1, the JAK inhibitor dramatically suppressed the differentiation of Th17 cells generated with IL-6 and IL-23 (Fig. 4, A and B). Under these conditions, neutralizing TGF-β1 did not affect the action of CP-690,550. In contrast, when adding TGF-β1 to the IL-6/IL-23 condition, the JAK inhibitor enhanced IL-17A expression (Fig. 4B). Interestingly, Rorc expression was inhibited in all conditions in the presence of CP-690,550, even if increased IL-17A expression was observed (Fig. 4C). CP-690,550 also blocked the expression of Ahr, which is primarily induced in the presence of TGF-β1 (51). As shown previously, Il23r expression was dramatically induced by IL-6 and IL-23 in the absence of TGF-β1 (51). Importantly, CP-690,550 completely abrogated the expression of IL-23R, which strictly depends on STAT3 activation (51) (Fig. 4C).
As previously shown, Th17 cells generated with IL-6, IL-1β and either TGF-β1 or IL-23 produce not only IL-17A but also IL-17F and IL-22, all of which can contribute to the pathogenicity of these cells (51). As shown in Fig. 5A, CP-690,550 effectively blocked the expression of IL-17A, IL-17F and IL-22 when Th17 cells were generated in the absence of TGF-β1. In contrast, the JAK inhibitor did not affect IL-17A or IL-17F expression when Th17 cells were induced in the presence of TGF-β1, but IL-22-production was affected. IL-21 is another key cytokine produced by both Th17 cells and follicular Th cells. Of note, its production was efficiently blocked by CP-690,550 regardless of how the Th17 cells were generated (Fig. 5B).
Figure 5.

CP-690,550 inhibits the differentiation of IL-23-induced Th17 cells and associated cytokines. Sorted naïve CD4+ T cells were activated in serum-free media in the absence or presence of CP-690,550 with anti-CD3/anti-CD28 antibodies and the combination of IL-6, IL-1β and either TGF-β1 or IL-23. After 4 days of culture cells were restimulated with PMA/ionomycin and the expression of IL-17A, IL-17F and IL-22 was assessed by flow cytometry (A). Expression of Il21 (B) and Tbx21 (C) was determined by quantitative RT-PCR. Results were normalized using β-actin transcripts and represent relative expression (mean ± SEM, B, *P<0.001, C, *P<0.01).
Increasingly, it has been recognized that Th cells that arise in the setting of autoimmunity can produce both IL-17A and IFN-γ; this has long been observed with human IL-17-producing cells (54, 55). Similarly, T cells found at sites of autoimmune lesions express both Rorγt and T-bet (55). Importantly, Th17 cells generated in the absence of TGF-β also express both Rorγt and T-bet (51), and CP-690,550 blocked T-bet expression in these cells (Fig. 5C). Thus, although CP-690,550 can enhance IL-17 production in TGF-β1-induced Th17 cells, it suppresses IL-17 production in pathogenic IL-23-induced Th17 cells, and also inhibits expression of Rorγt, T-bet and IL-23R.
CP-690,550 rapidly suppresses CIA, inflammatory biomarkers and STAT1-dependent gene expression
Therapies that selectively target T cell activation or differentiation can block CIA when used prophylactically during immunization, but may be less effective in established disease where innate immune mechanisms are also prominent (56). The efficacy of anti-TNF therapies in rheumatoid arthritis further underscores the role of innate cells in chronic arthritis. Therefore, we examined whether CP-690,550 could influence the course of established arthritis. Mice which had developed symptoms of arthritis by day 45 following collagen immunization were treated with CP-690,550 beginning on day 48. As shown in Fig. 6A, significant reduction in arthritis was apparent within 48 hours (day 50) of initiating treatment, and mice with continuous inhibitor treatment improved throughout the study. Significant expression of inflammatory mediators was noted in the plasma of vehicle-treated mice on day 48. Strikingly, many of these markers were reduced within 4 hours of initial CP-690,550 administration (Fig. 6B), suggesting a surprisingly rapid mode of action. It should be noted that in these studies we were unable to detect IL-17 in plasma. From our experience with the mouse CIA model, IL-17 is more readily detectable earlier in disease progression prior to development of arthritic symptoms (21). Since CP-690,550 potently suppressed STAT1 activation in T cells in response to IL-6, IFN-γ and IL-12, we sought to determine if the inhibitor would also reduce expression of canonical STAT1 target genes (57). Interestingly, prominent expression of many STAT1-responsive genes was evident in mice with arthritis and expression of these genes was rapidly suppressed by CP-690,550 as measured in the inflamed joints (Fig. 6C). Continuous CP-690,550 treatment further suppressed the expression of STAT1-responsive genes to near normal levels as disease resolved (data not shown).
Figure 6.
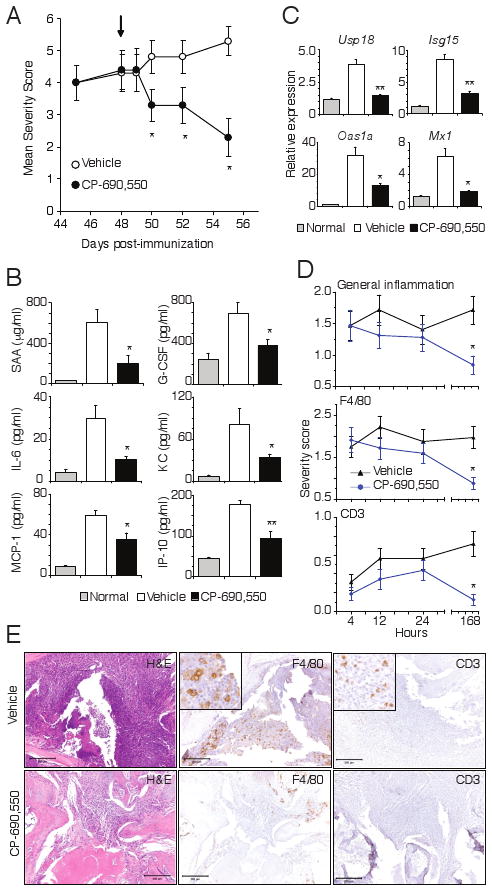
Rapid amelioration of established arthritis and inflammation by CP-690,550. Mice with fully developed CIA were orally administrated vehicle of CP-690,550 at 50 mg/kg b.i.d. beginning on day 48 post immunization (arrow). Data represent the mean ± SEM severity score from 8 mice in each group (A). Plasma concentrations of SAA, G-CSF, IL-6, CXCL1 (KC), CCL2 (MCP-1) and CXCL10 (IP-10) were measured 4 h after dosing on day 48 (B). Relative expression of STAT1-induced genes in paw tissue 4 h after dosing on day 48 (C). Usp18, ubiquitin specific peptidase 18; Isg15, interferon-stimulated gene 15; Oas1a, 2′-5′ oligoadenylate synthase 1A; Mx1, myxovirus resistance protein 1. Quantitative RT-PCR results were normalized to cyclophilin and represent mean ± SEM relative expression from non-diseased animals (normal), as well as vehicle and CP-690,550 treated mice with CIA (C). Cellular infiltration of inflamed paw tissue in mice treated as in A was assessed by histology and IHC at the indicated time points (D). Data represents the mean ± SEM score for all 4 paws from 8 mice per treatment group. For all panels, *P<0.01 and **P<0.001. Representative histology (H&E) and F4/80 or CD3 IHC of paw tissue sections collected 7 days after onset of treatment with vehicle or CP-690,550 (E).
To ensure that the observed transcript suppression was not the result of alterations of tissue infiltrating cells, we examined the inflammatory infiltration in paw tissues from mice treated by the same regimen. As shown in Fig. 6D using histopathology as well as macrophage and T cell IHC assessment of joint tissue, there was no decrease in inflammatory cell infiltrates within the first 24 hours of CP-690,550 treatment. These results confirmed the rapid suppression of STAT1 signaling pathways and demonstrated that the beneficial effects of JAK inhibition were not due to leukocyte depletion. Consistent with CP-690,550 effects on the arthritis severity score, histopathology and IHC assessment did, however, reveal significantly reduced inflammation after 7 days of treatment (Fig. 6, D and E).
Efficacy in mouse CIA correlates with inhibition of both JAK1 and JAK3
To assess the relative JAK inhibition by CP-690,550 in vivo, we measured STAT phosphorylation in ex vivo cytokine stimulated whole blood from mice, which had been treated orally with the inhibitor. For these experiments IL-6 signaling through STAT1 was used as a measure of JAK1/JAK2 activity, whereas IL-15 and GM-CSF stimulation of STAT5 were used to assess JAK1/JAK3 and JAK2 signaling pathways, respectively. As shown in Fig. 7A, mice administered a single oral dose of CP-690,550 had similar inhibition of JAK1/JAK2 and JAK1/JAK3 pathways, and significantly reduced suppression of the JAK2 pathway, confirming that in vivo CP-690,550 administration inhibits cytokine receptor signaling pathways activating STAT1 to a similar extent as γc-cytokine signaling pathways.
Figure 7.

CP-690,550 blocks innate JAK signaling in vivo. Mice were administered single doses of CP-690,550, whole blood was collected after 1 hour (∼Cmax) and stimulated ex vivo with the indicated cytokines. STAT phosphorylation expressed as percent of control cells (from non-treated mice) is plotted against plasma drug exposure in the same sample (A). Each data point represents an individual mouse from 1 of 4 separate experiments. EC50 corresponded to 470 nM, 273 nM and 6656 nM for JAK1/JAK2, JAK1/JAK3 and JAK2 inhibition, respectively. JAK inhibition following chronic dosing in mice induced with CIA (B). Data respresent the mean ± SEM JAK inhibition or mean AUC for efficacy from 1 of 2 separate studies involving 5 mice at each dose. Plasma cytokine levels were assessed 1, 2 and 6 hours after a 10 μg i.p. administration of LPS to DBA/1J mice pre-treated for 1 h with or without 5 mg/kg CP-690,550 (C). Time courses of TNF, IL-6, IL-12p70, IL-10, IFN-γ and IL-1β production in plasma are shown. Data represent plasma cytokine concentrations (mean ± SEM) from 1 of 2 separate studies with similar results involving 8 mice at each dose.
The same method was used to evaluate CP-690,550 inhibition of γc-cytokine and STAT1-dependent signaling pathways in the context of inflammatory disease. Cytokine signaling was examined in mice which had been treated orally with various doses of CP-690,550 for 5 weeks as treatment for CIA (Fig. 7B), and the results revealed that inhibition of both JAK1/JAK2 and JAK1/JAK3 pathways correlated with efficacy, while there was little or no inhibition of JAK2 at therapeutic doses. These results suggested that the anti-inflammatory activity of CP-690,550 is mediated by its potent inhibition of both JAK1 and JAK3 activity.
CP-690,550 rapidly blocks innate responses in vivo
The rapid suppression of inflammatory cytokines and STAT1-dependent gene expression observed in mice with CIA following CP-690,550 treatment suggested that in addition to suppressing T cell function the JAK inhibitor might also be affecting innate immune responses. To investigate this possibility, we examined the effect of CP-690,550 on the acute response to LPS in vivo, a model known to be dependent upon IFN-γ and STAT1 (58, 59). We found that a single dose of the JAK inhibitor suppressed TNF and IL-6 production along with other inflammatory cytokines (Fig. 7C), confirming a rapid anti-inflammatory mode of action. Interestingly, the production of IL-10 was enhanced by the treatment, consistent with the reported STAT1-mediated repression of this anti-inflammatory cytokine (60). Collectively, these data indicated that the immunosuppressive effects of CP-690,550 appear to be mediated by blockade of innate, as well as, adaptive immune responses.
Discussion
CP-690,550 is currently being studied in a variety of autoimmune diseases. In this study, we show that the inhibitor blocks signaling by JAK3-dependent γc cytokine receptors, as well as by other cytokine receptors that signal through JAK1. Accordingly, we found that CP-690,550 interfered with Th1 and Th2 differentiation, and also impaired the production of inflammatory Th17 cells generated in response to IL-1β, IL-6 and IL-23. In contrast, the JAK inhibitor enhanced production of IL-17A in cells cultured with IL-6 and TGF-β1. These effects were associated with amelioration of murine arthritis, which correlated with reduced expression of STAT1-dependent genes. Furthermore, CP-690,550 also blocked cytokine production in a sepsis model suggesting that the mechanism of action of this drug involves blocking the action of cytokines during innate and adaptive responses.
Despite its advanced stage of clinical development, the mode of action by which CP-690,550 exerts efficacy in RA and other autoimmune settings remains unresolved (15, 22, 23). In contrast to its activity against isolated kinases, CP-690,550 demonstrates functional specificity for JAK1 and JAK3 over other JAK family members in cells, although the basis of this apparent discrepancy has not been determined (18, 21). Since many of the cytokines involved in RA and other autoimmune diseases signal through receptors associated with JAKs, the question arises as to how the effects of CP-690,550 relate to the apparent efficacy of the drug in the setting of autoimmune disease. A central component of the pathophysiology of RA and psoriasis is the action of autoreactive T cells and the inflammatory cytokines that act upon them (61-63). As was expected, CP-690,550 potently inhibited γc-cytokine signaling pathways in the current studies by targeting JAK1 and JAK3 in T cells. Similar results have been observed in JAK1 and JAK3 deficient cells (2, 10) and with JAK1-selective inhibitors (unpublished results) suggesting that blockade of either or both of these kinases can modulate γc-cytokine receptor signals. A recent study has also demonstrated that a selective JAK3 inhibitor, WYE-151650, is effective in collagen-induced arthritis (24).
Neither the clinical efficacy of CP-690,550 nor the potential efficacy of other JAK inhibitors is likely to be explained by inhibition of γc-cytokine receptor signaling alone. By such a mechanism, the differentiation of naive T cells to Th2 effector cells would be inhibited, but Th2 cells are likely not relevant to the pathogenesis of CIA in mice or RA and psoriasis in humans (41, 51). Surprisingly, CP-690,550 also prevented Th1 differentiation. While previous observations have indicated that cellular JAK3-deficiency or inhibition of JAK3 can suppress Th1 differentiation (64), our data suggest a different mechanism since CP-690,550 suppressed expression of the Th1-associated transcription factor T-bet. Th1 differentiation is driven by IL-12 and IFN-γ and by the activation of STAT1 and T-bet (42, 65). Our results indicate that CP-690,550 has only a modest effect on IL-12-induced STAT4 activation while profoundly inhibiting STAT1 activation in T cells induced either by IL-12 or IFN-γ. Indeed, the inhibition of IFN-γ signaling alone could likely account for the observed Th1 suppression as demonstrated by the effect of anti-IFN-γ neutralizing antibodies. The consequences of CP-690,550 treatment on Th1 differentiation and STAT1 signaling could also explain efficacy of the inhibitor in a mouse Graft-versus-Host Disease (GVHD) model, where Th1 responses were limited by CP-690,550 without affecting cell proliferation (66).
While blocking Th1 responses can be highly efficient in GVHD and transplant rejection, this mechanism alone would likely be less successful in autoimmune diseases in which Th17 cells also play a major role. Thus, using inhibitors that target not only JAK3 but also JAK1 or JAK2 and subsequently affect the differentiation of Th1 as well as Th17 cells could be of benefit in autoimmune settings. The generation of Th17 cells is regulated by multiple factors. While IL-6 and TGF-β1 can efficiently induce IL-17 production, IL-6 together with IL-23 and IL-1β, in the absence of TGFβ-1, can also induce IL-17 in naïve Th cells (50, 51, 67). Indeed, we have shown recently that Th17 cells generated in the absence of TGF-β are more pathogenic in vivo than those generated in the presence of this cytokine (51). Moreover, we have found that the balance between STAT3 and STAT5 activation can have opposing regulatory effects on IL-17 expression (68). Results from the present studies demonstrate that CP-690,550, most likely by inhibiting STAT5, increases IL-17 expression when Th17 cells are generated with TGF-β and IL-6. In contrast, in the absence of TGF-β-signaling CP-690,550 blocked IL-17 expression.
While the regulation of IL-17A and IL-17F expression are more complex (68), the expression of IL-23R and IL-22 are strictly dependent on STAT3 activation (51). We show in these studies that CP-690,550 interferes with IL-23 action by blocking upregulation of its receptor and subsequent IL-17 induction. Moreover, CP-690,550 inhibited IL-23R expression under either Th17 condition. Similarly, the JAK inhibitor abrogated STAT3-mediated IL-22 and IL-21 expression in Th17 cells, and also inhibited RORγt and T-bet expression. Thus, CP-690,550 potently suppresses the generation of pathogenic Th17 cells with an IL-23/STAT3-signature. Inhibitory effects on Th17-associated cytokines have also been suggested for the JAK1/JAK2 inhibitor INCB028050 (25).
This mode of action of CP-690,550 may be of interest in a number of autoimmune diseases where interfering with IL-23 signaling attenuates disease (41, 69-72). Thus, it may very well be that a clinically important action of CP-690,550 is to block the combined actions of IL-23.
On the other hand, IL-6 has wide-ranging biological activities in various target cells. In addition to promoting Th17 differentiation, it regulates immune responses, the acute phase reaction, hematopoiesis, and bone metabolism (73, 74). IL-6-deficient mice are protected from experimental autoimmune diseases such as CIA (34). Additionally, elevated serum IL-6 levels have been observed in patients with inflammatory diseases such as RA and Crohn's disease, and tocilizumab, a humanized anti-IL-6R antibody that blocks IL-6 signaling, has shown clinical efficacy in these indications, ameliorating inflammation and normalizing acute phase protein levels (75). Our data indicate that CP-690,550 interferes with production of IL-6 and also blocks IL-6 signaling, which could be explained by effects of the inhibitor on JAK1 and/or JAK2. Thus, an additional mechanism underlying CP-690,550 efficacy in RA is likely mediated through effects on IL-6.
We were surprised by the rapid effects of CP-690,550 on established disease in the mouse CIA model. Indeed, effects of the inhibitor were observable within hours of initiating treatment. Despite the inhibitory consequences of CP-690,550 on Th cell differentiation, it seemed unlikely that this could induce such rapid effects in vivo. Rather, the rapid suppression of inflammatory responses suggested that blockade of innate immune mechanisms might represent part of the salutatory effects of JAK inhibition. This led us to examine the efficacy of the JAK inhibitor in the sepsis model. Importantly, we found that CP-690,550 had no direct effect on TLR4 signaling in vitro, as we did not observe inhibition of LPS induced TNF or IL-6 production from human PBMC (data not shown). Rather, the suppression of acute TNF responses in vivo following LPS administration is more consistent with inhibition of IFN-γ signaling by blockade of JAK1, since both STAT1-deficient and IFN-γR-deficient mice are resistant to LPS-induced endotoxemic shock (58, 59,). In contrast, IFN-γ priming of macrophages has been shown to enhance both LPS-stimulated TNF production in vivo (76) and STAT1 expression (77), and it has been suggested that IFN-γ activation of STAT1 may alter signaling pathways downstream of anti-inflammatory cytokines such as IL-10 or TGF-β, leading to antagonism of their suppressive function (78). If this were the case, CP-690,550 suppression of STAT1-responsive genes could override the effect of priming. IL-10 responses to LPS are enhanced in mice made deficient for IFN-α/β/γ or STAT1 (60), suggesting that STAT1 is a negative regulator of IL-10 gene expression. Our observations were consistent with this hypothesis, as we observed enhanced IL-10 levels in LPS treated mice given the JAK inhibitor. Another possible contribution to CP-690,550 suppression of LPS responses in vivo could involve blockade of IL-15 signaling since both IL-15 deficiency and anti-IL-15 neutralizing antibody have been shown to suppress LPS-induced endotoxemia in vivo (79). While there is no doubt that IL-15 signaling is potently inhibited by CP-690,550, this mechanism can not completely explain the results from the current study since blockade of IL-15 signaling would not be expected to affect IL-10 in this model.
The simultaneous control of signaling pathways involved in innate and adaptive immune responses by CP-690,550 may explain why this JAK inhibitor has produced rapid clinical improvement in RA patients who have previously failed other disease-modifying anti-rheumatic drug therapies or TNF antagonists (22). Based on the present data, it appears that the efficacy of CP-690,550 is likely based on its ability to block multiple cytokines and break the cycle of inflammation. Clearly, it will be important to try to understand which critical cytokines are blocked in humans undergoing JAK inhibitor treatment and the extent to which signaling is abrogated. As such, our findings have implications for the possible utility of CP-690,550 in a wide variety of inflammatory disorders.
Acknowledgments
This study was funded by NIAMS and Pfizer, Inc.
References
- 1.Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26:6724–6737. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]
- 2.Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 4.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 5.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O'Shea JJ, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377:65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–683. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 7.Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 8.Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, Yamashita M, Numata A, Takase K, Kobayashi S, Shibata S, Asano Y, Gondo H, Sekiguchi K, Nakayama K, Nakayama T, Okamura T, Okamura S, Niho Y. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13:561–571. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 9.Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev. 2005;203:38–47. doi: 10.1111/j.0105-2896.2005.00227.x. [DOI] [PubMed] [Google Scholar]
- 10.Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson EM, Jr, Schreiber RD. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–383. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 11.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 12.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–142. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghoreschi K, Laurence A, O'Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. 2009;10:356–360. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardanani A. JAK2 inhibitor therapy in myeloproliferative disorders: rationale, preclinical studies and ongoing clinical trials. Leukemia. 2008;22:23–30. doi: 10.1038/sj.leu.2404948. [DOI] [PubMed] [Google Scholar]
- 15.van Gurp E, Weimar W, Gaston R, Brennan D, Mendez R, Pirsch J, Swan S, Pescovitz MD, Ni G, Wang C, Krishnaswami S, Chow V, Chan G. Phase 1 dose-escalation study of CP-690 550 in stable renal allograft recipients: preliminary findings of safety, tolerability, effects on lymphocyte subsets and pharmacokinetics. Am J Transplant. 2008;8:1711–1718. doi: 10.1111/j.1600-6143.2008.02307.x. [DOI] [PubMed] [Google Scholar]
- 16.Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, Kennedy D, Estrov Z, Cortes J, Verstovsek S. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010;115:1131–1136. doi: 10.1182/blood-2009-10-246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R, Tefferi A. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, McCurdy SP, Kudlacz EM, Conklyn MJ, Elliott EA, Koslov ER, Fisher MB, Strelevitz TJ, Yoon K, Whipple DA, Sun J, Munchhof MJ, Doty JL, Casavant JM, Blumenkopf TA, Hines M, Brown MF, Lillie BM, Subramanyam C, Shang-Poa C, Milici AJ, Beckius GE, Moyer JD, Su C, Woodworth TG, Gaweco AS, Beals CR, Littman BH, Fisher DA, Smith JF, Zagouras P, Magna HA, Saltarelli MJ, Johnson KS, Nelms LF, Des Etages SG, Hayes LS, Kawabata TT, Finco-Kent D, Baker DL, Larson M, Si MS, Paniagua R, Higgins J, Holm B, Reitz B, Zhou YJ, Morris RE, O'Shea JJ, Borie DC. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- 19.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 20.Changelian PS, Moshinsky D, Kuhn CF, Flanagan ME, Munchhof MJ, Harris TM, Whipple DA, Doty JL, Sun J, Kent CR, Magnuson KS, Perregaux DG, Sawyer PS, Kudlacz EM. The specificity of JAK3 kinase inhibitors. Blood. 2008;111:2155–2157. doi: 10.1182/blood-2007-09-115030. [DOI] [PubMed] [Google Scholar]
- 21.Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, Gross CJ, Dowty ME, Ramaiah SK, Hirsch JL, Saabye MJ, Barks JL, Kishore N, Morris DL. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm (Lond) 2010;7:41. doi: 10.1186/1476-9255-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, Krishnaswami S, Burgos-Vargas R, Wilkinson B, Zerbini CA, Zwillich SH. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 23.Boy MG, Wang C, Wilkinson BE, Chow VF, Clucas AT, Krueger JG, Gaweco AS, Zwillich SH, Changelian PS, Chan G. Double-blind, placebo-controlled, dose-escalation study to evaluate the pharmacologic effect of CP-690,550 in patients with psoriasis. J Invest Dermatol. 2009;129:2299–2302. doi: 10.1038/jid.2009.25. [DOI] [PubMed] [Google Scholar]
- 24.Lin TH, Hegen M, Quadros E, Nickerson-Nutter CL, Appell KC, Cole AG, Shao Y, Tam S, Ohlmeyer M, Wang B, Goodwin DG, Kimble EF, Quintero J, Gao M, Symanowicz P, Wrocklage C, Lussier J, Schelling SH, Hewet AG, Xuan D, Krykbaev R, Togias J, Xu X, Harrison R, Mansour T, Collins M, Clark JD, Webb ML, Seidl KJ. Selective functional inhibition of JAK-3 is sufficient for efficacy in collagen-induced arthritis in mice. Arthritis Rheum. 2010;62:2283–2293. doi: 10.1002/art.27536. [DOI] [PubMed] [Google Scholar]
- 25.Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, Metcalf B, Friedman SM, Vaddi K. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–5307. doi: 10.4049/jimmunol.0902819. [DOI] [PubMed] [Google Scholar]
- 26.Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221. doi: 10.1111/j.1749-6632.2009.05133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang KS, Burow A, Kurrer M, Lang PA, Recher M. The role of the innate immune response in autoimmune disease. J Autoimmun. 2007;29:206–212. doi: 10.1016/j.jaut.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 28.Milici AJ, Kudlacz EM, Audoly L, Zwillich S, Changelian P. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther. 2008;10:R14. doi: 10.1186/ar2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bendele A, McAbee T, Sennello G, Frazier J, Chlipala E, McCabe D. Efficacy of sustained blood levels of interleukin-1 receptor antagonist in animal models of arthritis: comparison of efficacy in animal models with human clinical data. Arthritis Rheum. 1999;42:498–506. doi: 10.1002/1529-0131(199904)42:3<498::AID-ANR15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.LaBranche TP, Hickman-Brecks CL, Meyer DM, Storer CE, Jesson MI, Shevlin KM, Happa FA, Barve RA, Weiss DJ, Minnerly JC, Racz JL, Allen PM. Characterization of the KRN cell transfer model of rheumatoid arthritis (KRN-CTM), a chronic yet synchronized version of the K/BxN mouse. Am J Pathol. 2010;177:1388–1396. doi: 10.2353/ajpath.2010.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson SE, Shah N, Bajer AA, LeBien TW. IL-7 activates the phosphatidylinositol 3-kinase/AKT pathway in normal human thymocytes but not normal human B cell precursors. J Immunol. 2008;180:8109–8117. doi: 10.4049/jimmunol.180.12.8109. [DOI] [PubMed] [Google Scholar]
- 32.Ettinger R, Kuchen S, Lipsky PE. Interleukin 21 as a target of intervention in autoimmune disease. Ann Rheum Dis. 2008;67 3:iii83–86. doi: 10.1136/ard.2008.098400. [DOI] [PubMed] [Google Scholar]
- 33.Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, Schrader S, Burkhead S, Heipel M, Brandt C, Kuijper JL, Kramer J, Conklin D, Presnell SR, Berry J, Shiota F, Bort S, Hambly K, Mudri S, Clegg C, Moore M, Grant FJ, Lofton-Day C, Gilbert T, Rayond F, Ching A, Yao L, Smith D, Webster P, Whitmore T, Maurer M, Kaushansky K, Holly RD, Foster D. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 34.Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–997. doi: 10.1016/S0140-6736(08)60453-5. [DOI] [PubMed] [Google Scholar]
- 36.Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, Takada H, Hara T, Kawamura N, Ariga T, Kaneko H, Kondo N, Tsuge I, Yachie A, Sakiyama Y, Iwata T, Bessho F, Ohishi T, Joh K, Imai K, Kogawa K, Shinohara M, Fujieda M, Wakiguchi H, Pasic S, Abinun M, Ochs HD, Renner ED, Jansson A, Belohradsky BH, Metin A, Shimizu N, Mizutani S, Miyawaki T, Nonoyama S, Karasuyama H. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Watford WT, O'Shea JJ. Human tyk2 kinase deficiency: another primary immunodeficiency syndrome. Immunity. 2006;25:695–697. doi: 10.1016/j.immuni.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Villarino AV, Tato CM, Stumhofer JS, Yao Z, Cui YK, Hennighausen L, O'Shea JJ, Hunter CA. Helper T cell IL-2 production is limited by negative feedback and STAT-dependent cytokine signals. J Exp Med. 2007;204:65–71. doi: 10.1084/jem.20061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao X, Constant S, Jorritsma P, Bottomly K. Strength of TCR signal determines the costimulatory requirements for Th1 and Th2 CD4+ T cell differentiation. J Immunol. 1997;159:5956–5963. [PubMed] [Google Scholar]
- 40.Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol. 2008;582:154–161. doi: 10.1016/j.ejphar.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 41.van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:549–553. doi: 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- 42.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O'Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 45.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 46.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 47.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 48.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 50.Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL, Van Kaer L, Shi Y, Das G. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206:2407–2416. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 53.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 54.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol. 2009;5:325–331. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 55.Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, Duquette P, Prat A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 56.Drexler SK, Kong PL, Wales J, Foxwell BM. Cell signalling in macrophages, the principal innate immune effector cells of rheumatoid arthritis. Arthritis Res Ther. 2008;10:216. doi: 10.1186/ar2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 58.Car BD, Eng VM, Schnyder B, Ozmen L, Huang S, Gallay P, Heumann D, Aguet M, Ryffel B. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J Exp Med. 1994;179:1437–1444. doi: 10.1084/jem.179.5.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamezaki K, Shimoda K, Numata A, Matsuda T, Nakayama K, Harada M. The role of Tyk2, Stat1 and Stat4 in LPS-induced endotoxin signals. Int Immunol. 2004;16:1173–1179. doi: 10.1093/intimm/dxh118. [DOI] [PubMed] [Google Scholar]
- 60.VanDeusen JB, Shah MH, Becknell B, Blaser BW, Ferketich AK, Nuovo GJ, Ahmer BM, Durbin J, Caligiuri MA. STAT-1-mediated repression of monocyte interleukin-10 gene expression in vivo. Eur J Immunol. 2006;36:623–630. doi: 10.1002/eji.200535241. [DOI] [PubMed] [Google Scholar]
- 61.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 62.Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, van der Zee R, Biedermann T, Prinz J, Mack M, Mrowietz U, Christophers E, Schlondorff D, Plewig G, Sander CA, Rocken M. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003;9:40–46. doi: 10.1038/nm804. [DOI] [PubMed] [Google Scholar]
- 63.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 64.Shi M, Lin TH, Appell KC, Berg LJ. Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity. 2008;28:763–773. doi: 10.1016/j.immuni.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 66.Park HB, Oh K, Garmaa N, Seo MW, Byoun OJ, Lee HY, Lee DS. CP-690550, a Janus Kinase Inhibitor, Suppresses CD4+ T-Cell-Mediated Acute Graft-Versus-Host Disease by Inhibiting the Interferon-gamma Pathway. Transplantation. 2010 doi: 10.1097/TP.0b013e3181f24e59. [DOI] [PubMed] [Google Scholar]
- 67.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O'Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–1674. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 71.Tato CM, Cua DJ. Reconciling id, ego, and superego within interleukin-23. Immunol Rev. 2008;226:103–111. doi: 10.1111/j.1600-065X.2008.00715.x. [DOI] [PubMed] [Google Scholar]
- 72.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O'Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–119. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 74.Fonseca JE, Santos MJ, Canhao H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538–542. doi: 10.1016/j.autrev.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 75.Ohsugi Y, Kishimoto T. The recombinant humanized anti-IL-6 receptor antibody tocilizumab, an innovative drug for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2008;8:669–681. doi: 10.1517/14712598.8.5.669. [DOI] [PubMed] [Google Scholar]
- 76.Williams JG, Jurkovich GJ, Hahnel GB, Maier RV. Macrophage priming by interferon gamma: a selective process with potentially harmful effects. J Leukoc Biol. 1992;52:579–584. doi: 10.1002/jlb.52.6.579. [DOI] [PubMed] [Google Scholar]
- 77.Hu X, Park-Min KH, Ho HH, Ivashkiv LB. IFN-gamma-primed macrophages exhibit increased CCR2-dependent migration and altered IFN-gamma responses mediated by Stat1. J Immunol. 2005;175:3637–3647. doi: 10.4049/jimmunol.175.6.3637. [DOI] [PubMed] [Google Scholar]
- 78.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fehniger TA, Yu H, Cooper MA, Suzuki K, Shah MH, Caligiuri MA. Cutting edge: IL-15 costimulates the generalized Shwartzman reaction and innate immune IFN-gamma production in vivo. J Immunol. 2000;164:1643–1647. doi: 10.4049/jimmunol.164.4.1643. [DOI] [PubMed] [Google Scholar]